
Newly Identified Deficiencies in the Multiple Sclerosis Central Nervous System and Their Impact on the Remyelination Failure


Abstract
:1. Introduction
2. Basic Concepts Regarding the Biochemistry and Functions of CNS Myelin and the MS Remyelinating Process
3. The Role of EGF in the Genesis and Maintenance of CNS Myelin
4. CNS EGF Changes in MS and Experimental MS-like Models
5. Cbl in MS CNS
6. Role off PrPCs in CNS Myelin and MS SC
7. The Horns of the Dilemma of MS Pathogenesis: “To Be or Not to Be an Autoimmune Disease—That Is the Question”
7.1. Mitochondrial Abnormalities in MS
7.2. Neurosteroid Abnormalities
7.3. MS Epigenetic Abnormalities in OPC→ODC Cell Lineage
miRNA Abnormalities
8. Conclusions: “Ad Impossibilia Nemo Tenetur” (“Nobody Is Compelled to Do What Is Impossible”)
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| AST | Astrocyte |
| Cbl | Cobalamin |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| EAE | Experimental allergic encephalomyelitis |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| Erk | Extracellular signal-related kinase |
| GFAP | Glial fibrillary acidic protein |
| GS | Glutamine synthetase |
| HB | Heparin-binding |
| IGF | Insulin-like growth factor |
| MAG | Myelin associated glycoprotein |
| MBP | Myelin basic protein |
| miRNA | micro-RNA |
| MOG | Myelin oligodendrocyte-specific glycoprotein |
| mTORC | Mammalian target of rapamycin complex |
| NEU | Neuron |
| NF-κB | Nuclear factor kappa-light chain of B cell |
| NRG | Neuregulin |
| NSC | Neural stem cell |
| ODC | Oligodendrocyte |
| OPC | Oligodendrocyte precursor cell |
| PDGF | Platelet-derived growth factor |
| PLP | Proteolipid protein |
| PP | Primary progressive |
| PrPC | Normal cellular prion protein |
| PrPSC | Scrapie prion |
| RR | Relapsing and remitting |
| SC | Spinal cord |
| Shh | Sonic hedgehog |
| SP | Secondary progressive |
| SVZ | Subventricular zone |
| tHCYS | total Homocysteine |
| TNF | Tumour necrosis factor |
References
- Moore, G.R.W.; Stadelman-Nessler, C. Demyelinating Diseases. In Greenfield’s Neuropathology, 9th ed.; Love, S., Budka, H., Ironside, J.W., Perry, A., Eds.; CRC Press: Boca Raton, FL, USA, 2015; Volume 2, pp. 1297–1412. [Google Scholar]
- Wingerchuk, D.M.; Lucchinetti, C.F.; Noseworthy, J.H. Multiple sclerosis: Current pathophysiological concepts. Lab. Investig. 2001, 81, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.W.; Trapp, B.D. Neuropathobiology of multiple sclerosis. Neurol. Clin. 2005, 23, 107–129. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Brück, W.; Lassmann, H. Neuroimmunologic Mechanisms in the Etiology of Multiple Sclerosis. In Neuroinflammation. Mechanisms and Management, 2nd ed.; Wood, P.L., Ed.; Humana Press: Totowa, NJ, USA, 2003; pp. 359–377. [Google Scholar]
- Lucchinetti, C.; Parisi, J.; Brück, W. The pathology of multiple sclerosis. Neurol. Clin. 2005, 23, 77–105. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.C.; Douglas, J.N.; Meyers, L.; Lee, S.; Shin, Y.; Gardner, L.A. Neurodegeneration in multiple sclerosis involves multiple pathogenic mechanisms. Degen. Neurol. Neuromus. Dis. 2014, 4, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waxman, S.G. Ions, energy and axonal injury: Towards a molecular neurology of multiple sclerosis. Trends Mol. Med. 2006, 12, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Brück, W.; Stadelmann, C. The spectrum of multiple sclerosis: New lesson from pathology. Curr. Opin. Neurol. 2005, 18, 221–224. [Google Scholar] [CrossRef]
- Bø, L.; Esiri, M.; Evangelou, N.; Kuhlman, T. Demyelination and Remyelination in Multiple Sclerosis. In Myelin Repair and Neuroprotection in Multiple Sclerosis, 1st ed.; Duncan, I.D., Franklin, R.J.M., Eds.; Springer: New York, NY, USA, 2013; pp. 23–45. [Google Scholar]
- Lassmann, H. Pathophysiological Basis of Autoimmune-Initiated/Mediated Neurodegeneration. In Translational Methods for Multiple Sclerosis Research; Groppa, S., Meuth, S.G., Eds.; Humana Press: New York, NY, USA, 2021; pp. 3–12. [Google Scholar]
- Pérez-Pérez, S.; Dominguez-Mozo, M.I.; Garcia-Martínez, M.Á.; Ballester-González, R.; Gañán, I.; Arroyo, R.; Alvarez-Lafuente, R. Epstein-Barr virus load correlates with multiple sclerosis-associated retrovirus envelope expression. Biomedicines 2022, 10, 387. [Google Scholar] [CrossRef]
- Pimentel, E. Epidermal Growth Factor. In Handbook of Growth Factors; CRC Press: Boca Raton, FL, USA, 1994; Volume 2, pp. 97–185. [Google Scholar]
- Carpenter, G. Epidermal Growth Factor. In Tissue Growth Factors; Baserga, R., Ed.; Springer: Berlin, Germany, 1981; pp. 89–132. [Google Scholar]
- Iwakura, Y.; Nawa, H. Erb1-4-dependent EGF/neuregulin signals and their cross talk in the central nervous system: Pathological implications in schizophrenia and Parkinson’s disease. Front. Cell. Neurosci. 2013, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Hirata, Y.; Uchihashi, M.; Nakajima, H.; Fujita, T.; Matsukura, S. Presence of human epidermal growth factor in human cerebrospinal fluid. J. Clin. Endocrinol. Metab. 1982, 55, 1174–1177. [Google Scholar] [CrossRef]
- Fallon, J.H.; Seroogy, K.B.; Morrison, R.S.; Bradshaw, R.A.; Knaver, D.J.; Cunnigham, D.D. Epidermal growth factor immunoreactive material in the central nervous system: Location and development. Science 1984, 224, 1107–1109. [Google Scholar] [CrossRef]
- Lazar, L.; Blum, M. Regional distribution and developmental expression of epidermal growth factor and transforming growth factor-α mRNA in mouse brain by a quantitative nuclease protection assay. J. Neurosci. 1992, 12, 1688–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, Y.-S.; Iwakura, Y.; Takei, N.; Nawa, H. Differential distribution of peptides in the epidermal growth factor family and phosphorylation of ErbB1 receptor in adult rat brain. Neurosci. Lett. 2005, 390, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Harris, R.C. Epidermal growth factor, from gene organization to bedside. Sem. Cell. Dev. Biol. 2014, 28, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Landeghem, L.; Chevalier, J.; Mahé, M.M.; Wedel, T.; Urvil, P.; Derkinderen, P.; Savidge, T.; Neunlist, M. Enteric glia promote intestinal mucosal healing via activation of focal adhesion kinase and release of proEGF. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G976–G987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neunlist, M.; Van Landeghem, L.; Mahé, M.M.; Derkinderen, P.; Bruley des Varannes, S.; Rolli-Derkinderen, M. The digestive neuronal-glial-epithelial unit: A new actor in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 90–100. [Google Scholar] [CrossRef]
- Scalabrino, G.; Lorenzini, E.C.; Monzio-Compagnoni, M.; Colombi, R.P.; Chiodini, E.; Buccellato, F.R. Subacute combined degeneration in the spinal cords of totally gastrectomized rats. Ornithine decarboxylase induction, cobalamin status, and astroglial reaction. Lab. Investig. 1995, 72, 114–123. [Google Scholar]
- Pan, W.; Kastin, A.J. Transport of Cytokines and Neurotrophins across the Blood-Brain Barrier and Their Regulation after Spinal Cord Injury. In Blood-Spinal Cord and Brain Barriers in Health and Disease; Sharma, H.S., Westman, J., Eds.; Elsevier-Academic Press: San Diego, CA, USA, 2004; pp. 395–407. [Google Scholar]
- Pan, W.; Kastin, A.J. Entry of EGF into brain is rapid and saturable. Peptides 1999, 20, 1091–1098. [Google Scholar] [CrossRef]
- Nagano, T.; Namba, H.; Abe, Y.; Aoki, H.; Takei, N.; Nawa, H. In vivo administration of epidermal growth factor and its homologue attenuates developmental maturation of functional excitatory synapses in cortical GABAergic neurons. Eur. J. Neurosci. 2007, 25, 380–390. [Google Scholar] [CrossRef]
- Johanson, C.E.; McMillan, P.N.; Palm, D.E.; Stopa, E.G.; Doberstein, C.E.; Duncan, J.A. Volume Transmission-Mediated Protective Impact of Choroid Plexus-Cerebrospinal Fluid Growth Factors on Forebrain Ischemic Injury. In Blood-Spinal Cord and Brain Barriers in Health and Disease; Sharma, H.S., Westman, J., Eds.; Elsevier-Academic Press: San Diego, CA, USA, 2004; pp. 361–384. [Google Scholar]
- Scalabrino, G. The multi-faceted basis of vitamin B12 (cobalamin) neurotrophism in adult central nervous system: Lessons learned from its deficiency. Prog. Neurobiol. 2009, 88, 203–220. [Google Scholar] [CrossRef]
