
Vitamin D Impacts on Skeletal Muscle Dysfunction in Patients with COPD Promoting Mitochondrial Health

 , , , , and
, , , , and
Abstract
:1. Introduction
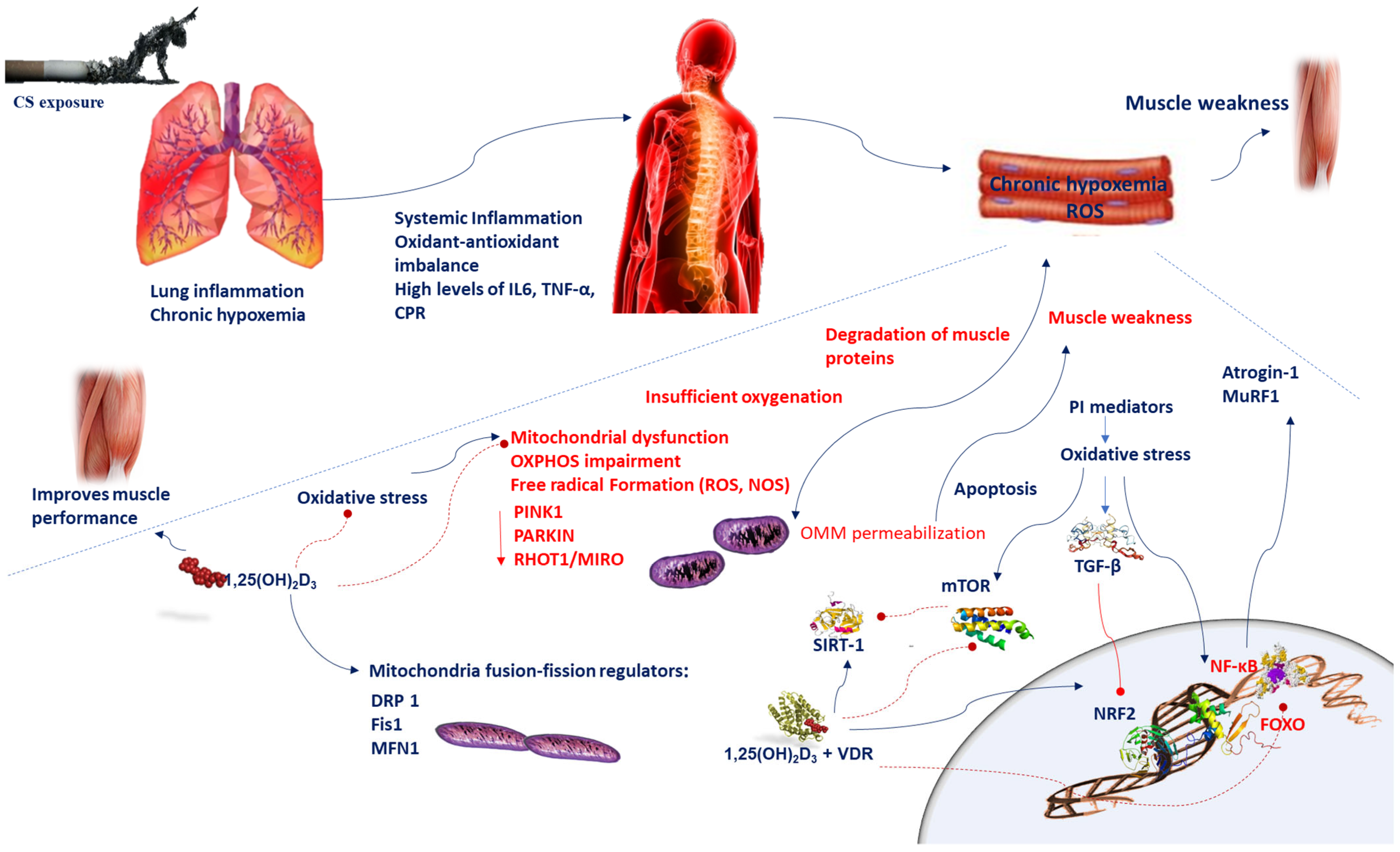
2. Mechanisms Mediating Muscular Wasting in COPD Patients
3. Vitamin D Metabolism and Biological Function in the Muscle
4. Vitamin D Deficiency and Muscle Weakness
5. Mitochondria, Oxidative Stress, and Muscular Wasting in COPD Patients
6. Mitochondrial Alterations and Muscular Wasting in COPD Patients
7. Role of Vitamin D in Anti-Oxidative Mechanisms Implicated in Muscular Wasting in COPD Patients
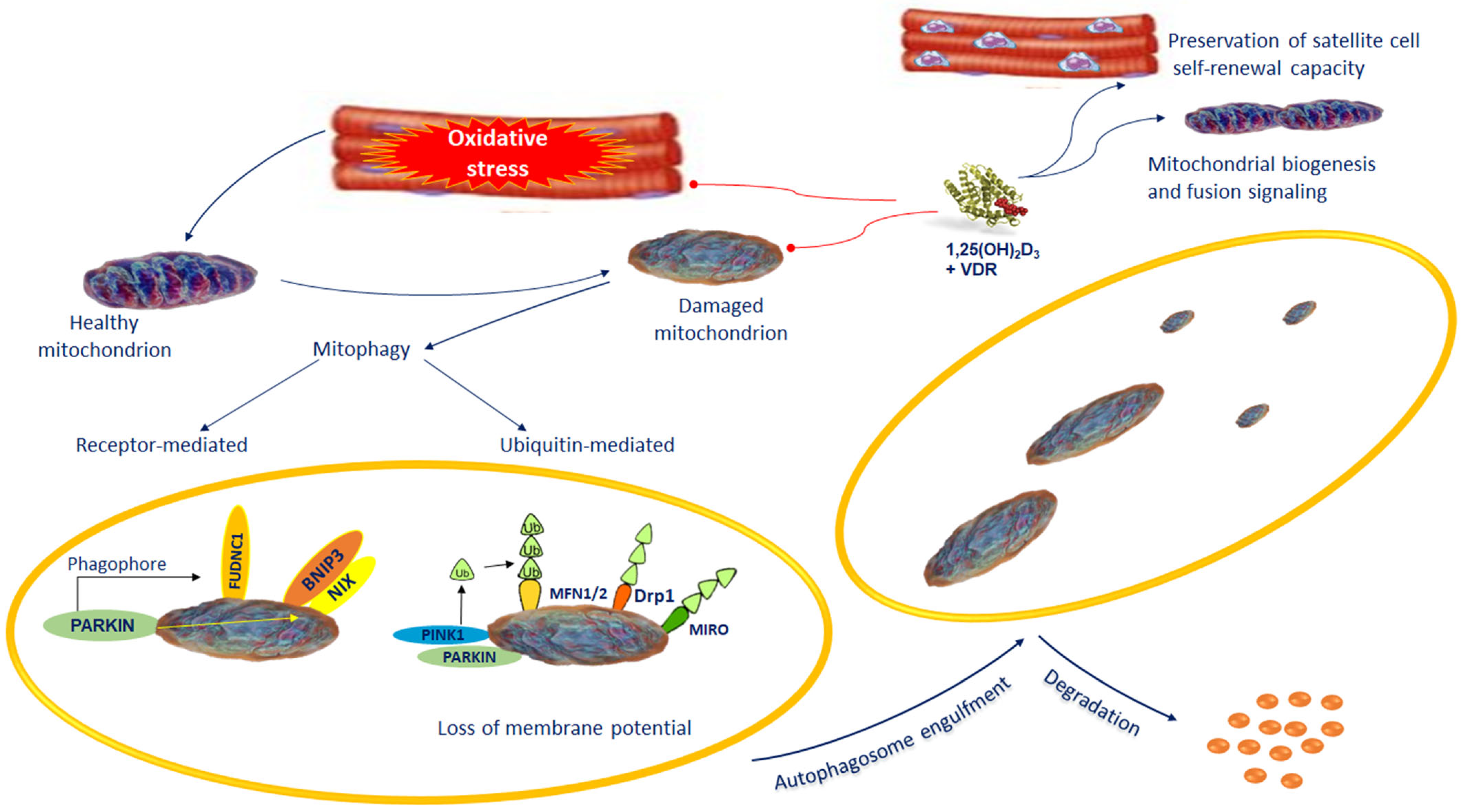
8. Mitophagy Alterations and Muscular Wasting in COPD Patients
9. Role of Vitamin D in Mitochondrial Biogenesis and Dysfunction in Muscular Wasting of COPD Patients
10. Concluding Remarks and Future Prospective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barnes, P.J.; Burney, P.G.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Primers 2015, 1, 15076. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Halpin, D.M.G.; Han, M.K.; Miravitlles, M.; Singh, D.; Grönke, L.; Voß, F.; Martinez, F.J. Composite endpoints in COPD: Clinically important deterioration in the UPLIFT trial. Respir. Res. 2020, 21, 177. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, L.; Iliescu, A.; Dinu, H.; Badea, R.; Savulescu, S.; Huidu, S.; Berteanu, M. Disability in COPD and Chronic Heart Failure Is the Skeletal Muscle the Final Common Pathway? Maedica (Bucur) 2013, 8, 206–213. [Google Scholar] [PubMed]
- Maltais, F.; Decramer, M.; Casaburi, R.; Barreiro, E.; Burelle, Y.; Debigaré, R.; Dekhuijzen, P.N.; Franssen, F.; Gayan-Ramirez, G.; Gea, J.; et al. ATS/ERS Ad Hoc Committee on Limb Muscle Dysfunction in COPD. An official American Thoracic Society/European Respiratory Society statement: Update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2014, 189, e15–e62. [Google Scholar] [CrossRef] [Green Version]
- Natanek, S.A.; Gosker, H.R.; Slot, I.G.; Marsh, G.S.; Hopkinson, N.S.; Man, W.D.; Tal-Singer, R.; Moxham, J.; Kemp, P.R.; Schols, A.M.; et al. Heterogeneity of quadriceps muscle phenotype in chronic obstructive pulmonary disease (Copd); implications for stratified medicine? Muscle Nerve 2013, 48, 488–497. [Google Scholar] [CrossRef]
- Jaitovich, A.; Barreiro, E. Skeletal Muscle Dysfunction in Chronic Obstructive Pulmonary Disease. What We Know and Can Do for Our Patients. Am. J. Respir. Crit. Care Med. 2018, 198, 175–186. [Google Scholar] [CrossRef]
- Khan, D.M.; Ullah, A.; Randhawa, F.A.; Iqtadar, S.; Butt, N.F.; Waheed, K. Role of Vitamin D in reducing number of acute exacerbations in Chronic Obstructive Pulmonary Disease (COPD) patients. Pak. J. Med. Sci. 2017, 33, 610–614. [Google Scholar] [CrossRef]
- Li, X.; He, J.; Yu, M.; Sun, J. The efficacy of vitamin D therapy for patients with COPD: A meta-analysis of randomized controlled trials. Ann. Palliat. Med. 2020, 9, 286–297. [Google Scholar] [CrossRef]
- Jolliffe, D.A.; Greenberg, L.; Hooper, R.L.; Mathyssen, C.; Rafiq, R.; de Jongh, R.T.; Camargo, C.A.; Griffiths, C.J.; Janssens, W.; Martineau, A.R. Vitamin D to prevent exacerbations of COPD: Systematic review and meta-analysis of individual participant data from randomised controlled trials. Thorax 2019, 74, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Redzic, M.; Lewis, R.M.; Thomas, D.T. Relationship between 25-hydoxyvitamin D, muscle strength, and incidence of injury in healthy adults: A systematic review. Nutr. Res. 2013, 33, 251–258. [Google Scholar] [CrossRef]
- Latham, C.M.; Brightwell, C.R.; Keeble, A.R.; Munson, B.D.; Thomas, N.T.; Zagzoog, A.M.; Fry, C.S.; Fry, J.L. Vitamin D Promotes Skeletal Muscle Regeneration and Mitochondrial Health. Front. Physiol. 2021, 12, 660498. [Google Scholar] [CrossRef] [PubMed]
