
Human iPSC Modeling of Genetic Febrile Seizure Reveals Aberrant Molecular and Physiological Features Underlying an Impaired Neuronal Activity

 , , , , , , , , , and
, , , , , , , , , and
Abstract
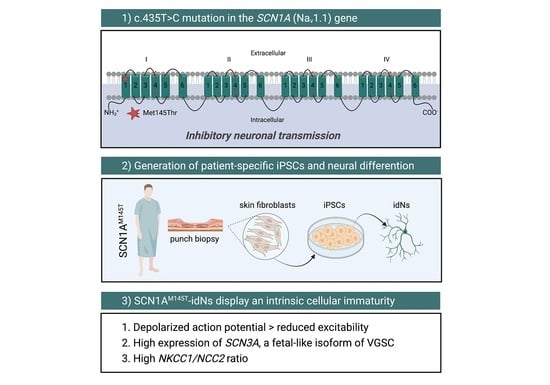
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
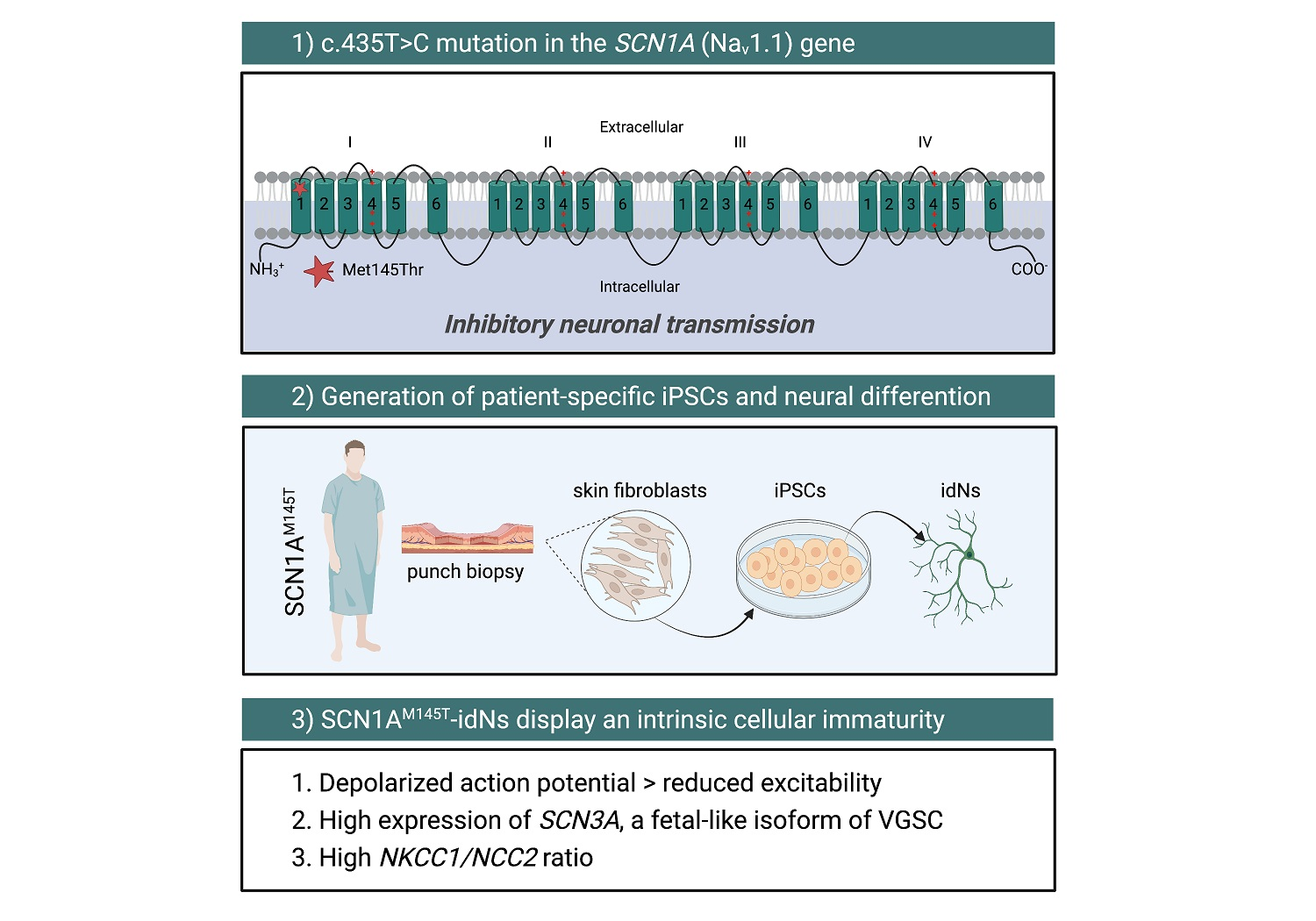
2.1. Clinical Features and iPSCs Generation from a Patient with Missense Mutation in the SCN1A Gene
2.2. Generation of iPSCs-Derived Neurons (idNs)
2.3. RNA Extraction and qRT-PCR Analysis
2.4. cDNA Sequencing
2.5. Western Blot Analysis
2.6. Immunofluorescence
2.7. Patch-Clamp Recordings on idNs
2.8. Membrane Preparation from idNs
2.9. Xenopus Laevis Oocytes Injection and Voltage-Clamp Recordings
2.10. Statistical Analysis
3. Results
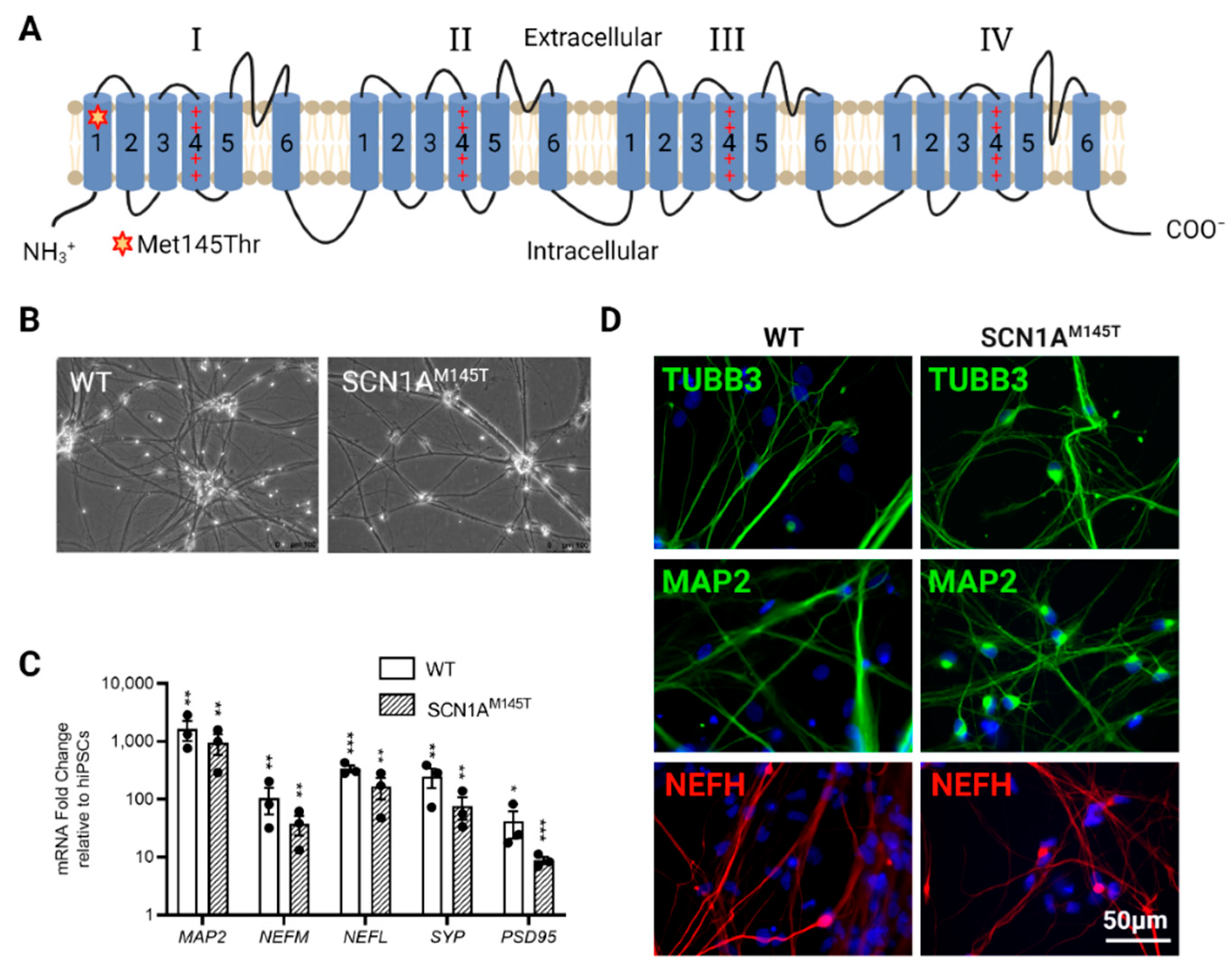
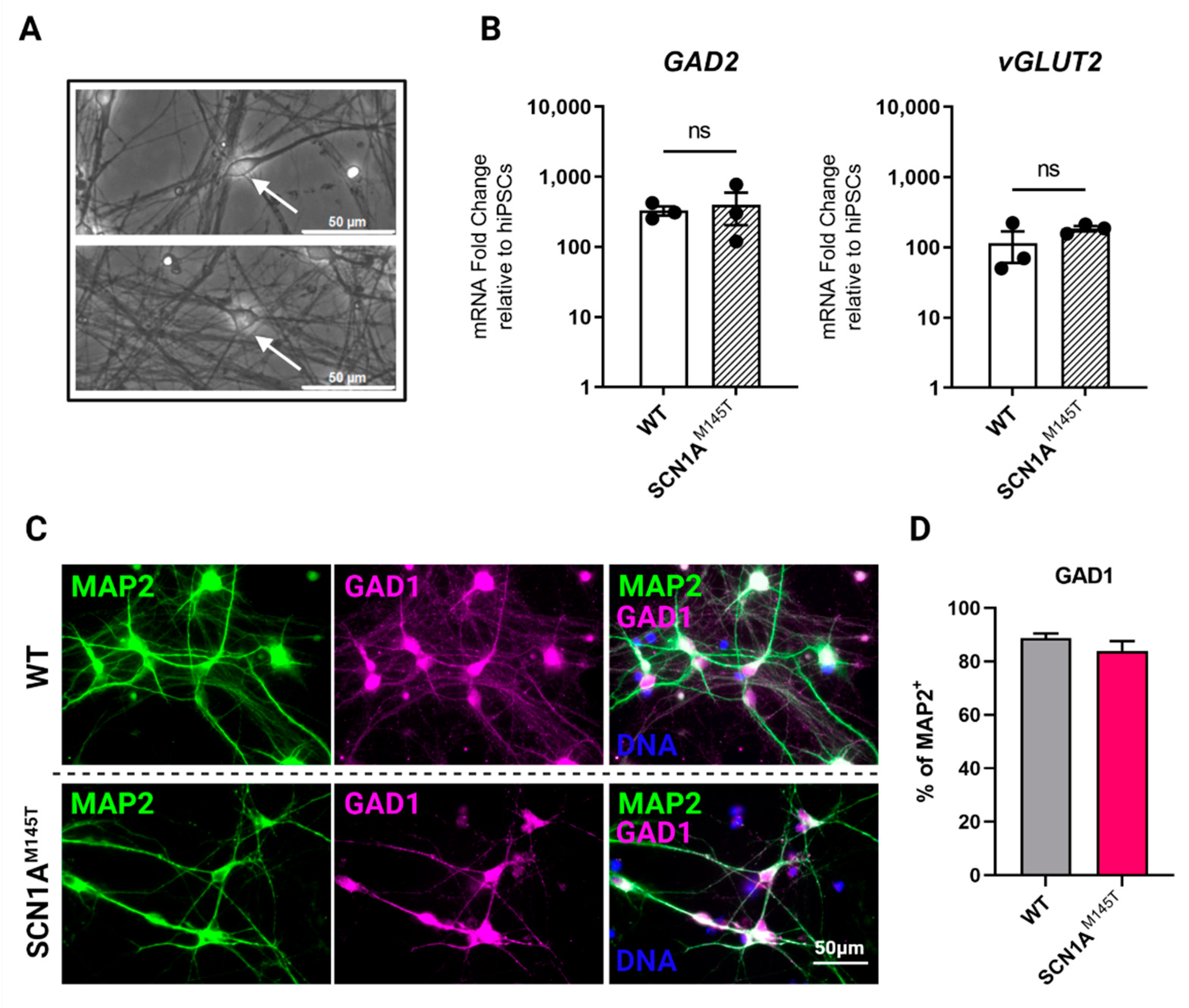
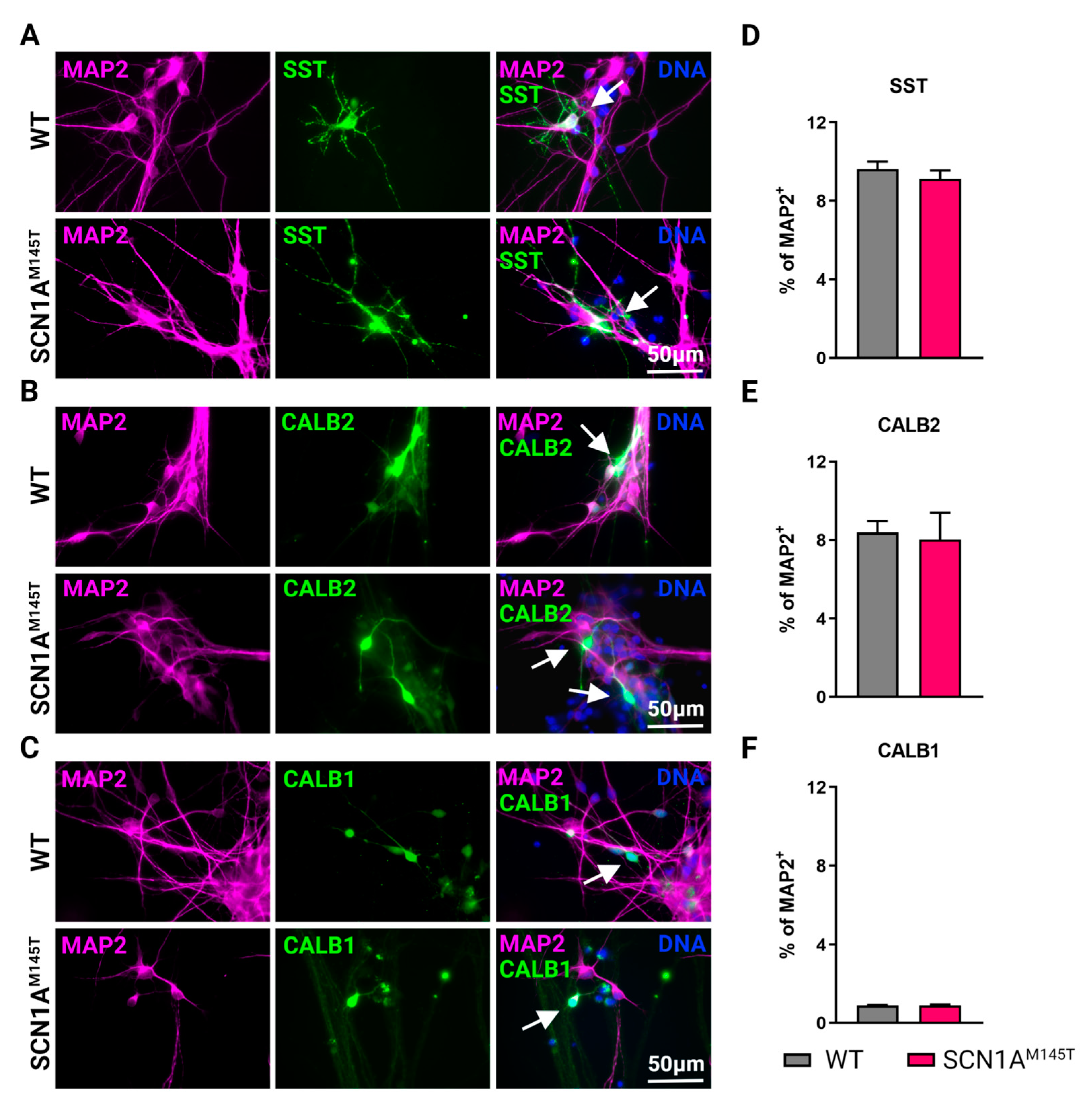
3.1. Generation of SCN1AM145T and Control idNs



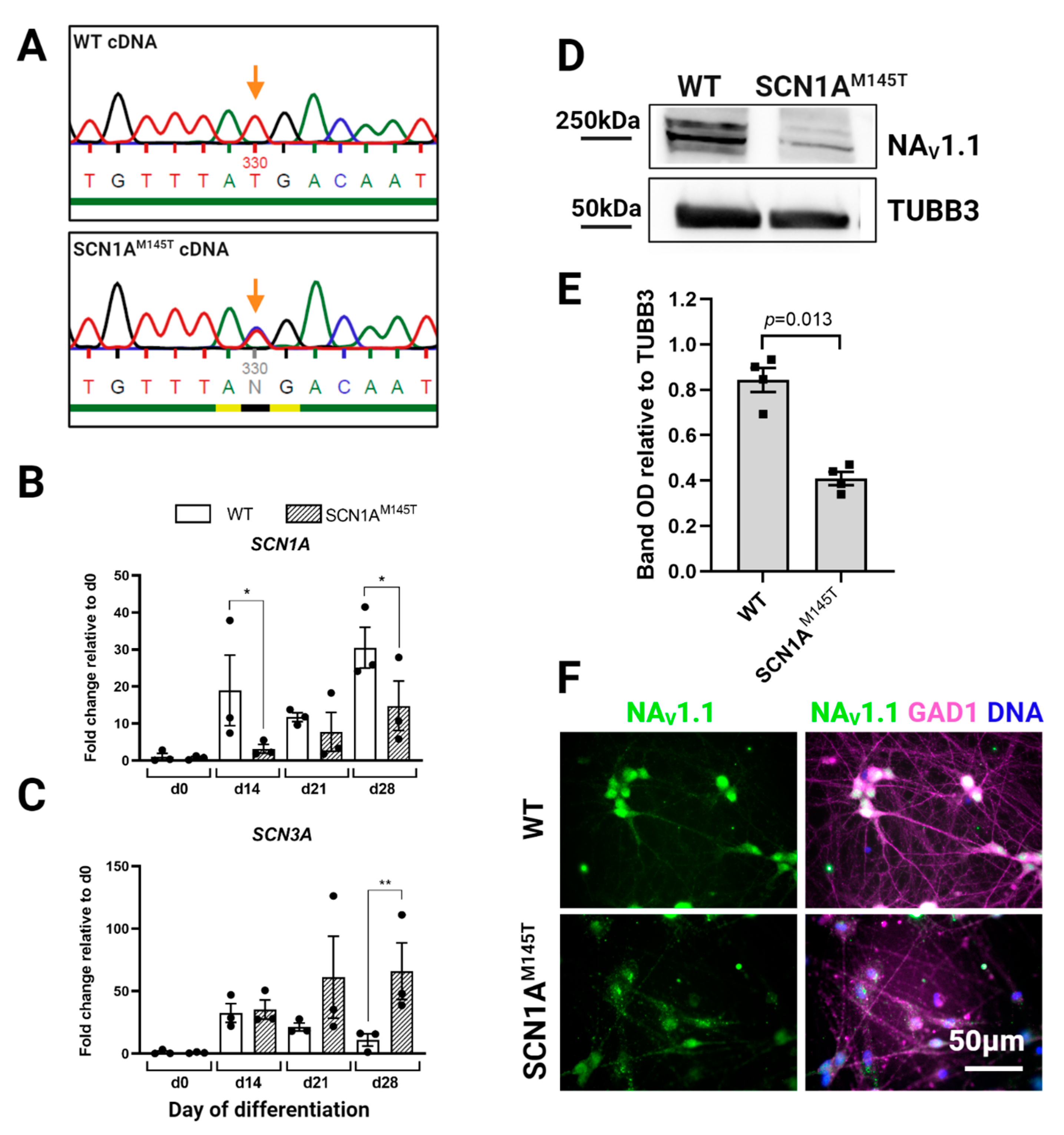
3.2. Expression of SCN1A Gene and NaV1.1 Protein in idNs
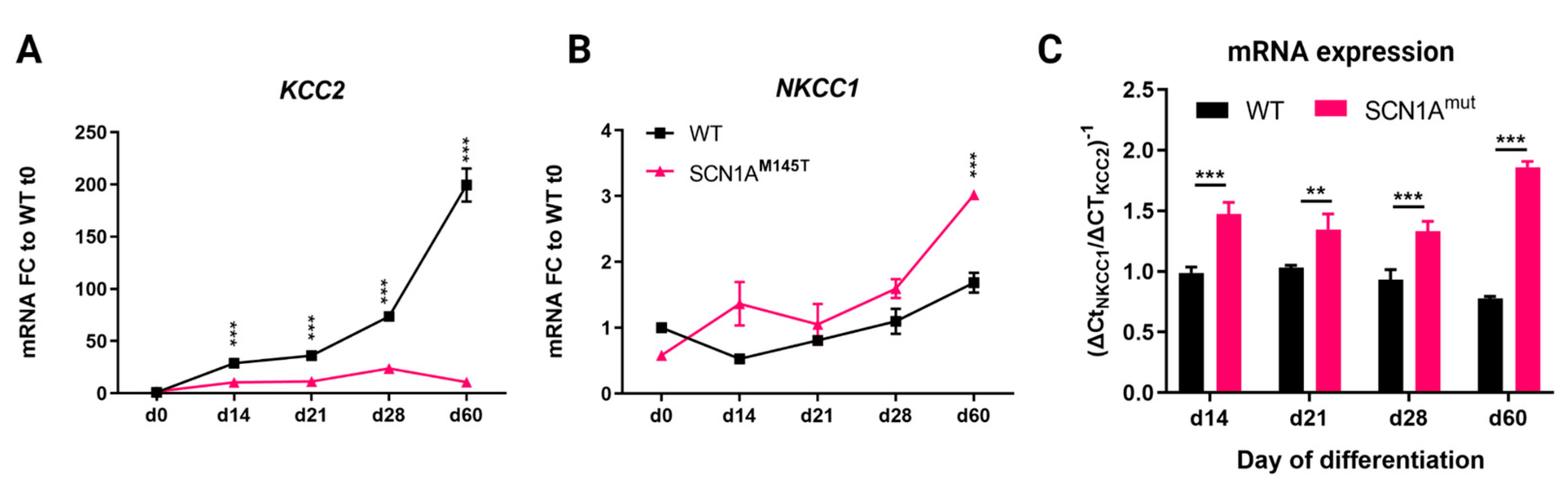
3.3. Expression of Chloride Cotransporters in idNs
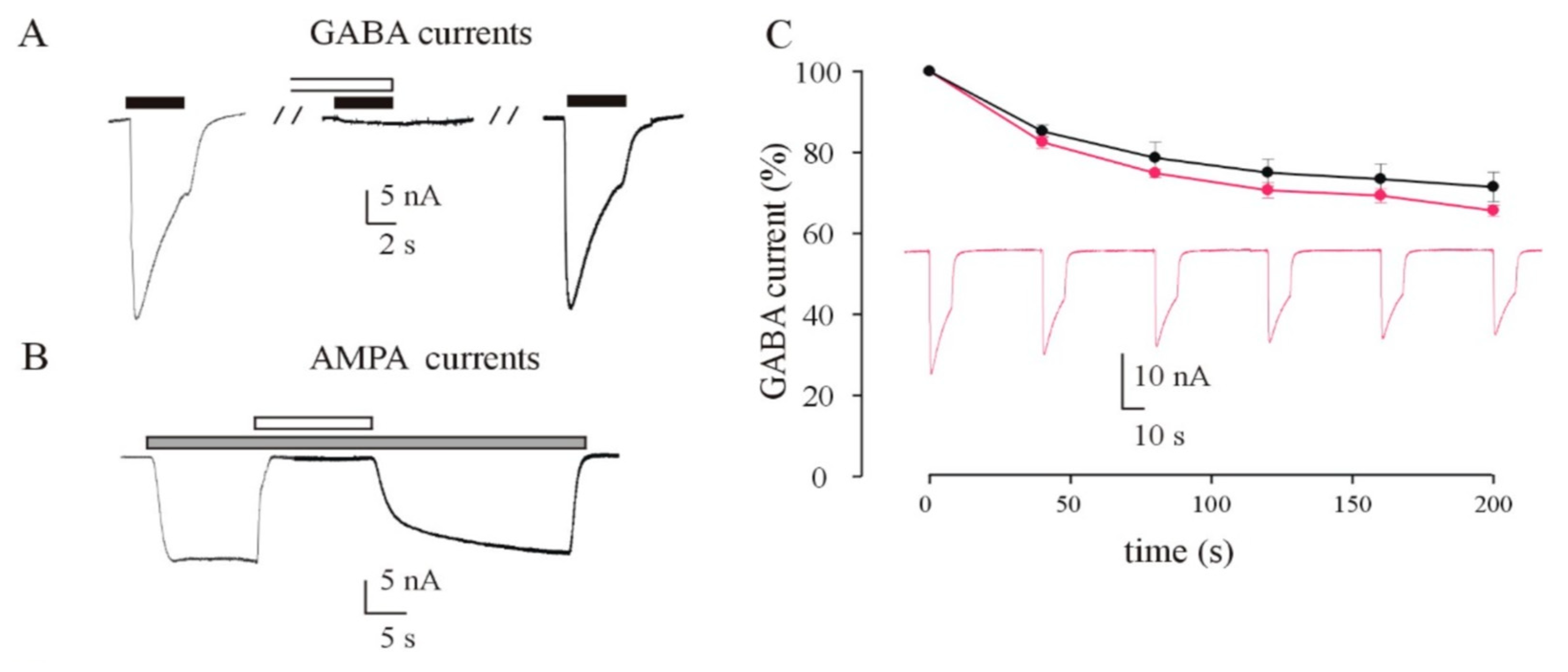
3.4. Recording of GABA and AMPA Currents by Injection of idNs Membranes in Xenopus Oocytes
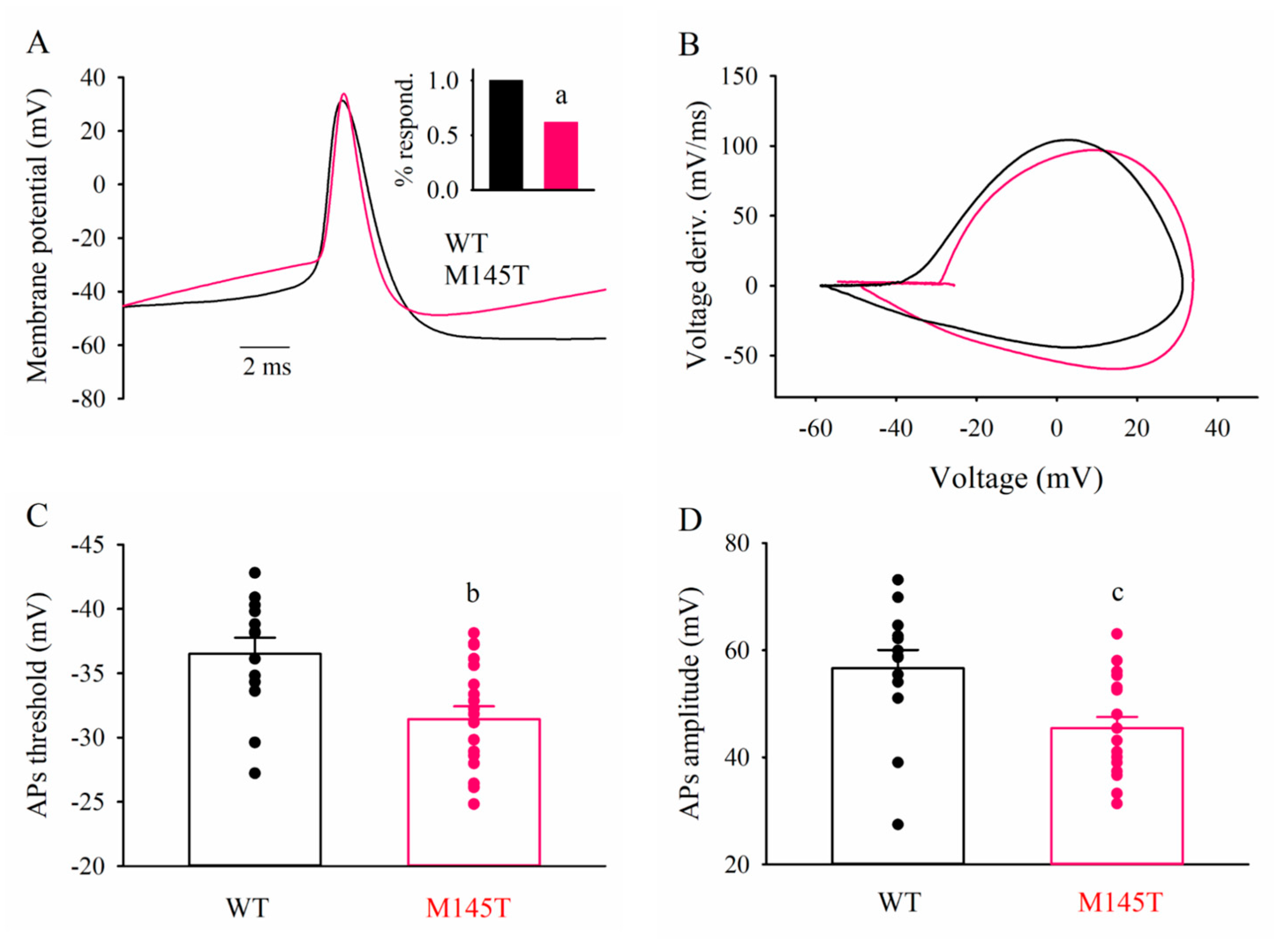
3.5. Patch-clamp recordings of WT and SCN1AM145T idNs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Camfield, P.; Camfield, C. Incidence, Prevalence and Aetiology of Seizures and Epilepsy in Children. Epileptic Disorders 2015, 17, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Saghazadeh, A.; Mastrangelo, M.; Rezaei, N. Genetic Background of Febrile Seizures. Rev. Neurosci. 2014, 25, 129–161. [Google Scholar] [CrossRef] [PubMed]
- de Tisi, J.; Bell, G.S.; Peacock, J.L.; McEvoy, A.W.; Harkness, W.F.; Sander, J.W.; Duncan, J.S. The Long-Term Outcome of Adult Epilepsy Surgery, Patterns of Seizure Remission, and Relapse: A Cohort Study. Lancet 2011, 378, 1388–1395. [Google Scholar] [CrossRef] [Green Version]
- Marini, C.; Scheffer, I.E.; Nabbout, R.; Suls, A.; De Jonghe, P.; Zara, F.; Guerrini, R. The Genetics of Dravet Syndrome: Genetics of Dravet Syndrome. Epilepsia 2011, 52, 24–29. [Google Scholar] [CrossRef]
- Escayg, A.; Heils, A.; MacDonald, B.T.; Haug, K.; Sander, T.; Meisler, M.H. A Novel SCN1A Mutation Associated with Generalized Epilepsy with Febrile Seizures Plus—And Prevalence of Variants in Patients with Epilepsy. Am. J. Hum. Genet. 2001, 68, 866–873. [Google Scholar] [CrossRef] [Green Version]