- Fenech, M. Folate (vitamin B9) and vitamin B12 and their function in the maintenance of nuclear and mitochondrial genome integrity. Mut. Res. 2012, 733, 21–33. [Google Scholar] [CrossRef]
- Scalabrino, G. Vitamin-regulated cytokines and growth factors in the central nervous system and elsewhere. J. Neurochem. 2009, 111, 1309–1326. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol. 2006, 5, 949–960. [Google Scholar] [CrossRef]
- Wolffenbuttel, B.H.R.; Wouters, H.J.C.M.; Heiner-Fokkema, M.R.; van der Klauw, M.M. The many faces of cobalamin (Vitamin B12) deficiency. Mayo Clin. Proc. Inn. Qual. Out. 2019, 3, 200–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, S.N.; Singh, P.; Steinbusch, H.W.M.; Vamanu, E.; Ashraf, G.; Singh, M.P. The role of vitamins in neurodegenerative disease: An update. Biomedicines 2021, 9, 1284. [Google Scholar] [CrossRef]
- Linden, R.; Martins, V.R.; Prado, M.A.M.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the prion protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef]
- Castle, A.R.; Gill, A.C. Physiological functions of the cellular prion protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Linden, R. The biological function of the prion protein: A cell surface scaffold of signalling modules. Front. Mol. Neurosci. 2017, 10, 77. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.A. Cellular biology of prion diseases. Clin. Microbiol. Rev. 1999, 12, 429–444. [Google Scholar] [CrossRef] [Green Version]
- Mehrpour, M.; Codogno, P. Prion protein: From physiology to cancer biology. Cancer Lett. 2010, 290, 1–23. [Google Scholar] [CrossRef]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prio protein (PrPc): Its physiological function and role in disease. Biochim. Biophys. Acta 2007, 1172, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Rutishauser, D.; Mertz, K.D.; Moos, R.; Brunner, E.; Rülicke, T.; Calella, A.M.; Aguzzi, A. The comprehensive native interactome of a fully functional tagged prion protein. PLoS ONE 2009, 4, e4446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiesa, R. The elusive role of the prion protein and the mechanism of toxicity in prion disease. PLoS Pathog. 2015, 11, e1004745. [Google Scholar] [CrossRef] [PubMed]
- Scalabrino, G.; Veber, D. Cobalamin and normal prions: A new horizon for cobalamin neurotrophism. Biochemie 2013, 95, 1041–1046. [Google Scholar] [CrossRef]
- Baumann, F.; Tolnay, M.; Brabeck, C.; Pahnke, J.; Kloz, U.; Niemann, H.H.; Heikenwalder, M.; Rülike, T.; Brücke, A.; Aguzzi, A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007, 26, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Mitteregger, G.; Vosko, M.; Krebs, B.; Xiang, W.; Kohlmannsperger, V.; Nölting, S.; Hamann, G.F.; Kretzschmar, H.A. The role of octapeptide region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007, 17, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Piccardo, P.; Barmada, S.J.; Ghetti, B.; Harris, D.A. Prion protein with an octapeptide insertion has impaired neuroprotective activity in transgenic mice. EMBO J. 2007, 26, 2777–2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalabrino, G.; Veber, D. Myelin damage due to local quantitative abnormalities in normal prion levels: Evidence from subacute combined degeneration and multiple sclerosis. J. Neurol. 2014, 261, 1451–1460. [Google Scholar] [CrossRef]
- Veber, D.; Scalabrino, G. Are PrPcs involved in some human myelin diseases? Relating experimental studies to human pathology. J. Neurol. Sci. 2015, 359, 396–403. [Google Scholar] [CrossRef]
- Watts, J.C.; Bourkas, M.E.C.; Arshad, H. The function of the cellular prion protein in health and disease. Acta Neuropathol. 2018, 135, 159–178. [Google Scholar] [CrossRef]
- Scalabrino, G.; Veber, D.; Tredici, G. Relationship between cobalamin, epidermal growth factor, and normal prions in the myelin maintenance of central nervous system. Int. J. Biochem. Cell Biol. 2014, 55, 232–241. [Google Scholar] [CrossRef]
- Scalabrino, G. Epidermal growth factor in the CNS: A beguiling journey from integrated cell biology to multiple sclerosis, An extensive translational overview. Cell. Mol. Neurobiol. 2020, 42, 891–916. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the central nervous system: Structure, function, and pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Kidd, G.J. Structure of the Myelinated Axon. In Myelin Biology and Disorders; Lazzarini, R.A., Ed.; Elsevier-Academic Press: San Diego, CA, USA, 2004; Volume 1, pp. 3–27. [Google Scholar]
- Aggarwal, S.; Yurlova, L.; Simons, M. Central nervous system myelin: Structure, synthesis, and assembly. Trends Cell Biol. 2011, 21, 585–593. [Google Scholar] [CrossRef]
- Trapp, B.D.; Pfeiffer, S.E.; Anitei, M.; Kidd, G.J. Cell Biology of Myelin Assembly. In Myelin Biology and Disorders; Lazzarini, R.A., Ed.; Elsevier-Academic Press: San Diego, CA, USA, 2004; Volume 1, pp. 29–55. [Google Scholar]
- Sherman, D.L.; Brophy, P.J. Mechanisms of axon ensheathment and myelin growth. Nat. Rev. Neurosci. 2005, 6, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.M.; Marta, C.B.; Bansal, R.; Pfeiffer, S.E. The Transport, Assembly, and Function of Myelin Lipids. In Myelin Biology and Disorders; Lazzarini, R.A., Ed.; Elsevier-Academic Press: San Diego, CA, USA, 2004; Volume 1, pp. 57–88. [Google Scholar]
- Hughes, E.G.; Appel, B. The cell biology of CNS myelination. Curr. Opin. Neurobiol. 2016, 39, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Nave, K.-A. Oligodendrocytes: Myelination and Axonal Support. Cold Sping Harbor Persp. Biol. 2016, 8, a020479. [Google Scholar] [CrossRef]
- Miron, V.E.; Kuhlmann, T.; Antel, J.P. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim. Biophys. Acta 2011, 1812, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Tognatta, R.; Karl, M.T.; Fyffe-Maricich, S.L.; Popratiloff, A.; Garrison, E.D.; Schenck, J.K.; Abu-Rub, M.; Miller, R.H. Astrocytes are required for oligodendrocyte survival and maintenance of myelin compaction and integrity. Front. Cell. Neurosci. 2020, 14, 74. [Google Scholar] [CrossRef] [Green Version]
- Nutma, E.; van Gent, D.; Amor, S.; Peferoen, L.A.N. Astrocyte and oligodendrocyte cross-talk in the central nervous system. Cells 2020, 9, 600. [Google Scholar] [CrossRef] [Green Version]
- Barnett, S.C.; Linington, C.L. Myelination: Do astrocytes play a role? Neuroscientist 2012, 20, 1–9. [Google Scholar] [CrossRef]
- Emery, B. Regulation of oligodendrocyte differentiation and myelination. Science 2010, 330, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Gonzales, I.; Miron, V.E. Astrocytes in myelination and remyelination. Neurosci. Lett. 2019, 713, 134532. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.M.; Purger, D.; Mount, C.W.; Goldstein, A.K.; Lin, G.L.; Wood, L.S.; Inema, I.; Miller, S.E.; Bieri, G.; Zuchero, J.B.; et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014, 344, 1252304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miron, V.E. Microglia-driven regulation of oligodendrocyte lineage cells, myelination, and remyelination. J. Leukoc. Biol. 2017, 101, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Kotter, M.R.; Li, W.W.; Zhao, C.; Franklin, R.J.M. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci. 2006, 26, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetanova, I.D.; Nave, K.-A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 2013, 23, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Scherer, S.S.; Arroyo, E.J.; Peles, E. Functional Organization of the Nodes of Ranvier. In Myelin Biology and Disorders; Lazzarini, R.A., Ed.; Elsevier-Academic Press: San Diego, CA, USA, 2004; Volume 1, pp. 89–116. [Google Scholar]
- Nave, K.-A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 2010, 11, 275–283. [Google Scholar] [CrossRef]
- Waxman, S.G.; Bangalore, L. Electrophysiological Consequences of Myelination. In Myelin Biology and Disorders; Lazzarini, R.A., Ed.; Elsevier-Academic Press: San Diego, CA, USA, 2004; Volume 1, pp. 117–141. [Google Scholar]
- Simons, M.; Trajkovic, K. Neuron-glia communication in the control of oligodendrocyte function and myelin biogenesis. J. Cell Sci. 2006, 119, 4381–4389. [Google Scholar] [CrossRef] [Green Version]