- Hornikx, M.; Van Remoortel, H.; Lehouck, A.; Mathieu, C.; Maes, K.; Gayan-Ramirez, G.; Decramer, M.; Troosters, T.; Janssens, W. Vitamin D supplementation during rehabilitation in COPD: A secondary analysis of a randomized trial. Respir. Res. 2012, 13, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezk, N.A.S.A.; Aly, N.Y.A.; Hewidy, A.A.H. Effect of vitamin D replacement in chronic obstructive pulmonary disease patients with vitamin D deficiency. Egypt J. Chest Dis. Tuberc. 2015, 64, 353–357. [Google Scholar] [CrossRef] [Green Version]
- Heulens, N.; Korf, H.; Janssens, W. Innate immune modulation in chronic obstructive pulmonary disease: Moving closer toward vitamin D therapy. J. Pharmacol. Exp. Ther. 2015, 353, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casabona, A.; Valle, M.S.; Laudani, L.; Crimi, C.; Russo, C.; Malaguarnera, L.; Crimi, N.; Cioni, M. Is the Power Spectrum of Electromyography Signal a Feasible Tool to Estimate Muscle Fiber Composition in Patients with COPD? J. Clin. Med. 2021, 10, 3815. [Google Scholar] [CrossRef]
- Sharanya, A.; Ciano, M.; Withana, S.; Kemp, P.R.; Polkey, M.I.; Sathyapala, S.A. Sex differences in COPD-related quadriceps muscle dysfunction and fibre abnormalities. Chron. Respir. Dis. 2019, 16, 1479973119843650. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, M.K.; Sibley, K.M.; Lakhani, B.; Romano, J.; Mathur, S.; Goldstein, R.S.; Brooks, D. Impairments in systems underlying control of balance in COPD. Chest 2012, 141, 1496–1503. [Google Scholar] [CrossRef]
- Nantsupawat, N.; Lane, P.; Siangpraipunt, O.; Gadwala, S.; Nugent, K. Gait Characteristics in Patients with Chronic Obstructive Pulmonary Disease. J. Prim. Care Community Health. 2015, 6, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Valle, M.S.; Casabona, A.; Di Fazio, E.; Crimi, C.; Russo, C.; Malaguarnera, L.; Crimi, N.; Cioni, M. Impact of chronic obstructive pulmonary disease on passive viscoelastic components of the musculoarticular system. Sci. Rep. 2021, 11, 18077. [Google Scholar] [CrossRef]
- De Boer, M.D.; Selby, A.; Atherton, P.; Smith, K.; Seynnes, O.R.; Maganaris, C.N.; Maffulli, N.; Movin, T.; Narici, M.V.; Rennie, M.J. The temporal responses of protein synthesis, gene expression and cell signaling in human quadriceps muscle and patellar tendon to disuse. J. Physiol. 2007, 585, 241–251. [Google Scholar] [CrossRef]
- Swallow, E.B.; Reyes, D.; Hopkinson, N.S.; Man, W.D.; Porcher, R.; Cetti, E.J.; Moore, A.J.; Moxham, J.; Polkey, M.I. Quadriceps strength predicts mortality in patients with moderate to severe chronic obstructive pulmonary disease. Thorax 2007, 62, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhdar, R.; Rabinovich, R.A. Can muscle protein metabolism be specifically targeted by nutritional support and exercise training in chronic obstructive pulmonary disease? J. Thorac. Dis. 2018, 10 (Suppl. S12), S1377–S1389. [Google Scholar] [CrossRef] [PubMed]
- Nishiki, K.; Nojiri, M.; Kato, R.; Shinomiya, S.; Oikawa, T.; Ishizaki, T.; Toga, H.; Mizuno, S. Serum Creatinine/Cystatin C Ratio Associated with Cross-Sectional Area of Erector Spinae Muscles and Pulmonary Function in Patients with Chronic Obstructive Pulmonary Disease. Int. J. Chronic Obstr. Pulm. Dis. 2021, ume 16, 3513–3524. [Google Scholar] [CrossRef]
- Owens, D.J.; Sharples, A.P.; Polydorou, I.; Alwan, N.; Donovan, T.; Tang, J.; Fraser, W.D.; Cooper, R.G.; Morton, J.P.; Stewart, C.; et al. A systems-based investigation into vitamin D and skeletal muscle repair, regeneration, and hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E1019–E1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, M.S.; Russo, C.; Casabona, A.; Crimi, N.; Crimi, C.; Colaianni, V.; Cioni, M.; Malaguarnera, L. Anti-inflammatory role of vitamin D in muscle dysfunctions of patients with COPD: A comprehensive review. Minerva Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Pojednic, R.M.; Ceglia, L. The emerging biomolecular role of vitamin D in skeletal muscle. Exerc. Sport Sci. Rev. 2014, 42, 76–81. [Google Scholar] [CrossRef]