- Mantegazza, M.; Gambardella, A.; Rusconi, R.; Schiavon, E.; Annesi, F.; Cassulini, R.R.; Labate, A.; Carrideo, S.; Chifari, R.; Canevini, M.P.; et al. Identification of an Nav1.1 Sodium Channel (SCN1A) Loss-of-Function Mutation Associated with Familial Simple Febrile Seizures. Proc. Natl. Acad. Sci. USA 2005, 102, 18177–18182. [Google Scholar] [CrossRef] [Green Version]
- Kasperavičiūtė, D.; Catarino, C.B.; Matarin, M.; Leu, C.; Novy, J.; Tostevin, A.; Leal, B.; Hessel, E.V.S.; Hallmann, K.; Hildebrand, M.S.; et al. Epilepsy, Hippocampal Sclerosis and Febrile Seizures Linked by Common Genetic Variation around SCN1A. Brain 2013, 136, 3140–3150. [Google Scholar] [CrossRef]
- Dutton, S.B.B.; Dutt, K.; Papale, L.A.; Helmers, S.; Goldin, A.L.; Escayg, A. Early-Life Febrile Seizures Worsen Adult Phenotypes in Scn1a Mutants. Exp. Neurol. 2017, 293, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Scheffer, I.E.; Zhang, Y.-H.; Jansen, F.E.; Dibbens, L. Dravet Syndrome or Genetic (Generalized) Epilepsy with Febrile Seizures Plus? Brain Dev. 2009, 31, 394–400. [Google Scholar] [CrossRef]
- Orban, M.; Goedel, A.; Haas, J.; Sandrock-Lang, K.; Gärtner, F.; Jung, C.B.; Zieger, B.; Parrotta, E.; Kurnik, K.; Sinnecker, D.; et al. Functional Comparison of Induced Pluripotent Stem Cell- and Blood-Derived GPIIbIIIa Deficient Platelets. PLoS ONE 2015, 10, e0115978. [Google Scholar] [CrossRef] [Green Version]
- Parrotta, E.I.; Scalise, S.; Scaramuzzino, L.; Cuda, G. Stem Cells: The Game Changers of Human Cardiac Disease Modelling and Regenerative Medicine. Int. J. Mol. Sci. 2019, 20, 5760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Angelis, M.T.; Santamaria, G.; Parrotta, E.I.; Scalise, S.; Lo Conte, M.; Gasparini, S.; Ferlazzo, E.; Aguglia, U.; Ciampi, C.; Sgura, A.; et al. Establishment and Characterization of Induced Pluripotent Stem Cells (IPSCs) from Central Nervous System Lupus Erythematosus. J. Cell. Mol. Med. 2019, 23, 7382–7394. [Google Scholar] [CrossRef] [PubMed]
- Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Grillone, K.; Lo Conte, M.; Esposito, C.; Aguglia, U.; Ferlazzo, E.; Perrotti, N.; Malatesta, P.; et al. Generation of Human Induced Pluripotent Stem Cell Lines (UNIMGi003-A and UNIMGi004-A) from Two Italian Siblings Affected by Unverricht-Lundborg Disease. Stem Cell Res. 2021, 53, 102329. [Google Scholar] [CrossRef] [PubMed]
- Colosimo, E.; Gambardella, A.; Mantegazza, M.; Labate, A.; Rusconi, R.; Schiavon, E.; Annesi, F.; Cassulini, R.R.; Carrideo, S.; Chifari, R.; et al. Electroclinical Features of a Family with Simple Febrile Seizures and Temporal Lobe Epilepsy Associated with SCN1A Loss-of-Function Mutation. Epilepsia 2007, 48, 1691–1696. [Google Scholar] [CrossRef]