- Young, K.M.; Psachoulia, K.; Tripathi, R.B.; Dunn, S.-J.; Cossell, L.; Attwell, D.; Tohama, K.; Richardson, W.D. Oligodendrocyte dynamics in the healthy adult CNS: Evidence for myelin remodelling. Neuron 2013, 77, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Bercury, K.K.; Macklin, W.B. Dynamics and mechanism of CNS myelination. Dev. Cell. 2015, 32, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomassy, G.S.; Dershowitz, L.B.; Arlotta, P. Diversity matters: A revised guide to myelination. Trends Cell Biol. 2016, 26, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.-J.; Redmond, S.A.; Chan, J.R. Remodeling myelination: Implications for mechanisms of neural plasticity. Nat. Neurosci. 2016, 19, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, A.; Dyck, S.M.; Karim-Abdolrezaee, K. Myelin damage and repair on pathologic CNS: Challenges and prospects. Front. Mol. Neurosci. 2015, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nave, K.-A.; Werner, H.B. Myelination of the nervous system: Mechanisms and functions. Annu. Rev. Cell Dev. Biol. 2014, 30, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Cayre, M.; Falque, M.; Mercier, O.; Magalon, K.; Durbec, P. Myelin repair: From animal model to humans. Front. Cell. Neurosci. 2021, 15, 604865. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, K.A.; Nanescu, S.E.; Psachoulia, K.; Huang, J.K. Oligodendrocyte regeneration: Its significance in myelin replacement and neuroprotection in multiple sclerosis. Neuropharmacology 2016, 110, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Piaton, G.; Gould, R.M.; Lubetzki, C. Axon-oligodendrocyte interactions during developmental myelination, demyelination and repair. J. Neurochem. 2010, 114, 1243–1260. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.-A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, R.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis. A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Irvine, K.A.; Blakemore, W.F. Remyelination protects axons from demyelination-associated axon degeneration. Brain 2008, 131, 1464–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, R.J.M.; Zhao, C.; Lubetzki, C.; ffrench-Constant, C. Endogenous Remyelination in the CNS. In Myelin Repair and Neuroprotection in Multiple Sclerosis; Duncan, I.D., Franklin, R.J.M., Eds.; Springer: Berlin, Germany, 2013; pp. 71–92. [Google Scholar]
- Campbell, G.R.; Ohno, N.; Turnbull, D.M.; Mahad, D.J. Mitochondrial changes within axons in multiple sclerosis: An update. Curr. Opin. Neurol. 2012, 25, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinks, G.L.; Franklin, R.J. Distinctive patterns of PDGF-A, FGF-2, IGF-1 and TGF-β1 gene expression during remyelination of experimentally-induced spinal cord demyelination. Mol. Cell. Neurosci. 1999, 14, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Fancy, S.P.J.; Chan, J.R.; Baranzini, S.E.; Franklin, R.J.M.; Rowitch, D.H. Myelin regeneration: A recapitulation of development? Annu. Rev. Neurosci. 2011, 34, 21–43. [Google Scholar] [CrossRef]
- Hanafy, K.A.; Sloane, J.A. Regulation of remyelination in multiple sclerosis. FEBS Lett. 2011, 585, 3821–3828. [Google Scholar] [CrossRef] [Green Version]
- Werkman, I.L.; Lentferink, D.H.; Baron, W. Macroglial diversity: White and grey areas and relevance to remyelination. Cell. Mol. Life Sci. 2021, 78, 143–171. [Google Scholar] [CrossRef]
- Clemente, D.; Ortega, M.C.; Melero-Jerez, C.; de Castro, F. The effect of glia-glia interactions on oligodendrocyte precursor cell biology during development and in demyelinating diseases. Front. Cell. Neurosci. 2013, 7, 268. [Google Scholar] [CrossRef] [Green Version]
- Höftberger, R.; Lassmann, H. Inflammatory demyelinating diseases of the central nervous system. In Handbook of Clinical Neurology, Neuropathology; Kovacs, G.G., Alafuzoff, I., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 263–283. [Google Scholar]
- Frohman, E.M.; Racke, M.R.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Yeung, M.S.Y.; Djelloul, M.; Steiner, E.; Bernard, S.; Salehpour, M.; Possnert, G.; Brundin, L.; Frisén, J. Oligodendrocyte generation dynamics in multiple sclerosis. Nature 2019, 566, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, C.; Andrews, G.; Tamaoki, T.; Fausto, N. Alpha-Fetoprotein and albumin mRNA levels in liver regeneration and carcinogenesis. J. Biol. Chem. 1983, 258, 4901–4906. [Google Scholar] [CrossRef]
- Gao, C.; Peng, J. All routes lead to Rome: Multifaceted origin of hepatocytes during liver regeneration. Cell Regen. 2021, 10, 2. [Google Scholar] [CrossRef]
- Ritter, O.; Neyses, L. The molecular basis of myocardial hypertrophy and heart failure. Trends Mol. Med. 2003, 9, 313–321. [Google Scholar] [CrossRef]
- Lyon, R.C.; Zanella, F.; Omens, J.H.; Sheikh, F. Mechanotransduction in cardiac hypertrophy and failure. Circ. Res. 2015, 116, 1462–1476. [Google Scholar] [CrossRef] [Green Version]
- Brück, W.; Kuhlmann, T.; Stadelmann, C. Remyelination in multiple sclerosis. J. Neurol. Sci. 2003, 206, 181–185. [Google Scholar] [CrossRef]
- El Waly, B.; Macchi, M.; Cayre, M.; Durbec, P. Oligodendrogenesis in the normal and pathological central nervous system. Front. Neurosci. 2014, 8, 145. [Google Scholar] [CrossRef] [Green Version]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar]
- Patel, J.R.; Klein, R.S. Mediators of oligodendrocyte differentiation during remyelination. FEBS Lett. 2011, 585, 3730–3737. [Google Scholar] [CrossRef] [Green Version]
- Parras, C.; Marie, C.; Zhao, C.; Lu, Q.R. Chromatin remodelers in oligodendroglia. Glia 2020, 68, 1604–1618. [Google Scholar] [CrossRef] [PubMed]
- Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; van den Hove, D.; Vanmierlo, T. From OPC to oligodendrocyte: An epigenetic journey. Cells 2019, 8, 1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbaz, B.; Popko, B. Molecular control of oligodendrocyte development. Trends Neurosci. 2019, 42, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Piaton, G.; Williams, A.; Seilhean, D.; Lubetzki, C. Remyelination in multiple sclerosis. In Progress in Brain Research; Verhaagen, J., Huitinga, I., Wijnholds, J., Bergen, A.B., Boer, G.J., Swaab, D.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 175, pp. 453–464. [Google Scholar]
- Wegner, M. A matter of identity: Transcriptional control in oligodendrocytes. J. Mol. Neurosci. 2008, 35, 3–12. [Google Scholar] [CrossRef]
- Copray, S.; Huynh, J.L.; Sher, F.; Casaccia-Bonnefil, P.; Boddeke, E. Epigenetic mechanisms facilitating oligodendrocyte development, maturation, and aging. Glia 2009, 57, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Casaccia, P. Epigenetic regulation of oligodendrocyte identity. Trends Neurosci. 2010, 33, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Moyon, S.; Hernandez, M.; Casaccia, P. Epigenetic control of oligodendrocyte development: Adding new players to old keepers. Curr. Opin. Neurobiol. 2016, 39, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Brück, W.; Lucchinetti, C.; Lassmann, H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006, 129, 3165–3172. [Google Scholar] [CrossRef] [Green Version]
- Patani, R.; Balaratnam, M.; Vora, A.; Reynolds, R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol. Appl. Neurobiol. 2007, 33, 277–287. [Google Scholar] [CrossRef]
- Bramow, S.; Frischer, J.M.; Lassmann, H.; Koch-Henriksen, N.; Lucchinetti, C.F.; Sørensen, P.S.; Laursen, H. Demyelination versus remyelination in progressive multiple sclerosis. Brain 2010, 133, 2983–2998. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, B.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiCicco-Bloom, E.; Townes-Anderson, E.; Black, I.B. Neuroblast mitosis in dissociated culture: Regulation and relationship to differentiation. J. Cell Biol. 1990, 110, 2073–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, R.C.; Dorn, H.H.; Kufta, C.V.; Friedman, E.; Dubois-Dalcq, E. Pre-oligodendrocytes from adult human CNS. J. Neurosci. 1992, 12, 1538–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Los, C.; Alvarez-Buylla, A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc. Natl. Acad. Sci. USA 1993, 90, 2074–2077. [Google Scholar] [CrossRef] [Green Version]
- Gensert, J.M.; Goldman, J.E. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron 1997, 19, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.C.; Ge, B.; Duncan, I.D. Adult brain retains the potential to generate oligodendroglial progenitors with extensive myelination capacity. Proc. Natl. Acad. Sci. USA 1999, 96, 4089–4094. [Google Scholar] [CrossRef] [Green Version]