- DeLuca, H.F. Overview of general physiologic features and functions of vitamin D. Am. J. Clin. Nutr. 2004, 80 (Suppl. S6), 1689S–1696S. [Google Scholar] [CrossRef] [Green Version]
- Ginde, A.A.; Wolfe, P.; Jr Camargo, C.A.; Schwartz, R.S. Defining vitamin D status by secondary hyperparathyroidism in the U.S. population. J. Endocrinol. Investig. 2012, 35, 42–48. [Google Scholar] [CrossRef]
- Valle, M.S.; Russo, C.; Malaguarnera, L. Protective role of vitamin D against oxidative stress in diabetic retinopathy. Diabetes Metab. Res. Rev. 2021, 37, e3447. [Google Scholar] [CrossRef]
- Olsson, K.; Saini, A.; Strömberg, A.; Alam, S.; Lilja, M.; Rullman, E.; Gustafsson, T. Evidence for Vitamin D Receptor Expression and Direct Effects of 1α,25(OH)2D3 in Human Skeletal Muscle Precursor Cells. Endocrinology 2016, 157, 98–111. [Google Scholar] [CrossRef] [Green Version]
- Bellido, T.; Boland, R. Effects of 1,25-dihydroxy-vitamin D3 on phosphate accumulation by myoblasts. Horm. Metab. Res. 1991, 23, 113–116, Erratum in Horm. Metab. Res. 1991, 23, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, L.A.; King, K.K.; Ferrini, M.G.; Norris, K.C.; Artaza, J.N. 1,25(OH)2vitamin D3 stimulates myogenic differentiation by inhibiting cell proliferation and modulating the expression of promyogenic growth factors and myostatin in C2C12 skeletal muscle cells. Endocrinology 2011, 152, 2976–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, M.; Begerow, B.; Minne, H.W. Vitamin D and muscle function. Osteoporos Int. 2002, 13, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D status: Measurement, interpretation, and clinical application. Ann. Epidemiol. 2009, 19, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Shuler, F.D.; Wingate, M.K.; Moore, G.H.; Giangarra CGiangarra, C. Sports health benefits of vitamin d. Sports Health 2012, 4, 496–501. [Google Scholar] [CrossRef] [Green Version]
- Ranathunga, R.M.T.K.; Hill, T.R.; Mathers, J.C.; Francis, R.M.; Prentice, A.; Schoenmakers, I.; Aspray, T.J. Vitamin D in Older People Study group. No effect of monthly supplementation with 12000 IU, 24000 IU or 48000 IU vitamin D3 for one year on muscle function: The vitamin D in older people study. J. Steroid. Biochem Mol. Biol. 2019, 190, 256–262. [Google Scholar] [CrossRef]
- Barker, T.; Henriksen, V.T.; Martins, T.B.; Hill, H.R.; Kjeldsberg, C.R.; Schneider, E.D.; Dixon, B.M.; Weaver, L.K. Higher serum 25-hydroxyvitamin D concentrations associate with a faster recovery of skeletal muscle strength after muscular injury. Nutrients 2013, 5, 1253–1275. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, P.B.; Joseph, C.; Angioi, M. Effects of vitamin D supplementation on upper and lower body muscle strength levels in healthy individuals. A systematic review with meta-analysis. J. Sci. Med. Sport. 2015, 18, 575–580. [Google Scholar] [CrossRef]
- Ceglia, L. Vitamin D and its role in skeletal muscle. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 628–633. [Google Scholar] [CrossRef] [Green Version]
- Sakai, S.; Suzuki, M.; Tashiro, Y.; Tanaka, K.; Takeda, S.; Aizawa, K.; Hirata, M.; Yogo, K.; Endo, K. Vitamin D receptor signaling enhances locomotive ability in mice. J. Bone Miner. Res. 2015, 30, 128–136. [Google Scholar] [CrossRef]