- Ruffolo, G.; Martinello, K.; Labate, A.; Cifelli, P.; Fucile, S.; Di Gennaro, G.; Quattrone, A.; Esposito, V.; Limatola, C.; Giangaspero, F.; et al. Modulation of GABAergic Dysfunction Due to SCN1A Mutation Linked to Hippocampal Sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1726–1731. [Google Scholar] [CrossRef]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced Sodium Current in GABAergic Interneurons in a Mouse Model of Severe Myoclonic Epilepsy in Infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef]
- Cheah, C.S.; Yu, F.H.; Westenbroek, R.E.; Kalume, F.K.; Oakley, J.C.; Potter, G.B.; Rubenstein, J.L.; Catterall, W.A. Specific Deletion of NaV1.1 Sodium Channels in Inhibitory Interneurons Causes Seizures and Premature Death in a Mouse Model of Dravet Syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 14646–14651. [Google Scholar] [CrossRef] [Green Version]
- Palma, E.; Esposito, V.; Mileo, A.M.; Di Gennaro, G.; Quarato, P.; Giangaspero, F.; Scoppetta, C.; Onorati, P.; Trettel, F.; Miledi, R.; et al. Expression of Human Epileptic Temporal Lobe Neurotransmitter Receptors in Xenopus Oocytes: An Innovative Approach to Study Epilepsy. Proc. Natl. Acad. Sci. USA 2002, 99, 15078–15083. [Google Scholar] [CrossRef] [Green Version]
- Higurashi, N.; Uchida, T.; Lossin, C.; Misumi, Y.; Okada, Y.; Akamatsu, W.; Imaizumi, Y.; Zhang, B.; Nabeshima, K.; Mori, M.X.; et al. A Human Dravet Syndrome Model from Patient Induced Pluripotent Stem Cells. Mol. Brain 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Paşca, S.P.; Portmann, T.; Goold, C.; Worringer, K.A.; Guan, W.; Chan, K.C.; Gai, H.; Vogt, D.; Chen, Y.-J.J.; et al. A Deleterious Nav1.1 Mutation Selectively Impairs Telencephalic Inhibitory Neurons Derived from Dravet Syndrome Patients. eLife 2016, 5, e13073. [Google Scholar] [CrossRef]
- Liu, Y.; Lopez-Santiago, L.F.; Yuan, Y.; Jones, J.M.; Zhang, H.; O’Malley, H.A.; Patino, G.A.; O’Brien, J.E.; Rusconi, R.; Gupta, A.; et al. Dravet Syndrome Patient-Derived Neurons Suggest a Novel Epilepsy Mechanism. Ann. Neurol. 2013, 74, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalise, S.; Scaramuzzino, L.; Lucchino, V.; Esposito, C.; Malatesta, P.; Grillone, K.; Perrotti, N.; Cuda, G.; Parrotta, E.I. Generation of IPSC Lines from Two Patients Affected by Febrile Seizure Due to Inherited Missense Mutation in SCN1A Gene. Stem Cell Res. 2020, 49, 102083. [Google Scholar] [CrossRef] [PubMed]