- Nait-Oumesmar, B.; Decker, L.; Lachapelle, F.; Avellana-Adalid, V.; Bachelin, C.; Baron-Van Evercooren, A. Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur. J. Neurosci. 1999, 11, 4357–4366. [Google Scholar] [CrossRef]
- Horner, P.J.; Power, A.E.; Kempermann, G.; Kuhn, H.G.; Palmer, T.D.; Winkler, J.; Thal, L.J.; Gage, F.H. Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J. Neurosci. 2000, 20, 2218–2228. [Google Scholar] [CrossRef]
- Chang, A.; Nishiyama, A.; Peterson, J.; Prineas, J.; Trapp, B.D. NG-2 positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J. Neurosci. 2000, 20, 6404–6412. [Google Scholar] [CrossRef] [Green Version]
- Doetsch, F. A niche for adult neural stem cells. Curr. Opin. Genet. Dev. 2003, 13, 543–550. [Google Scholar] [CrossRef]
- Xing, Y.L.; Röth, P.T.; Stratton, J.A.S.; Chuang, B.H.A.; Danne, J.; Ellis, S.L.; Ng, W.S.; Kilpatrick, T.J.; Merson, T.D. Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J. Neurosci. 2014, 34, 14128–14146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fancy, S.P.J.; Zhao, C.; Franklin, R.J.M. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol. Cell. Neurosci. 2004, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.J.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-Resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell. 2010, 6, 578–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmur, R.; Kessler, J.A.; Zhu, G.; Gokhan, S.; Mehler, M.F. Differentiation of oligodendroglial progenitors derived from cortical multipotent cells requires extrinsic signals including activation of gp130/LIFβ receptors. J. Neurosci. 1998, 18, 9800–9811. [Google Scholar] [CrossRef] [PubMed]
- Gago, N.; Avellana-Adalid, V.; Baron-Van Evercooren, A.; Schumacher, M. Control of cell survival and proliferation of postnatal PSA-NCAM+ progenitors. Mol. Cell. Neurosci. 2003, 22, 162–178. [Google Scholar] [CrossRef]
- Nunes, M.C.; Roy, N.S.; Keyoung, H.M.; Goodman, R.R.; McKhann, G., 2nd; Jiang, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Identification and isolation of multipotent neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003, 9, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, B.A.; Weiss, S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev. Biol. 1996, 175, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Galvez-Contreras, A.Y.; Quiñones-Hinojosa, A.; Gonzalez-Perez, O. The role of EGFR and ErbB family related proteins in the oligodendrocyte specification in germinal niches of the adult mammalian brain. Front. Cell. Neurosci. 2013, 7, 258. [Google Scholar] [CrossRef]
- Doetsch, F.; Caillé, I.; Lim, D.A.; García-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Laywell, E.D.; Rakic, P.; Kukekov, V.G.; Holland, E.C.; Steindeler, D.A. Identification of a multipotent astrocytic stem cell in the immature and adult mouse brain. Proc. Natl. Acad. Sci. USA 2000, 97, 13883–13888. [Google Scholar] [CrossRef] [Green Version]
- Akkermann, R.; Beyer, F.; Küry, P. Heterogenous populations of neural stem cells contribute to myelin repair. Neural Regen. Res. 2017, 12, 509–517. [Google Scholar] [PubMed]
- Foerster, S.; Hill, M.F.E.; Franklin, R.J.M. Diversity in the oligodendrocyte lineage: Plasticity or heterogeneity? Glia 2019, 67, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Sampaio-Baptista, C.; Johansen-Berg, H. White matter plasticity in the adult brain. Neuron 2017, 96, 1239–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santa-Olalla, J.; Covarrubias, L. Epidermal growth factor (EGF), transforming growth factor-α (TGF-α), and basic fibroblast growth factor (bFGF) differentially influence neural precursor cells of mouse embryonic mesencephalon. J. Neurosci. Res. 1995, 42, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Craig, C.C.; Tropepe, V.; Morshead, C.M.; Reynolds, B.A.; Weiss, S.; van der Kooy, D. In vivo growth factor expansion of endogenous subependymal neural precursor cell populations in the adulte mouse brain. J. Neurosci. 1996, 16, 2649–2658. [Google Scholar] [CrossRef] [Green Version]
- Martens, D.J.; Tropepe, V.; van der Kooy, D. Separate proliferation kinetics of fibroblast growth factor-responsive and epidermal growth factor-responsive neural stem cells within the embryonic forebrain germinal zone. J. Neurosci. 2000, 20, 1085–1095. [Google Scholar] [CrossRef]
- Nait-Oumesmar, B.; Picard-Riera, N.; Kerninon, C.; Decker, L.; Seilhean, D.; Höglinger, G.U.; Hirsch, E.C.; Reynolds, R.; Baron-Van Evercooren, A. Activation of the subventricular zone in multiple sclerosis: Evidence for early glial progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 4694–4699. [Google Scholar] [CrossRef] [Green Version]
- Brousse, B.; Magalon, K.; Durbec, P.; Cayre, M. Region and dynamic specificities of adult neural stem cells and oligodendrocyte precursors in myelin regeneration in the mouse brain. Biol. Open 2015, 4, 980–992. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.B.; Rivers, L.E.; Young, K.M.; Jamen, F.; Richardson, W.D. NG2 Glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. J. Neurosci. 2010, 30, 16383–16390. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Goldman, J.E. Remyelination by Endogenous Glia. In Myelin Biology and Disorders; Lazzarini, R., Ed.; Elsevier-Academic Press: San Diego, CA, USA, 2004; Volume 1, pp. 173–196. [Google Scholar]
- Cantarella, C.; Cayre, M.; Magalon, K.; Durbec, P. Intranasal HB-EGF administration favors adult SVZ cell mobilization to demyelinated lesions in mouse corpus callosum. Devel. Neurobiol. 2008, 68, 223–236. [Google Scholar] [CrossRef]
- Pichard-Riera, N.; Decker, L.; Delarasse, C.; Goude, K.; Nait-Oumesmar, B.; Liblau, R.; Pham-Dinh, D.; Baron-VanEvercooren, A. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc. Natl. Acad. Sci. USA 2002, 99, 13211–13216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayre, M.; Bancila, M.; Virard, I.; Borges, A.; Durbec, P. Migrating and myelinating potential of subventricular zone neural progenitor cells in white matter tracts of the adult rodent brain. Mol. Cell. Neurosci. 2006, 31, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Mecha, M.; Feliú, A.; Carrillo-Salinas, F.J.; Mestre, L.; Guaza, C. Mobilization of progenitors in the subventricular zone to undergo oligodendrogenesis in the Theiler’s virus model of multiple sclerosis: Implications for remyelination at lesions sites. Exp. Neurol. 2013, 250, 348–352. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Perez, O.; Quiñones-Hinojosa, A. Dose-dependent of EGF on migration and differentiation of adult subventricular zone astrocytes. Glia 2010, 58, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Perez, O.; Alvarez-Buylla, A. Oligodendrogenesis in the subventricular zone and the role of epidermal growth factor. Brain Res. Rev. 2011, 67, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Solanky, M.; Maeda, Y.; Ming, X.; Husar, W.; Li, W.; Cook, S.; Dowling, P. Proliferating oligodendrocytes are present in both active and chronic inactive multiple sclerosis plaques. J. Neurosci. Res. 2001, 65, 308–317. [Google Scholar] [CrossRef]
- Colognato, H.; Tzvetanova, I.D. Glia unglued: How signals from the extracellular matrix regulate the development of myelinating glia. Dev. Neurobiol. 2011, 71, 924–955. [Google Scholar] [CrossRef] [Green Version]
- Mitew, S.; Hay, C.M.; Peckham, H.; Xiao, J.; Koenning, M.; Emery, B. Mechanisms regulating the development of oligodendrocytes and central nervous system. Neuroscience 2014, 276, 29–47. [Google Scholar] [CrossRef]
- Boggs, J.M. Myelin basic protein: A multifunctional protein. Cell. Mol. Life Sci. 2006, 63, 1945–1961. [Google Scholar] [CrossRef]
- Emery, B.; Lu, Q.R. Transcriptional and epigenetic regulation of oligodendrocyte development and myelination in the central nervous system. Cold Spring Harb. Perspect. Biol. 2015, 7, a020461. [Google Scholar] [CrossRef] [Green Version]
- Hughes, E.G.; Stockton, M.E. Premyelinating oligodendrocytes: Mechanisms underlying cell survival and integration. Front. Cell Dev. Biol. 2021, 9, 714169. [Google Scholar] [CrossRef] [PubMed]