- Van Schoor, N.M.; de Jongh, R.T.; Daniels, J.M.; Heymans, M.W.; Deeg, D.J.; Lips, P. Peak expiratory flow rate shows a gender-specific association with vitamin D deficiency. J. Clin. Endocrinol. Metab. 2012, 97, 2164–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girgis, C.M.; Cha, K.M.; Houweling, P.J.; Rao, R.; Mokbel, N.; Lin, M.; Clifton-Bligh, R.J.; Gunton, J.E. Vitamin D Receptor Ablation and Vitamin D Deficiency Result in Reduced Grip Strength, Altered Muscle Fibers, and Increased Myostatin in Mice. Calcif. Tissue Int. 2015, 97, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Endo, I.; Inoue, D.; Mitsui, T.; Umaki, Y.; Akaike, M.; Yoshizawa, T.; Kato, S.; Matsumoto, T. Deletion of vitamin D receptor gene in mice results in abnormal skeletal muscle development with deregulated expression of myoregulatory transcription factors. Endocrinology 2003, 144, 5138–5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romme, E.A.; Rutten, E.P.; Smeenk, F.W.; Spruit, M.A.; Menheere, P.P.; Wouters, E.F. Vitamin D status is associated with bone mineral density and functional exercise capacity in patients with chronic obstructive pulmonary disease. Ann. Med. 2013, 45, 91–96. [Google Scholar] [CrossRef]
- Ryan, Z.C.; Craig, T.A.; Folmes, C.D.; Wang, X.; Lanza, I.R.; Schaible, N.S.; Salisbury, J.L.; Nair, K.S.; Terzic, A.; Sieck, G.C.; et al. 1α,25-Dihydroxyvitamin D3 Regulates Mitochondrial Oxygen Consumption and Dynamics in Human Skeletal Muscle Cells. J. Biol. Chem. 2016, 291, 514–528. [Google Scholar] [CrossRef] [Green Version]
- Beaudart, C.; Buckinx, F.; Rabenda, V.; Gillain, S.; Cavalier, E.; Slomian, J.; Petermans, J.; Reginster, J.Y.; Bruyère, O. The effects of vitamin D on skeletal muscle strength, muscle mass, and muscle power: A systematic review and meta-analysis of randomized controlled trials. J. Clin. Endocrinol. Metab. 2014, 99, 4336–4345. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.S.; Shrikrishna, D.; Kelly, J.L.; Kemp, S.V.; Hart, N.; Moxham, J.; Polkey, M.I.; Kemp, P.; Hopkinson, N.S. Vitamin D and skeletal muscle strength and endurance in COPD. Eur. Respir. J. 2013, 41, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhao, D.; Shah, S.Z.A.; Zhang, X.; Lai, M.; Yang, D.; Wu, X.; Guan, Z.; Li, J.; Zhao, H.; et al. OPA1 overexpression ameliorates mitochondrial cristae remodeling, mitochondrial dysfunction, and neuronal apoptosis in prion diseases. Cell Death Dis. 2019, 10, 710. [Google Scholar] [CrossRef]
- Montecinos-Franjola, F.; Ramachandran, R. Imaging Dynamin-Related Protein 1 (Drp1)-Mediated Mitochondrial Fission in Living Cells. Methods Mol. Biol. 2020, 2159, 205–217. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 2016, 6, 422. [Google Scholar] [CrossRef]
- Barreiro, E.; Peinado, V.I.; Galdiz, J.B.; Ferrer, E.; Marin-Corral, J.; Sánchez, F.; Gea, J.; Barberà, J.A. ENIGMA in COPD Project. Cigarette smoke-induced oxidative stress: A role in chronic obstructive pulmonary disease skeletal muscle dysfunction. Am. J. Respir. Crit. Care Med. 2010, 182, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.A.M.; Roche, N.; Oliver, B.G.; Mattos, W.; Barnes, P.J.; Fan Chung, K. Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers regulation by interleukin-10. Am. J. Respir. Crit. Care Med. 2000, 162, 1355–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. Modulating mitochondrial intracellular location as a redox signal. Sci. Signal. 2012, 5, pe39. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Kritchevsky, S.B.; Baumgartner, R.N.; Atkinson, H.H.; Penninx, B.W.; Lenchik, L.; Palla, S.L.; Ambrosius, W.T.; Tracy, R.P.; Pahor, M. Sarcopenia, obesity, and inflammation—Results from the Trial of Angiotensin Converting Enzyme Inhibition and Novel Cardiovascular Risk Factors study. Am. J. Clin. Nutr. 2005, 82, 428–434. [Google Scholar] [CrossRef]
- De la Fuente, M.; Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026. [Google Scholar] [CrossRef]
- Gonzalez, N.C.; Wood, J.G. Alveolar hypoxia-induced systemic inflammation: What low PO(2) does and does not do. Adv. Exp. Med. Biol. 2010, 662, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Puig-Vilanova, E.; Rodriguez, D.A.; Lloreta, J.; Ausin, P.; Pascual-Guardia, S.; Broquetas, J.; Roca, J.; Gea, J.; Barreiro, E. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic. Biol. Med. 2015, 79, 91–108. [Google Scholar] [CrossRef]