- Parrotta, E.I.; Scalise, S.; Taverna, D.; De Angelis, M.T.; Sarro, G.; Gaspari, M.; Santamaria, G.; Cuda, G. Comprehensive Proteogenomic Analysis of Human Embryonic and Induced Pluripotent Stem Cells. J. Cell. Mol. Med. 2019, 23, 5440–5453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Ruffolo, G.; Di Bonaventura, C.; Cifelli, P.; Roseti, C.; Fattouch, J.; Morano, A.; Limatola, C.; Aronica, E.; Palma, E.; Giallonardo, A.T. A Novel Action of Lacosamide on GABAA Currents Sets the Ground for a Synergic Interaction with Levetiracetam in Treatment of Epilepsy. Neurobiol. Dis. 2018, 115, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.X.; Yuan, Q.; Tan, S.; Xiao, Y.; Wang, D.; Khoo, A.T.T.; Sani, L.; Tran, H.-D.; Kim, P.; Chiew, Y.S.; et al. Direct Induction and Functional Maturation of Forebrain GABAergic Neurons from Human Pluripotent Stem Cells. Cell Rep. 2016, 16, 1942–1953. [Google Scholar] [CrossRef] [Green Version]
- Heighway, J.; Sedo, A.; Garg, A.; Eldershaw, L.; Perreau, V.; Berecki, G.; Reid, C.A.; Petrou, S.; Maljevic, S. Sodium Channel Expression and Transcript Variation in the Developing Brain of Human, Rhesus Monkey, and Mouse. Neurobiol. Dis. 2022, 164, 105622. [Google Scholar] [CrossRef]
- Rahmati, N.; Hoebeek, F.E.; Peter, S.; De Zeeuw, C.I. Chloride Homeostasis in Neurons With Special Emphasis on the Olivocerebellar System: Differential Roles for Transporters and Channels. Front. Cell. Neurosci. 2018, 12, 101. [Google Scholar] [CrossRef] [Green Version]
- Blaesse, P.; Airaksinen, M.S.; Rivera, C.; Kaila, K. Cation-Chloride Cotransporters and Neuronal Function. Neuron 2009, 61, 820–838. [Google Scholar] [CrossRef] [Green Version]
- Schulte, J.T.; Wierenga, C.J.; Bruining, H. Chloride Transporters and GABA Polarity in Developmental, Neurological and Psychiatric Conditions. Neurosci. Biobehav. Rev. 2018, 90, 260–271. [Google Scholar] [CrossRef]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-Temporal Transcriptome of the Human Brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Yang, K.; Zheng, C.; Liu, Q.; Chang, Y.; Kerrigan, J.F.; Wu, J. Functional Rundown of Gamma-Aminobutyric AcidA Receptors in Human Hypothalamic Hamartomas. Ann. Neurol. 