- Menn, B.; Garcia-Verdugo, J.M.; Yaschine, C.; Gonzalez-Perez, O.; Rowitch, D.; Alvarez-Buylla, A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J. Neurosci. 2006, 26, 7907–7918. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, O.; Romero-Rodriguez, R.; Soriano-Navarro, M.; Garcia-Valdugo, J.M.; Alvarez-Buylla, A. EGF induces the progeny of subventricular zone type B cells to migrate and differentiate into oligodendrocytes. Stem Cells 2009, 27, 2032–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.; Rizvi, T.A.; Ratner, N.; Gallo, V. Overexpression of the epidermal growth factor receptor confers migratory properties to nonmigratory postnatal neural progenitors. J. Neurosci. 2005, 25, 11092–11106. [Google Scholar] [CrossRef]
- Grinspan, J. Cells and signalling in oligodendrocyte development. J. Neuropath. Exp. Neurol. 2002, 61, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Bankston, A.N.; Mandler, M.D.; Feng, Y. Oligodendroglia and neurotrophic factors in neurodegeneration. Neurosci. Bull. 2013, 29, 216–228. [Google Scholar] [CrossRef]
- Lopez Juarez, A.; He, D.; Lu, Q.R. Oligodendrocyte progenitor programming and reprogramming: Toward myelin regeneration. Brain Res. 2016, 1638, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in development, myelin generation and beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [Green Version]
- Temple, S.; Alvarez-Buylla, A. Stem cells in the adult mammalian central nervous system. Curr. Opin. Neurobiol. 1999, 9, 135–141. [Google Scholar] [CrossRef]
- Akkermann, R.; Jadasz, J.J.; Azim, K.; Küry, P. Taking advantage of nature’s gift: Can endogenous neural stem cells improve myelin regeneration? Int. J. Mol. Sci. 2016, 17, 1985. [Google Scholar] [CrossRef] [Green Version]
- Raabe, T.D.; Deadwyler, G.; Varga, J.W.; Devries, G.H. Localization of neuregulin isoforms and erbB receptors in myelinating glial cells. Glia 2004, 45, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Deadwyler, G.D.; Pouly, S.; Antel, J.P.; Devries, G.H. Neuregulins and erbB receptor expression in adult human oligodendrocytes. Glia 2000, 32, 304–312. [Google Scholar] [CrossRef]
- Mayoral, S.R.; Chan, J.R. The environment rules: Spatiotemporal regulation of oligodendrocyte differentiation. Curr. Opin. Neurobiol. 2016, 39, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; He, Y.; Richardson, W.D.; Casaccia, P. Two-tier transcriptional control of oligodendrocyte differentiation. Curr. Opin. Neurobiol. 2009, 19, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.S.; Abdullah, S.L.; Brown, A.; Arulpragasam, A.; Crocker, S.J. How factors secreted from astrocytes impact myelin repair. J. Neurosci. Res. 2001, 89, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Kiray, H.; Lindsay, S.L.; Hosseinzadeh, S.; Barnett, S.C. The multifaceted role of astrocytes in regulating myelination. Exp. Neurol. 2016, 283, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, J.R.; Menzies, S.L. Cellular management of iron in the brain. J. Neurol. Sci. 1995, 134, 33–44. [Google Scholar] [CrossRef]
- Li, J.; Khankan, R.R.; Caneda, C.; Godoy, M.I.; Haney, M.S.; Krawczyk, M.C.; Bassik, M.C.; Sloan, S.A.; Zhang, Y. Astrocyte-to-astrocyte contact and a positive feedback loop of growth factor signaling regulate astrocyte maturation. Glia 2019, 67, 1571–1597. [Google Scholar] [CrossRef]
- Hughes, A.N. Glial cells promote myelin formation and elimination. Front. Cell Dev. Biol. 2021, 9, 661486. [Google Scholar] [CrossRef]
- Grove, J.; Gomez, J.; Kentroti, S.; Vernadakis, A. Plasticity of astrocytes derived from aged mouse cerebral hemispheres: Changes with cell passage and immortalization. Brain Res. Bull. 1996, 39, 211–217. [Google Scholar] [CrossRef]
- Anlauf, E.; Derouiche, A. Glutamine synthetase as an astrocytic marker: Its cell type and vescicle localization. Front. Endocrinol. 2013, 4, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shousboe, A.; Bak, L.K.; Madsen, K.K.; Waagepetersen, H.S. Amino Acid Neurotransmitter Synthesis and Removal. In Neuroglia, 3rd ed.; Kettenmann, H., Ranson, B.R., Eds.; Oxford University Press: Oxford, UK; New York, NY, USA, 2013; pp. 443–456. [Google Scholar]
- D’Amelio, F.; Eng, L.F.; Gibbs, M.A. Glutamine synthetase immunoreactivity is present in oligodendroglia of various regions of the central nervous system. Glia 1990, 3, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Dhaher, R.; Parent, M.; Hu, Q.-X.; Hassel, B.; Siu-Pok, Y.; Hyder, F.; Gruenbaum, S.E.; Eid, T.; Danbolt, N.C. Selective deletion of glutamine synthetase in the mouse cerebral cortex induces glial dysfunction and vascular impairment that precede epilepsy and neurodegeneration. Neurochem. Int. 2019, 123, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Eid, T.; Hassel, B.; Danbolt, N.C. Novel aspects of glutamine synthetase in ammonia homeostasis. Neurochem. Int. 2020, 140, 104809. [Google Scholar] [CrossRef]
- Haim, L.B.; Schirmer, L.; Zulji, A.; Sabeur, K.; Tiret, B.; Ribon, M.; Chang, S.; Lamers, W.H.; Boillée, S.; Chaumeil, M.M.; et al. Evidence for glutamine synthetase function in mouse spinal cord. Glia 2021, 69, 2812–2827. [Google Scholar] [CrossRef]
- Gautier, H.O.B.; Evans, K.A.; Volbracht, K.; James, R.; Sitnikov, S.; Lundgaard, I.; James, F.; Lao-Peregrin, C.; Reynolds, R.; Franklin, R.J.M.; et al. Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat. Commun. 2015, 6, 8518. [Google Scholar] [CrossRef] [Green Version]
- Wake, H.; Lee, P.R.; Fields, R.D. Control of local protein synthesis and initial events in myelination by action potentials. Science 2011, 333, 1647–1651. [Google Scholar] [CrossRef] [Green Version]
- Habermacher, C.; Angulo, M.C.; Benamer, N. Glutamate versus GABA in neuron-oligodendroglia communication. Glia 2019, 67, 2092–2106. [Google Scholar] [CrossRef]
- Han, V.K.M.; Smith, A.; Myint, W.; Nygard, K.; Bradshaw, S. Mitogenic activity of epidermal growth factor on newborn rat astroglia: Interaction with insulin-like growth factors. Endocrinology 1992, 131, 1134–1142. [Google Scholar] [CrossRef]
- Chernausek, S.D. Insulin-like growth factor-I (IGF-I) production by astroglial cells: Regulation and importance for epidermal growth factor-induced cell replication. J. Neurosci. Res. 1993, 34, 189–197. [Google Scholar] [CrossRef]
- Saneto, R.P.; Low, K.G.; Melner, M.H.; de Vellis, J. Insulin/insulin-like growth factor I and other epigenetic modulators of myelin basic protein expression in isolated oligodendrocyte progenitor cells. J. Neurosci. Res. 1988, 21, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.; Reynolds, R. Oligodendrocyte lineage. In Myelin Biology and Disorders; Lazzarini, R.A., Ed.; Elsevier-Academic Press: San Diego, CA, USA, 2004; pp. 289–310. [Google Scholar]
- Hsieh, J.; Aimone, J.B.; Kaspar, B.K.; Kuwabara, T.; Nakashima, K.; Gage, F.H. IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J. Cell. Biol. 2004, 164, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wake, H.; Ortiz, F.C.; Woo, D.H.; Lee, P.R.; Angulo, M.C.; Fields, R.D. Nonsynaptic junctions on myelinating glia promote preferential myelination of electrically active axons. Nat. Comm. 2015, 6, 7844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, F.C.; Habermacher, C.; Graciarena, M.; Houry, P.-Y.; Nishiyama, A.; Nait Oumesmar, B.; Angulo, M.C. Neuronal activity in vivo enhances functional myelin repair. JCI Insight 2019, 4, e123434. [Google Scholar] [CrossRef] [Green Version]
- Canoll, P.D.; Musacchio, J.M.; Hardy, R.; Reynolds, R.; Marchionni, M.A.; Salzer, J.L. GGF/Neuregulin is a neuronal signal that promotes the proliferation and survival and inhibits the differentiation of oligodendrocyte progenitors. Neuron 1996, 17, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Roger, P.P.; Van Heuverswyn, B.; Lambert, C.; Reuse, S.; Vassart, G.; Dumont, J.E. Antagonistic effect of thyrotropin and epidermal growth factor on thyroglobulin mRNA level in cultured thyroid cells. Eur. J. Biochem. 1985, 152, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Johe, K.K.; Hazel, T.G.; Muller, T.; Dugich-Djordjevic, M.M.; Mc Kay, R.D.G. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. 1996, 10, 3129–3140. [Google Scholar] [CrossRef] [Green Version]
- Nicolay, D.J.; Doucette, J.R.; Nazarali, A.J. Transcriptional control of oligodendrogenesis. Glia 2007, 55, 1287–1299. [Google Scholar] [CrossRef]
- Lee, J.Y.; Petratos, S. Thyroid hormone signalling in oligodendrocytes: From extracellular transport to intracellular signal. Mol. Neurobiol. 2016, 53, 6568–6583. [Google Scholar] [CrossRef]