- O’Donnell, C.; Newbold, P.; White, P.; Thong, B.; Stone, H.; Stockley, R.A. 3-Chlorotyrosine in sputum of COPD patients: Relationship with airway inflammation. COPD 2010, 7, 411–417. [Google Scholar] [CrossRef]
- Caramori, G.; Casolari, P.; Barczyk, A.; Durham, A.L.; Di Stefano, A.; Adcock, I. COPD immunopathology. Semin. Immunopathol. 2016, 38, 497–515. [Google Scholar] [CrossRef] [Green Version]
- Remels, A.H.; Gosker, H.R.; Schrauwen, P.; Hommelberg, P.P.; Sliwinski, P.; Polkey, M.; Galdiz, J.; Wouters, E.F.; Langen, R.C.; Schols, A.M. TNF-alpha impairs regulation of muscle oxidative phenotype: Implications for cachexia? FASEB J. 2010, 24, 5052–5062. [Google Scholar] [CrossRef]
- Gorowiec, M.R.; Borthwick, L.A.; Parker, S.M.; Kirby, J.A.; Saretzki, G.C.; Fisher, A.J. Free radical generation induces epithelial-to-mesenchymal transition in lung epithelium via a TGF-β1-dependent mechanism. Free Radic. Biol. Med. 2012, 52, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Sukkar, M.B.; Khorasani, N.M.; Bhavsar, P.K.; Chung, K.F. TGF-β regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L295–L304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef]
- Tengan, C.H.; Moraes, C.T. NO control of mitochondrial function in normal and transformed cells. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Lark, D.S.; Torres, M.J.; Lin, C.T.; Ryan, T.E.; Anderson, E.J.; Neufer, P.D. Direct real-time quantification of mitochondrial oxidative phosphorylation efficiency in permeabilized skeletal muscle myofibers. Am. J. Physiol. Cell Physiol. 2016, 311, C239–C245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, C.S.; Cadenas, E. Nitric oxide and cell signaling pathways in mitochondrial-dependent apoptosis. Biol. Chem. 2002, 383, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Murano, G.; Wagner, H.; Nogueira, L.; Wagner, P.D.; Tang, A.; Dalton, N.D.; Gu, Y.; Peterson, K.L.; Breen, E.C. Impaired exercise capacity and skeletal muscle function in a mouse model of pulmonary inflammation. J. Appl. Physiol. 1985 2013, 114, 1340–1350. [Google Scholar] [CrossRef] [Green Version]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene. Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Lavrik, I.; Golks, A.; Krammer, P.H. Death receptor signaling. J. Cell. Sci. 2005, 118, 265–267. [Google Scholar] [CrossRef] [Green Version]
- Roberge, S.; Roussel, J.; Andersson, D.C.; Meli, A.C.; Vidal, B.; Blandel, F.; Lanner, J.T.; Le Guennec, J.Y.; Katz, A.; Westerblad, H.; et al. TNF-α-mediated caspase-8 activation induces ROS production and TRPM2 activation in adult ventricular myocytes. Cardiovasc. Res. 2014, 103, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Perez, C.; Roy, S.S.; Naghdi, S.; Lin, X.; Davies, E.; Hajnóczky, G. Bid-induced mitochondrial membrane permeabilization waves propagated by local reactive oxygen species (ROS) signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 4497–4502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remels, A.H.; Langen, R.C.; Schrauwen, P.; Schaart, G.; Schols, A.M.; Gosker, H.R. Regulation of mitochondrial biogenesis during myogenesis. Mol. Cell. Endocrinol. 2010, 315, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, S.; Bashir, M.H.; Clanton, T.L.; Powers, S.K.; Singhal, S. COPD elicits remodeling of the diaphragm and vastus lateralis muscles in humans. J. Appl. Physiol. 2013, 114, 1235–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, M.D.; Distefano, G.; Gagliano, C.; Rusciano, D.; Malaguarnera, L. Autophagy in Diabetic Retinopathy. Curr. Neuropharmacol. 2016, 14, 810–825. [Google Scholar] [CrossRef] [Green Version]