2011, 69, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Ragozzino, D.; Palma, E.; Di Angelantonio, S.; Amici, M.; Mascia, A.; Arcella, A.; Giangaspero, F.; Cantore, G.; Di Gennaro, G.; Manfredi, M.; et al. Rundown of GABA Type A Receptors Is a Dysfunction Associated with Human Drug-Resistant Mesial Temporal Lobe Epilepsy. Proc. Natl. Acad. Sci. USA 2005, 102, 15219–15223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, E.; Roseti, C.; Maiolino, F.; Fucile, S.; Martinello, K.; Mazzuferi, M.; Aronica, E.; Manfredi, M.; Esposito, V.; Cantore, G.; et al. GABAA-Current Rundown of Temporal Lobe Epilepsy Is Associated with Repetitive Activation of GABAA “Phasic” Receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 20944–20948. [Google Scholar] [CrossRef] [Green Version]
- Tidball, A.M.; Lopez-Santiago, L.F.; Yuan, Y.; Glenn, T.W.; Margolis, J.L.; Clayton Walker, J.; Kilbane, E.G.; Miller, C.A.; Martina Bebin, E.; Scott Perry, M.; et al. Variant-Specific Changes in Persistent or Resurgent Sodium Current in SCN8A-Related Epilepsy Patient-Derived Neurons. Brain 2020, 143, 3025–3040. [Google Scholar] [CrossRef]
- Whitaker, W.R.J.; Faull, R.L.M.; Waldvogel, H.J.; Plumpton, C.J.; Emson, P.C.; Clare, J.J. Comparative Distribution of Voltage-Gated Sodium Channel Proteins in Human Brain. Mol. Brain Res. 2001, 88, 37–53. [Google Scholar] [CrossRef]
- Duflocq, A.; Le Bras, B.; Bullier, E.; Couraud, F.; Davenne, M. Nav1.1 Is Predominantly Expressed in Nodes of Ranvier and Axon Initial Segments. Mol. Cell. Neurosci. 2008, 39, 180–192. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Nabbout, R. SCN1A-Related Phenotypes: Epilepsy and Beyond. Epilepsia 2019, 60, S17–S24. [Google Scholar] [CrossRef]
- Bartolini, E.; Campostrini, R.; Kiferle, L.; Pradella, S.; Rosati, E.; Chinthapalli, K.; Palumbo, P. Epilepsy and Brain Channelopathies from Infancy to Adulthood. Neurol. Sci. 2020, 41, 749–761. [Google Scholar] [CrossRef]
- Tang, B.; Dutt, K.; Papale, L.; Rusconi, R.; Shankar, A.; Hunter, J.; Tufik, S.; Yu, F.H.; Catterall, W.A.; Mantegazza, M.; et al. A BAC Transgenic Mouse Model Reveals Neuron Subtype-Specific Effects of a Generalized Epilepsy with Febrile Seizures Plus (GEFS+) Mutation. Neurobiol. Dis. 2009, 35, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Mashimo, T.; Ohmori, I.; Ouchida, M.; Ohno, Y.; Tsurumi, T.; Miki, T.; Wakamori, M.; Ishihara, S.; Yoshida, T.; Takizawa, A.; et al. A Missense Mutation of the Gene Encoding Voltage-Dependent Sodium Channel (Nav1.1) Confers Susceptibility to Febrile Seizures in Rats. J. Neurosci. 2010, 30, 5744–5753. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, U.B.S.; Liautard, C.; Kirschenbaum, D.; Pofahl, M.; Lavigne, J.; Liu, Y.; Theiss, S.; Slotta, J.; Escayg, A.; Dihné, M.; et al. Impaired Action Potential Initiation in GABAergic Interneurons Causes Hyperexcitable Networks in an Epileptic Mouse Model Carrying a Human NaV1.1 Mutation. J. Neurosci. 2014, 34, 14874–14889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Zhu, B.; Xie, Y.; Zeng, L.; Pham, A.T.; Neumann, J.C.; Safrina, O.; Benavides, D.R.; MacGregor, G.R.; Schutte, S.S.; et al. Interneuron Dysfunction in a New Mouse Model of SCN1A GEFS+. ENeuro 2021, 8, ENEURO.0394-20.2021. [Google Scholar] [CrossRef] [PubMed]