- Vancamp, P.; Butruille, L.; Demeneix, B.A.; Remaud, S. Thyroid hormone and neural stem cells: Repair potential following brain and spinal cord injury. Front. Neurosci. 2020, 14, 875. [Google Scholar] [CrossRef]
- Pagnin, M.; Kondos-Devcic, D.; Chincarini, G.; Cumberland, A.; Richardson, S.J.; Tolcos, M. Role of thyroid hormones in normal and abnormal central nervous system myelination in humans and rodents. Front. Neuroendocrinol. 2021, 61, 100901. [Google Scholar] [CrossRef] [PubMed]
- Grade, S.; Bernardino, L.; Malva, J.O. Oligodendrogenesis from neural stem cells: Perspectives for remyelinating strategies. Int. J. Dev. Neurosci. 2013, 31, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.J.; Steffenhagen, C.; Kremer, D.; Kandasamy, M.; Sandner, B.; Couillard-Despres, S.; Weidner, N.; Küry, P.; Aigner, L. Deciphering the oligodendrocyte program of neural progenitors: Cell intrinsic and extrinsic regulators. Stem Cells Dev. 2010, 19, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Long, K.L.P.; Breton, J.M.; Barraza, M.K.; Perloff, O.S.; Kaufer, D. Hormonal regulation of oligodendrogenesis I: Effects across the lifespan. Biomolecules 2021, 11, 283. [Google Scholar] [CrossRef]
- Breton, J.M.; Long, K.L.P.; Barraza, M.K.; Perloff, O.S.; Kaufer, D. Hormonal regulation of oligodendrogenesis II: Implications for myelin repair. Biomolecules 2021, 11, 290. [Google Scholar] [CrossRef]
- Webster, H.D. Growth factors and myelin regeneration in multiple sclerosis. Mult. Scler. 1997, 3, 113–120. [Google Scholar] [CrossRef]
- Franco, P.G.; Silvestroff, L.; Soto, E.F.; Pasquini, J.M. Thyroid hormones promote differentiation of oligodendrocyte progenitor cells and improve remyelination after cuprizone-induced demyelination. Exp. Neurol. 2008, 212, 458–467. [Google Scholar] [CrossRef]
- Zhang, M.; Zhan, X.L.; Ma, Z.Y.; Chen, X.S.; Cai, Q.Y.; Yao, Z.X. Thyroid hormone alleviates demyelination induced by cuprizone through its role in remyelination during the remission period. Exp. Biol. Med. 2015, 240, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Ma, Z.; Qin, H.; Yao, Z. Thyroid hormone potentially benefits multiple sclerosis via facilitating remyelination. Mol. Neurobiol. 2016, 53, 4406–4416. [Google Scholar] [CrossRef]
- Gallo, V.; Deneen, B. Glial development: The crossroads of regeneration and repair in the CNS. Neuron 2014, 83, 283–308. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.L.; Liu, X.; Hudson, L.D.; Webster, H.D. Insulin-like growth factor I treatment reduces demyelination and up-regulates gene expression of myelin-related proteins in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 1995, 92, 6190–6194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Yao, D.L.; Webster, H. Insulin-like growth factor I treatment reduces clinical effects and lesion severity in acute experimental autoimmune encephalomyelitis. Mult. Scler. J. 1995, 1, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.; Bucci, C. Role of EGFR in the nervous system. Cells 2020, 9, 1887. [Google Scholar] [CrossRef] [PubMed]
- Metha, V.B.; Besner, G.E. Heparin-binding epidermal growth factor-like growth factor inhibits cytokine-induced NF-kB activation and nitric oxide production via activation of the phosphatidylinositol 3-kinase pathway. J. Immunol. 2005, 175, 1911–1918. [Google Scholar]
- Okada, K.; Tanaka, H.; Temporin, K.; Okamoto, M.; Kuroda, Y.; Morimoto, H.; Murase, T.; Yoshikawa, H. Methylcobalamin increases Erk1/2 and Akt activities through the methylation cycle and promotes nerve regeneration in a rat sciatic nerve injury model. Exp. Neurol. 2010, 222, 191–203. [Google Scholar] [CrossRef]
- Taveggia, C.; Feltri, M.L.; Wrabetz, L. Signals to promote myelin formation and repair. Nat. Rev. Neurol. 2010, 6, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Gaesser, J.M.; Fyffe-Maricich, S.L. Intracellular signalling pathway regulation of myelination and remyelination in the CNS. Exp. Neurol. 2016, 283, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.; Rueger, M.A.; Bae, S.-K.; Kittappa, R.; McKay, R.D.G. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442, 823–826. [Google Scholar] [CrossRef]
- Ahrendsen, J.T.; Macklin, W. Signalling mechanism regulating myelination in the central nervous system. Neurosci. Bull. 2013, 29, 199–215. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signalling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, S.P.; Flores, A.I.; Wang, F.; Macklin, W.B. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J. Neurosci. 2009, 29, 6860–6870. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.I.; Narayanan, S.P.; Morse, E.N.; Shick, H.E.; Yin, X.; Kidd, G.; Avila, R.L.; Kirschner, D.A.; Macklin, W.B. Constitutively active Akt induces enhanced myelination in the CNS. J. Neurosci. 2008, 28, 7174–7183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaes, J.E.G.; Brandt, M.J.V.; Wanders, N.; Benders, M.J.N.L.; de Theije, C.G.M.; Gressens, P.; Nijboer, C.H. The impact of throphic and immunomodulatory factors on oligodendrocyte maturation: Potential treatments for encephalopathy of prematurity. Glia 2020, 69, 1311–1340. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Vallée, J.-N.; Guillevin, R.; Lecarpentier, Y. Interactions between the canonical WNT/Beta-catenin pathway and PPAR Gamma on neuroinflammation, demyelination, and remyelination in multiple sclerosis. Cell. Mol. Neurobiol. 2018, 38, 783–795. [Google Scholar] [CrossRef]
- Gao, W.-L.; Tian, F.; Zhang, S.-Q.; Zhang, H.; Yin, Z.-S. Epidermal growth factor increases the expression of Nestin in rat reactive astrocytes through the Ras-Raf-ERK pathway. Neurosci. Lett. 2014, 562, 54–59. [Google Scholar] [CrossRef]
- Jeffries, M.A.; Urbanek, K.; Torres, L.; Wendell, S.G.; Rubio, M.E.; Fyffe-Maricich, S.L. ERK1/2 Activation in pre-existing oligodendrocytes of adult mice drives new myelin synthesis and enhanced CNS function. J. Neurosci. 2016, 36, 9186–9200. [Google Scholar] [CrossRef]
- Alcover-Sanchez, B.; Garcia-Martin, G.; Wandosell, F.; Cubelos, B. R-Ras GTPases signaling role in myelin neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 5911. [Google Scholar] [CrossRef]
- Lee, H.K.; Chaboub, L.S.; Zhu, W.; Zollinger, D.; Rasband, M.N.; Fancy, S.P.J.; Deneen, B. Daam2-PIP5K is a regulatory pathway for Wnt signalling and therapeutic target for remyelination in the CNS. Neuron 2015, 85, 1227–1243. [Google Scholar] [CrossRef] [Green Version]
- Fancy, S.P.J.; Baranzini, S.E.; Zhao, C.; Yuk, D.-I.; Irvine, K.-A.; Kaing, S.; Sanai, N.; Franklin, R.J.M.; Rowitch, D.H. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009, 23, 1571–1585. [Google Scholar] [CrossRef] [Green Version]
- Fancy, S.P.J.; Kotter, M.R.; Harrington, E.P.; Huang, J.K.; Zhao, C.; Rowitch, D.H.; Franklin, R.J.M. Overcoming remyelination failure in multiple sclerosis and other myelin disorders. Exp. Neurol. 2010, 225, 18–23. [Google Scholar] [CrossRef]
- Ortega, F.; Gascón, S.; Masserdotti, G.; Deshpande, A.; Simon, C.; Fischer, J.; Dimou, L.; Lie, D.C.; Schroeder, T.; Berninger, B. Oligodendrogliogenic and neurogenic adult subependymal zone neural stem cells constitute distinct lineages and exhibit differential responsiveness to Wnt signalling. Nat. Cell Biol. 2013, 15, 602–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Li, Z.; Zhang, G.-X.; Guan, Y. Wnt signalling in remyelination in multiple sclerosis: Friend or foe? Mol. Neurobiol. 2014, 49, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sdrulla, A.D.; diSibio, G.; Bush, G.; Nofziger, D.; Hicks, C.; Weinmaster, G.; Barres, B.A. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 1998, 21, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Genoud, S.; Lappe-Siefke, C.; Goebbels, S.; Radtke, F.; Aguet, M.; Scherer, S.S.; Suter, U.; Nave, K.-A.; Mantei, N. Notch1 control oligodendrocyte differentiation in the spinal cord. J. Cell Biol. 2002, 158, 709–718. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Mattson, M.P.; Cheng, A. Notch: From neural development to neurological disorders. J. Neurochem. 2008, 107, 1471–1481. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Argaw, A.T.; Gurfein, B.T.; Zameer, A.; Snyder, B.J.; Ge, C.; Lu, Q.R.; Rowitch, D.H.; Raine, C.S.; Brosnan, C.F.; et al. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc. Natl. Acad. Sci. USA 2009, 106, 19162–19167. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-C.; Appel, B. Delta-Notch signalling regulates oligodendrocyte specification. Development 2003, 130, 3747–3755. [Google Scholar] [CrossRef] [Green Version]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived Endothelin-1 inhibits remyelination through Notch activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef] [Green Version]