- Nakamaru, Y.; Vuppusetty, C.; Wada, H.; Milne, J.C.; Ito, M.; Rossios, C.; Elliot, M.; Hogg, J.; Kharitonov, S.; Goto, H.; et al. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J. 2009, 23, 2810–2819. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Sundar, I.K.; Maremanda, K.P.; Rahman, I. Mitochondrial dysfunction is associated with Miro1 reduction in lung epithelial cells by cigarette smoke. Toxicol. Lett. 2019, 317, 92–101. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell. Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Aung, L.H.; Prabhakar, B.S.; Li, P. The mitochondrial ubiquitin ligase plays an anti-apoptotic role in cardiomyocytes by regulating mitochondrial fission. J. Cell. Mol. Med. 2016, 20, 2278–2288. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005, 19, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Lerner, C.A.; Sundar, I.K.; Rahman, I. Mitochondrial redox system, dynamics, and dysfunction in lung inflammaging and COPD. Int. J. Biochem. Cell. Biol. 2016, 81, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Aravamudan, B.; Kiel, A.; Freeman, M.; Delmotte, P.; Thompson, M.; Vassallo, R.; Sieck, G.C.; Pabelick, C.M.; Prakash, Y.S. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L840–L854. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Scorrano, L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim. Biophys. Acta 2013, 1833, 205–212. [Google Scholar] [CrossRef]
- Dzik, K.P.; Kaczor, J.J. Mechanisms of vitamin D on skeletal muscle function: Oxidative stress, energy metabolism and anabolic state. Eur. J. Appl. Physiol. 2019, 119, 825–839. [Google Scholar] [CrossRef] [Green Version]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef]
- Bhat, M.; Ismail, A. Vitamin D treatment protects against and reverses oxidative stress induced muscle proteolysis. J. Steroid. Biochem. Mol. Biol. 2015, 152, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Van der Meijden, K.; Bravenboer, N.; Dirks, N.F.; Heijboer, A.C.; den Heijer, M.; de Wit, G.M.; Offringa, C.; Lips, P.; Jaspers, R.T. Effects of 1,25(OH)2 D3 and 25(OH)D3 on C2C12 Myoblast Proliferation, Differentiation, and Myotube Hypertrophy. J. Cell. Physiol. 2016, 231, 2517–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seldeen, K.L.; Berman, R.N.; Pang, M.; Lasky, G.; Weiss, C.; MacDonald, B.A.; Thiyagarajan, R.; Redae, Y.; Troen, B.R.; Vitamin, D. Insufficiency Reduces Grip Strength, Grip Endurance and Increases Frailty in Aged C57Bl/6J Mice. Nutrients 2020, 12, 3005. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Q.; Liu, Y.; Shu, L.; Wang, N.; Wu, Y.; Sun, X.; Wang, L. Attenuation of hyperoxia-induced lung injury in neonatal rats by 1α,25-Dihydroxyvitamin D3. Exp. Lung Res. 2015, 41, 344–352. [Google Scholar] [CrossRef]
- Ke, C.Y.; Yang, F.L.; Wu, W.T.; Chung, C.H.; Lee, R.P.; Yang, W.T.; Subeq, Y.M.; Liao, K.W. Vitamin D3 Reduces Tissue Damage and Oxidative Stress Caused by Exhaustive Exercise. Int. J. Med. Sci. 2016, 13, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Srikuea, R.; Hirunsai, M.; Charoenphandhu, N. Regulation of vitamin D system in skeletal muscle and resident myogenic stem cell during development, maturation, and ageing. Sci. Rep. 2020, 10, 8239. [Google Scholar] [CrossRef]
- Xiang, Y.; Fu, L.; Xiang, H.X.; Zheng, L.; Tan, Z.X.; Wang, L.X.; Cao, W.; Xu, D.X.; Zhao, H. Correlations among Pulmonary DJ-1, VDR and Nrf-2 in patients with Chronic Obstructive Pulmonary Disease: A Case-control Study. Int. J. Med. Sci. 2021, 18, 2449–2456. [Google Scholar] [CrossRef]
- Chen, L.; Yang, R.; Qiao, W.; Zhang, W.; Chen, J.; Mao, L.; Goltzman, D.; Miao, D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell 2019, 18, e12951. [Google Scholar] [CrossRef]
- Mathyssen, C.; Aelbrecht, C.; Serré, J.; Everaerts, S.; Maes, K.; Gayan-Ramirez, G.; Vanaudenaerde, B.; Janssens, W. Local expression profiles of vitamin D-related genes in airways of COPD patients. Respir. Res. 2020, 21, 137. [Google Scholar] [CrossRef]