- Engel, J. The Current Place of Epilepsy Surgery. Curr. Opin. Neurol. 2018, 31, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Baxendale, S. The Cognitive Costs, Contraindications and Complications of Epilepsy Surgery in Adults. Curr. Opin. Neurol. 2020, 33, 207–212. [Google Scholar] [CrossRef]
- Löscher, W.; Klitgaard, H.; Twyman, R.E.; Schmidt, D. New Avenues for Anti-Epileptic Drug Discovery and Development. Nat. Rev. Drug Discov. 2013, 12, 757–776. [Google Scholar] [CrossRef]
- Löscher, W. Animal Models of Seizures and Epilepsy: Past, Present, and Future Role for the Discovery of Antiseizure Drugs. Neurochem. Res. 2017, 42, 1873–1888. [Google Scholar] [CrossRef]
- Aguilar-Castillo, M.J.; Cabezudo-García, P.; Ciano-Petersen, N.L.; García-Martin, G.; Marín-Gracia, M.; Estivill-Torrús, G.; Serrano-Castro, P.J. Immune Mechanism of Epileptogenesis and Related Therapeutic Strategies. Biomedicines 2022, 10, 716. [Google Scholar] [CrossRef]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-Chloride Cotransporters in Neuronal Development, Plasticity and Disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef] [Green Version]
- Ruffolo, G.; Cifelli, P.; Roseti, C.; Thom, M.; van Vliet, E.A.; Limatola, C.; Aronica, E.; Palma, E. A Novel GABAergic Dysfunction in Human Dravet Syndrome. Epilepsia 2018, 59, 2106–2117. [Google Scholar] [CrossRef] [Green Version]
- Ben-Ari, Y. The GABA Excitatory/Inhibitory Developmental Sequence: A Personal Journey. Neuroscience 2014, 279, 187–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musto, E.; Gardella, E.; Møller, R.S. Recent Advances in Treatment of Epilepsy-Related Sodium Channelopathies. Eur. J. Paediatr. Neurol. 2020, 24, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.S.; Kenny, C.J.; Ganesh, V.; Jang, A.; Borges-Monroy, R.; Partlow, J.N.; Hill, R.S.; Shin, T.; Chen, A.Y.; Doan, R.N.; et al. Sodium Channel SCN3A (NaV1.3) Regulation of Human Cerebral Cortical Folding and Oral Motor Development. Neuron 2018, 99, 905–913.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, P.; Opitz, T.; Steinbeck, J.A.; Ladewig, J.; Brüstle, O. A Rosette-Type, Self-Renewing Human ES Cell-Derived Neural Stem Cell with Potential for in Vitro Instruction and Synaptic Integration. PNAS 2009, 106, 3225–3230. [Google Scholar] [CrossRef] [Green Version]