- Gadea, A.; Aguirre, A.; Haydar, T.F.; Gallo, V. Endothelin-1 regulates oligodendrocyte development. J. Neurosci. 2009, 29, 10047–10062. [Google Scholar] [CrossRef]
- Dresselhaus, E.C.; Meffert, M.K. Cellular specificity of NF-kB function in the nervous system. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef] [PubMed]
- Blank, T.; Prinz, M. NF-kB signalling regulates myelination in the CNS. Front. Mol. Neurosci. 2014, 7, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibowitz, S.M.; Yan, J. NF-kB pathways in the pathogenesis of multiple sclerosis and the therapeutic implications. Front. Mol. Neurosci. 2016, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cui, C.; Ma, X.; Luo, W.; Zheng, S.G.; Qiu, W. Nuclear factor kB (NF-kB)-mediated inflammation in multiple sclerosis. Front. Immunol. 2020, 11, 391. [Google Scholar] [CrossRef]
- Mc Guire, C.; Prinz, M.; Beyaert, R.; van Loo, G. Nuclear factor kappa B (NF-kB) in multiple sclerosis pathology. Trends Mol. Med. 2013, 13, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Stine, S.; Lin, W. Role of nuclear factor kB in multiple sclerosis and experimental autoimmune encephalomyelitis. Neural Regen. Res. 2018, 13, 1507–1515. [Google Scholar] [PubMed]
- Bonetti, B.; Stegagno, C.; Cannella, B.; Rizzuto, N.; Moretto, G.; Raine, C.S. Activation of NF-kB and c-jun transcription factors in multiple sclerosis lesions. Implications for oligodendrocyte pathology. Am. J. Pathol. 1999, 155, 1433–1438. [Google Scholar] [CrossRef]
- Chitnis, T.; Imitola, J.; Khoury, S.J. Therapeutic strategies to prevent neurodegeneration and promote regeneration in multiple sclerosis. Curr. Drug Targets—Immune. Endocr. Metab. Dis. 2005, 5, 11–26. [Google Scholar] [CrossRef]
- Ludwin, S.K.; Rao, V.T.S.; Moore, C.S.; Antel, J.P. Astrocytes in multiple sclerosis. Mult. Scler. J. 2016, 22, 1114–1124. [Google Scholar] [CrossRef]
- Williams, A.; Piaton, G.; Lubetzki, C. Astrocytes—Friends or foes in multiple sclerosis? Glia 2007, 55, 1300–1312. [Google Scholar] [CrossRef]
- Nair, A.; Frederick, T.J.; Miller, S.D. Astrocytes in multiple sclerosis: A product of their environment. Cell. Mol. Life Sci. 2008, 65, 2702–2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosnan, C.F.; Raine, C.S. The astrocyte in multiple sclerosis revisited. Glia 2013, 61, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Park, C.; Pitt, D. The role of astrocytes in multiple sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Farez, M.F. The role of astrocytes in multiple sclerosis progression. Front. Neurol. 2015, 6, 180. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Eilam, R.; Arnon, R. Astrocytes in multiple sclerosis—Essential constituents with diverse multifaceted functions. Int. J. Mol. Sci. 2021, 22, 5904. [Google Scholar] [CrossRef]
- Powers, J. Pathology of myelin. Mol. Chem. Neuropathol. 1996, 27, 31–37. [Google Scholar] [CrossRef]
- Veber, D.; Mutti, E.; Tacchini, L.; Gammella, G.; Tredici, G.; Scalabrino, G. Indirect down-regulation of nuclear NF-kB levels by cobalamin in the spinal cotd and liver of the rat. J. Neurosci. Res. 2008, 86, 1380–1387. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Rothhammer, V. Protective functions of reactive astrocytes following central nervous system insult. Front. Immunol. 2020, 11, 573256. [Google Scholar] [CrossRef]
- Metha, V.B.; Besner, G.E. Inhibition of NF-kB activation and its target genes by heparin-binding epidermal growth factor-like growth factor. J. Immunol. 2003, 171, 6014–6022. [Google Scholar]
- Chang, S.Y.C.; Chan, J.R. Tapping into the glial reservoir: Cells committed to remaining uncommitted. J. Cell Biol. 2010, 188, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Kremer, D.; Aktas, O.; Hartung, H.-P.; Küry, P. The complex world of oligodendroglial differentiation inhibitors. Ann. Neurol. 2011, 69, 602–618. [Google Scholar] [CrossRef] [PubMed]
- Kotter, M.R.; Stadelmann, C.; Hartung, H.-P. Enhancing remyelination in disease—Can we wrap it up? Brain 2011, 134, 1882–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, C.S.; Franco, S.J.; Müller, U. Extracellular matrix: Functions in the nervous system. Cold Spring Harb. Perspect. Biol. 2011, 3, a005108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front. Pharm. 2012, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Mei, F.; Chong, S.Y.C.; Chan, J.R. Myelin-based inhibitors of oligodendrocyte myelination: Clues from axonal growth and regeneration. Neurosci. Bull. 2013, 29, 177–188. [Google Scholar] [CrossRef]
- Zuchero, J.B.; Barres, B.A. Intrinsic and extrinsic control of oligodendrocyte development. Curr. Opin. Neurobiol. 2013, 23, 914–920. [Google Scholar] [CrossRef] [Green Version]
- Maki, T.; Liang, A.C.; Miyamoto, N.; Lo, E.H.; Arai, K. Mechanisms of oligodendrocyte regeneration from ventricular-subventricular zone-derived progenitor cells in white matter diseases. Front. Cell. Neurosci. 2013, 7, 275. [Google Scholar] [CrossRef] [Green Version]
- Benarroch, E.E. Extracellular matrix in the CNS. Neurology 2015, 85, 1417–1427. [Google Scholar] [CrossRef]
- Pu, A.; Stephenson, E.L.; Yong, V.W. The extracellular matrix: Focus on oligodendrocyte biology and targeting CSPGs for remyelination therapies. Glia 2018, 66, 1809–1825. [Google Scholar] [CrossRef]
- Gruchot, J.; Weyers, V.; Göttle, P.; Förster, M.; Hartung, H.-P.; Küry, P.; Kremer, D. The molecular basis for remyelination failure in multiple sclerosis. Cells 2019, 8, 825. [Google Scholar] [CrossRef] [Green Version]
- Stassart, R.; Goebbels, S.; Nave, K.-A. Factors controlling myelin formation. In Neuroglia, 3rd ed.; Kettenmann, H., Ransom, B.R., Eds.; Oxford University Press: New York, NY, USA, 2013; pp. 555–572. [Google Scholar]
- de Jong, J.M.; Wang, P.; Oomkens, M.; Baron, W. Remodeling of the interstitial extracellular matrix in white matter multiple sclerosis lesions: Implications for remyelination (failure). J. Neurosci. Res. 2020, 98, 1370–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colognato, H.; Baron, W.; Avellana-Adalid, V.; Relvas, J.B.; Baron-Van Evercooren, A.; Georges-Labouesse, E.; ffrench-Constant, C. CNS integrins switch growth factor signalling to promote target-dependent survival. Nat. Cell Biol. 2002, 4, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Baron, W.; Colognato, H.; ffrench-Constant, C. Integrin-growth factor interactions as regulators of oligodendroglial development and function. Glia 2005, 49, 467–479. [Google Scholar] [CrossRef] [PubMed]
- ffrench-Constant, C. Integrins. In Myelin Biology and Disorders; Lazzarini, R.A., Ed.; Elsevier-Academic Press: San Diego, CA, USA, 2004; Volume 1, pp. 609–632. [Google Scholar]
- Suzuki, Y.; Yanagisawa, M.; Yagi, H.; Nakatami, Y.; Yu, R.K. Involvement of β1-integrin up-regulation in basic fibroblast growth factor- and epidermal growth factor-induced proliferation of mouse neuroepithelial cells. J. Biol. Chem. 2010, 285, 18443–18451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, C.S.; Nguyen, T.; Spencer, K.S.R.; Nishiyama, A.; Colognato, H.; Müller, U. β1 integrins are required for normal CNS myelination and promote AKT-dependent myelin outgrowth. Development 2009, 136, 2717–2724. [Google Scholar] [CrossRef] [Green Version]
- Campos, L.S.; Decker, L.D.; Taylor, V.; Skarnes, W. Notch, epidermal growth factor receptor, and β1-integrin pathways are coordinated in neural stem cells. J. Biol. Chem. 2006, 281, 5300–5309. [Google Scholar] [CrossRef] [Green Version]
- O’Meara, R.W.; Michalski, J.-P.; Kothary, R. Integrin signalling in oligodendrocytes and its importance in CNS myelination. J. Signal Transduc. 2011, 2011, 354091. [Google Scholar]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [Green Version]
- Moyon, S.; Dubessy, A.L.; Aigrot, M.S.; Trotter, M.; Huang, J.K.; Dauphinot, L.; Potier, M.C.; Kerninon, C.; Parsadaniantz, S.M.; Franklin, R.J.M.; et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J. Neurosci. 2015, 35, 4–20. [Google Scholar] [CrossRef]
- Crang, A.J.; Gilson, J.M.; Li, W.-W.; Blakemore, W.F. The remyelinating potential and in vitro differentiation of MOG-expressiong oligodendrocyte precursors isolated from the adult rat CNS. Eur. J. Neurosci. 2004, 20, 1445–1460. [Google Scholar] [CrossRef]