- Sinha, A.; Hollingsworth, K.G.; Ball, S.; Cheetham, T. Improving the vitamin D status of vitamin D deficient adults is associated with improved mitochondrial oxidative function in skeletal muscle. J. Clin. Endocrinol. Metab. 2013, 98, E509–E513. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Villalta, S.A.; Agrawal, D.K. FOXO1 Mediates Vitamin D Deficiency-Induced Insulin Resistance in Skeletal Muscle. J. Bone Miner. Res. 2016, 31, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manna, P.; Achari, A.E.; Jain, S.K. Vitamin D supplementation inhibits oxidative stress and upregulate SIRT1/AMPK/GLUT4 cascade in high glucose-treated 3T3L1 adipocytes and in adipose tissue of high fat diet-fed diabetic mice. Arch. Biochem. Biophys. 2017, 615, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta 2015, 1853, 2784–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Wang, K.Z.; Chu, C.T. After the banquet: Mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 2013, 9, 1663–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell. Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Langen, R.C.; Haegens, A.; Vernooy, J.H.; Wouters, E.F.; de Winther, M.P.; Carlsen, H.; Steele, C.; Shoelson, S.E.; Schols, A.M. NF-κB activation is required for the transition of pulmonary inflammation to muscle atrophy. Am. J. Respir. Cell Mol. Biol. 2012, 47, 288–297. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [Green Version]
- Salles, J.; Chanet, A.; Giraudet, C.; Patrac, V.; Pierre, P.; Jourdan, M.; Luiking, Y.C.; Verlaan, S.; Migné, C.; Boirie, Y.; et al. 1,25(OH)2-vitamin D3 enhances the stimulating effect of leucine and insulin on protein synthesis rate through Akt/PKB and mTOR mediated pathways in murine C2C12 skeletal myotubes. Mol. Nutr. Food Res. 2013, 57, 2137–2146. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.; Brunet, A.; Griffith, E.C.; Greenberg, M.E. The many forks in FOXO’s road. Sci. STKE 2003, 2003, re5. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The ubiquitin–proteasome system in regulation of the skeletal muscle homeostasis and atrophy: From basic science to disorders. J. Physiol. Sci. 2020, 70, 40. [Google Scholar] [CrossRef] [PubMed]
- Saline, M.; Badertscher, L.; Wolter, M.; Lau, R.; Gunnarsson, A.; Jacso, T.; Norris, T.; Ottmann, C.; Snijder, A. AMPK and AKT protein kinases hierarchically phosphorylate the N-terminus of the FOXO1 transcription factor, modulating interactions with 14-3-3 proteins. J. Biol. Chem. 2019, 294, 13106–13116. [Google Scholar] [CrossRef] [PubMed]
- An, B.S.; Tavera-Mendoza, L.E.; Dimitrov, V.; Wang, X.; Calderon, M.R.; Wang, H.J.; White, J.H. Stimulation of Sirt1-regulated FoxO protein function by the ligand-bound vitamin D receptor. Mol. Cell. Biol. 2010, 30, 4890–4900. [Google Scholar] [CrossRef] [Green Version]
- Camperi, A.; Pin, F.; Costamagna, D.; Penna, F.; Menduina, M.L.; Aversa, Z.; Zimmers, T.; Verzaro, R.; Fittipaldi, R.; Caretti, G.; et al. Vitamin D and VDR in cancer cachexia and muscle regeneration. Oncotarget 2017, 8, 21778–21793. [Google Scholar] [CrossRef] [Green Version]
- Correia, J.C.; Ferreira, D.M.; Ruas, J.L. Intercellular: Local and systemic actions of skeletal muscle PGC-1s. Trends Endocrinol. Metab. 2015, 26, 305–314. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Experimental Data | Model | Sample Size | Tissue | Approach | Reference |
|---|---|---|---|---|---|
| Evaluation of oxygen consumption, biogenesis, dynamics, and nuclear genes encoding variations. | Human | // | Muscle biopsies. Primary human skeletal muscle cells. | Supplementation of 1α,25-Dihydroxyvitamin D3 (10-8 M) for 48 h. VDR expression in human muscle cells and skeletal muscle homogenates. Effect of 1α,25(OH)2D mitochondrial oxygen consumption and in expression of: mitochondrial proteins that alter mitochondrial fusion; proteins associated with mitochondrial fission; phosphorylated pyruvate dehydrogenase and pyruvate dehydrogenase kinase 4; genes encoding mitochondrial proteins; and genes encoding cellular signaling and growth-regulatory pathways in adult human skeletal muscle cells. Knockdown of VDR with silencing RNA in skeletal muscle cells to detect the effects of 1α,25(OH)2D3 on OCR. | [45] |