- Chopra, R.; Isom, L.L. Untangling the Dravet Syndrome Seizure Network: The Changing Face of a Rare Genetic Epilepsy: The Paradox of Dravet Syndrome. Epilepsy Curr. 2014, 14, 86–89. [Google Scholar] [CrossRef] [Green Version]
- Cifelli, P.; Palma, E.; Roseti, C.; Verlengia, G.; Simonato, M. Changes in the Sensitivity of GABAA Current Rundown to Drug Treatments in a Model of Temporal Lobe Epilepsy. Front. Cell. Neurosci. 2013, 7, 108. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, A.; Labate, A.; Cifelli, P.; Ruffolo, G.; Mumoli, L.; Aronica, E.; Palma, E. Pharmacological Modulation in Mesial Temporal Lobe Epilepsy: Current Status and Future Perspectives. Pharmacol. Res. 2016, 113, 421–425. [Google Scholar] [CrossRef]
- Mazzuferi, M.; Palma, E.; Martinello, K.; Maiolino, F.; Roseti, C.; Fucile, S.; Fabene, P.F.; Schio, F.; Pellitteri, M.; Sperk, G.; et al. Enhancement of GABAA-Current Run-down in the Hippocampus Occurs at the First Spontaneous Seizure in a Model of Temporal Lobe Epilepsy. Proc. Natl. Acad. Sci. USA 2010, 107, 3180–3185. [Google Scholar] [CrossRef] [Green Version]
- Cifelli, P.; Di Angelantonio, S.; Alfano, V.; Morano, A.; De Felice, E.; Aronica, E.; Ruffolo, G.; Palma, E. Dissecting the Molecular Determinants of GABAA Receptors Current Rundown, a Hallmark of Refractory Human Epilepsy. Brain Sci. 2021, 11, 441. [Google Scholar] [CrossRef]
- Pitkänen, A.; Löscher, W.; Vezzani, A.; Becker, A.J.; Simonato, M.; Lukasiuk, K.; Gröhn, O.; Bankstahl, J.P.; Friedman, A.; Aronica, E.; et al. Advances in the Development of Biomarkers for Epilepsy. Lancet Neurol. 2016, 15, 843–856. [Google Scholar] [CrossRef]
- Stern, W.M.; Sander, J.W.; Rothwell, J.C.; Sisodiya, S.M. Impaired Intracortical Inhibition Demonstrated in Vivo in People with Dravet Syndrome. Neurology 2017, 88, 1659–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffolo, G.; Iyer, A.; Cifelli, P.; Roseti, C.; Mühlebner, A.; van Scheppingen, J.; Scholl, T.; Hainfellner, J.A.; Feucht, M.; Krsek, P.; et al. Functional Aspects of Early Brain Development Are Preserved in Tuberous Sclerosis Complex (TSC) Epileptogenic Lesions. Neurobiol. Dis. 2016, 95, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Deidda, G.; Parrini, M.; Naskar, S.; Bozarth, I.F.; Contestabile, A.; Cancedda, L. Reversing Excitatory GABAAR Signaling Restores Synaptic Plasticity and Memory in a Mouse Model of Down Syndrome. Nat. Med. 2015, 21, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R.; Marchetto, M.C.N.; Gage, F.H.; et al. KCC2 Rescues Functional Deficits in Human Neurons Derived from Patients with Rett Syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Vatine, G.D.; Barrile, R.; Workman, M.J.; Sances, S.; Barriga, B.K.; Rahnama, M.; Barthakur, S.; Kasendra, M.; Lucchesi, C.; Kerns, J.; et al. Human IPSC-Derived Blood-Brain Barrier Chips Enable Disease Modeling and Personalized Medicine Applications. Cell Stem Cell 2019, 24, 995–1005.e6. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, A.; Mzezewa, R.; Sukki, L.; Ryynänen, T.; Kreutzer, J.; Hyvärinen, T.; Vinogradov, A.; Aarnos, L.; Lekkala, J.; Kallio, P.; et al. A Modular Brain-on-a-Chip for Modelling Epileptic Seizures with Functionally Connected Human Neuronal Networks. Biosens. Bioelectron. 2020, 168, 112553. [Google Scholar] [CrossRef]
- Coluccio, M.L.; D’Attimo, M.A.; Cristiani, C.M.; Candeloro, P.; Parrotta, E.; Dattola, E.; Guzzi, F.; Cuda, G.; Lamanna, E.; Carbone, E.; et al. A Passive Microfluidic Device for Chemotaxis Studies. Micromachines 2019, 10, 551. [Google Scholar] [CrossRef] [Green Version]
- Guzzi, F.; Candeloro, P.; Coluccio, M.L.; Cristiani, C.M.; Parrotta, E.I.; Scaramuzzino, L.; Scalise, S.; Dattola, E.; D’Attimo, M.A.; Cuda, G.; et al. A Disposable Passive Microfluidic Device for Cell Culturing. Biosensors 2020, 10, 18. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalise, S.; Zannino, C.; Lucchino, V.; Lo Conte, M.; Scaramuzzino, L.; Cifelli, P.; D’Andrea, T.; Martinello, K.; Fucile, S.; Palma, E.; et al. Human iPSC Modeling of Genetic Febrile Seizure Reveals Aberrant Molecular and Physiological Features Underlying an Impaired Neuronal Activity. Biomedicines 2022, 10, 1075. https://doi.org/10.3390/biomedicines10051075
Scalise S, Zannino C, Lucchino V, Lo Conte M, Scaramuzzino L, Cifelli P, D’Andrea T, Martinello K, Fucile S, Palma E, et al. Human iPSC Modeling of Genetic Febrile Seizure Reveals Aberrant Molecular and Physiological Features Underlying an Impaired Neuronal Activity. Biomedicines. 2022; 10(5):1075. https://doi.org/10.3390/biomedicines10051075
Chicago/Turabian StyleScalise, Stefania, Clara Zannino, Valeria Lucchino, Michela Lo Conte, Luana Scaramuzzino, Pierangelo Cifelli, Tiziano D’Andrea, Katiuscia Martinello, Sergio Fucile, Eleonora Palma, and et al. 2022. "Human iPSC Modeling of Genetic Febrile Seizure Reveals Aberrant Molecular and Physiological Features Underlying an Impaired Neuronal Activity" Biomedicines 10, no. 5: 1075. https://doi.org/10.3390/biomedicines10051075