| Assessment of oxidative and nitrosative stress parameters. | Rat | // | Fasting blood samples analysis. C2C12 cell culture. | Supplementation of 1,25(OH)2D3 (1 nM and 10 nM) for 24 h. Effect of VDD in muscle oxidative stress in a rat model. Pre/post treatment of C2C12 muscle cells with vitamin D offers protection against oxidative stress induced muscle proteolysis. VDD increase in activities of the glutathione-dependent enzymes and decrease in SOD and catalase enzymes in the rat muscle. Pre/post treatment of C2C12 muscle cells with vitamin D correct total protein degradation, 20S proteasomal enzyme activity, muscle atrophy gene markers and expression of proteasome subunit genes induced by oxidative stress. | [91] |
| Serum and lung tissue analysis. | Human | 180 COPD patients | Human lung tissues and serum samples of COPD. | Level of pulmonary VDR-positive nuclei between COPD patients and control subjects Correlations of pulmonary function with pulmonary DJ-1, Nrf-2 and VDR in COPD patients. | [97] |
| Antioxidant and antiaging effects of 1,25Dihydroxyvitamin D by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. | Mouse | 120 | Skin, lung, liver, kidney, and spleen. | Two different supplementation: thrice weekly of 2.2 IU vitamin D/g or 1,25(OH)2D3 (1 μg/kg) until death. Effects of a high-calcium/phosphate diet, of 1,25(OH)2D3, and of antioxidant supplementation on lifespan, body weight, skin morphology; on oxidative stress, DNA damage, protein expression of oncogenes and tumor suppressive genes; and on cell proliferation and senescence in 1α(OH)ase−/− mice. | [98] |
| Improvement in parameters of mitochondrial function in vitamin-D-deficient individuals after vitamin D supplementation. | Human | 12 subjects with severe vitamin D deficiency | Serum samples | Effect of cholecalciferol therapy (20 000 IU supplementation on alternate days for 10–12 weeks) in muscle mitochondrial maximal oxidative phosphorylation after exercise in symptomatic, vitamin-D-deficient individuals. | [100] |
| FOXO1 activation in the skeletal muscle of global VDR-null mice. | Mouse | VDR−/− mice administered a diet enriched with calcium and phosphorus; SMVDR−/− mice generated by crossing VDRloxp/loxp mice with mice with muscle-specific Cre recombinase expression under the control of the myosin light chain 1f (MLC 1f) genomic locus; C2C12 muscle cells. | Treatment of C2C12 muscle cells with 1,25-dihydroxyvitamin D (100 nM for 48 h) to detect FOXO1 expression, nuclear translocation, and activity. Evaluation of FOXO1 activation in knockdown VDR mice. | [101] | |
| Effect of vitamin D supplementation on oxidative stress. | Mouse | Eight mice for each experimental group. | Adipocyte cell culture model | Supplementation of cholecalciferol (67 IU VD/kg daily for last 8 weeks) to detect the effects of 1,25(OH)2D3 supplementation in NOX4, Nrf2 SIRT-1 expression, ROS production, NF-κB and AMPK phosphorylation. | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, C.; Valle, M.S.; Casabona, A.; Spicuzza, L.; Sambataro, G.; Malaguarnera, L. Vitamin D Impacts on Skeletal Muscle Dysfunction in Patients with COPD Promoting Mitochondrial Health. Biomedicines 2022, 10, 898. https://doi.org/10.3390/biomedicines10040898
Russo C, Valle MS, Casabona A, Spicuzza L, Sambataro G, Malaguarnera L. Vitamin D Impacts on Skeletal Muscle Dysfunction in Patients with COPD Promoting Mitochondrial Health. Biomedicines. 2022; 10(4):898. https://doi.org/10.3390/biomedicines10040898
Chicago/Turabian StyleRusso, Cristina, Maria Stella Valle, Antonino Casabona, Lucia Spicuzza, Gianluca Sambataro, and Lucia Malaguarnera. 2022. "Vitamin D Impacts on Skeletal Muscle Dysfunction in Patients with COPD Promoting Mitochondrial Health" Biomedicines 10, no. 4: 898. https://doi.org/10.3390/biomedicines10040898