- Huang, Y.; Dreyfus, C.F. The role of growth factors as a therapeutic approach to demyelinating disease. Exp. Neurol. 2016, 283, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uccelli, A.; Laroni, A.; Ali, R.; Battaglia, M.A.; Blinkenberg, M.; Brundin, L.; Clanet, M.; Fernandez, O.; Marriott, J.; Muraro, P.; et al. Safety, tolerability, and activity of mesenchymal stem cells versus placebo in multiple sclerosis (MESEMS): A phase 2, randomised, double-blind crossover trial. Lancet Neurol. 2021, 20, 917–929. [Google Scholar] [CrossRef]
- Thomas, R.; Wynford-Thomas, R.; Robertson, N. Advances in the use of stem cell transplants in the treatment of multiple sclerosis. J. Neurol. 2022, 269, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- El Waly, B.; Cayre, M.; Durbec, P. Promoting myelin repair through in vivo neuroblast reprogramming. Stem Cell Rep. 2018, 10, 1492–1504. [Google Scholar] [CrossRef] [Green Version]
- Rivera, F.J.; Kraus, J.; Steffenhagen, C.; Küry, P.; Weidner, N.; Aigner, L. Remyelination in multiple sclerosis: The therapeutic potential of neural and mesenchimal stem/progenitor cells. Curr. Signal Transd. Ther. 2011, 6, 1–21. [Google Scholar] [CrossRef]
- Lubetzki, C.; Zalc, B.; Williams, A.; Stadelmann, C.; Stankoff, B. Remyelination in multiple sclerosis: From basic science to clinical translation. Lancet Neurol. 2020, 19, 678–688. [Google Scholar] [CrossRef]
- Scalabrino, G. New epidermal-growth-factor-related insights into the pathogenesis of multiple sclerosis: Is it also epistemology? Front. Neurol. 2021, 12, 754270. [Google Scholar] [CrossRef]
- Fisniku, L.K.; Chard, D.T.; Jackson, J.S.; Anderson, V.M.; Altmann, D.R.; Miszkiel, K.A.; Thompson, A.J.; Miller, D.H. Gray matter atrophy is related to long-term disability in multiple sclerosis. Ann. Neurol. 2008, 64, 247–254. [Google Scholar] [CrossRef]
- Fisher, E.; Lee, J.-C.; Nakamura, K.; Rudick, R.A. Gray matter atrophy in multiple sclerosis: A longitudinal study. Ann. Neurol. 2008, 64, 255–265. [Google Scholar] [CrossRef]
- Popescu, B.G.; Pirko, I.; Lucchinetti, C.F. Pathology of multiple sclerosis: Where do we stand? CONTINUUM: Lifelong Learn. Neurol. 2013, 19, 901–921. [Google Scholar] [CrossRef]
- Prins, M.; Schul, E.; Geurts, J.; van der Valk, P.; Drukarch, B.; van Dam, A.M. Pathological differences between white and grey matter multiple sclerosis lesions. Ann. N. Y. Acad. Sci. 2015, 1351, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Magliozzi, R.; Ciccarelli, O.; Geurts, J.J.G.; Reynolds, R.; Martin, R. Exploring the origins of grey matter damage in multiple sclerosis. Nat. Rev. Neurosci. 2015, 16, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Kearney, H.; Schneider, T.; Yiannakas, M.C. Spinal cord grey matter abnormalities are associated with secondary progression and physical disability in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Haider, L.; Zrzavy, T.; Hametner, S.; Höftberger, R.; Bagnato, F.; Grabner, G.; Trattnig, S.; Pfeifengring, S.; Brück, W.; Lassmann, H. The topography of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 2016, 139, 807–815. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, C.P.; Bö, L.; Owens, T.; Lowe, J.; Esiri, M.M.; Evangelou, N. Spinal cord gray matter demylination in multiple sclerosis—a novel pattern of residual plaque morphology. Brain Pathol. 2006, 16, 202–208. [Google Scholar] [CrossRef]
- Gilmore, C.P.; Donaldson, I.; Bö, L.; Owens, T.; Evangelou, N. Regional variations in the extent and pattern of gray matter demyelination in multiple sclerosis. A comparison between the cerebral cortex, cerebellar cortex, deep grey matter nuclei and the spinal cord. J. Neurol. Neurosurg. Psychiatry 2009, 80, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Albert, M.; Antel, J.; Brück, W.; Stadelmann, C. Extensive cortical remyelination in patients with chronic multiple sclerosis. Brain Pathol. 2007, 17, 129–138. [Google Scholar] [CrossRef]
- Strijbis, E.M.M.; Kooi, E.-J.; van der Valk, P.; Geurts, J.J.G. Cortical remyelination is heterogeneous in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2017, 76, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Werkman, I.L.; Dubbelaar, M.L.; van der Vlies, P.; de Boer-Bergsma, J.J.; Eggen, B.J.L.; Baron, W. Transcriptional heterogeneity between primary adult grey and white matter astrocytes underlie differences in modulation of in vitro myelination. J. Neuroinflam. 2020, 17, 373. [Google Scholar] [CrossRef]
- Chang, A.; Staugaitis, S.M.; Dutta, R.; Batt, C.E.; Easley, K.E.; Chomyk, A.M.; Yong, V.W.; Fox, R.J.; Kidd, G.T.; Trapp, B.D. Cortical remyelination: A new target for repair therapies in multiple sclerosis. Ann. Neurol. 2012, 72, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Bø, L.; Vedeler, C.A.; Nyland, H.; Trapp, B.D.; Mørk, S.J. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult. Scler. J. 2003, 9, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Rodriguez-Rodriguez, R.; Gaebler, A.; Casals, N.; Scheller, A.; Kuerschner, L. Astrocytes and oligodendrocytes in grey and white matter regions of the brain metabolize fatty acids. Sci. Rep. 2017, 7, 10779. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Miron, V.; Cuo, Q.; Wegner, C.; Antel, J.; Brück, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, S.; Steiner, I. Experimental allergic encephalomyelitis: A misleading model of multiple sclerosis. Ann. Neurol. 2005, 58, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L.; Zamvil, S.S. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005, 26, 565–571. [Google Scholar] [CrossRef]
- Steinman, L.; Zamvil, S.S. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann. Neurol. 2006, 60, 12–21. [Google Scholar] [CrossRef]
- Denic, A.; Johnson, A.J.; Bieber, A.J.; Warrington, A.E.; Rodriguez, M.; Pirko, I. The relevance of animal models in multiple sclerosis research. Pathophysiology 2011, 18, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. Animal models of multiple sclerosis: The good, the bad and the bottom line. Nat. Neurosci. 2012, 15, 1074–1077. [Google Scholar] [CrossRef]
- Pachner, A.R. Experimental models of multiple sclerosis. Curr. Opin. Neurol. 2011, 24, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Morales, Y.; Parisi, J.E.; Lucchinetti, C.F. The pathology of multiple sclerosis: Evidence for heterogeneity. Adv. Neurol. 2006, 98, 27–45. [Google Scholar]
- Hemmer, B.; Töpfner, N.; Hartung, H.-P. Immunopathogenesis of multiple sclerosis. In Neurobiology of Disease; Gilman, S., Ed.; Elsevier-Academic Press: Amsterdam, The Netherlands, 2007; pp. 197–204. [Google Scholar]
- Frisén, J. Neurogenesis and gliogenesis in nervous system plasticity and repair. Annu. Rev. Cell Devel. Biol. 2016, 32, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yoshida, S. Mechanisms of remyelination: Recent insight from experimental models. Biomol. Concepts 2014, 5, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Brück, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.F.; Alvarez, E. The immune-pathophysiology of multiple sclerosis. Neurol. Clin. 2022, 29, 257–278. [Google Scholar] [CrossRef] [Green Version]
- Ercolini, A.M.; Miller, S.D. Mechanisms of immunopathology in murine models of central nervous system demyelinating disease. J. Immunol. 2006, 176, 3293–3298. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T. The role of CD T cells in the pathogenesis of multiple sclerosis. Inter. Rev. Neurobiol. 2007, 79, 43–72. [Google Scholar]
- Dong, Y.; Yong, V.W. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat. Rev. Neurol. 2019, 15, 704–717. [Google Scholar] [CrossRef]
- Stadelmann, C.; Wegner, C.; Brück, W. Inflammation, demyelination, and degeneration—Recent insights from MS pathology. Biochim. Biophys. Acta 2011, 1812, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Reindl, M.; Waters, P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat. Rev. Neurol. 2019, 15, 89–102. [Google Scholar] [CrossRef]
- Cobo-Calvo, A.; d’Indy, H.; Ruiz, A.; Collongues, N.; Kremer, L.; Durand-Dubief, F.; Rollot, F.; Casey, R.; Vukusic, S.; De Seze, J.; et al. Frequency of myelin oligodendrocyte glycoprotein antibody in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuerten, S.; Lanz, T.V.; Lingampalli, N.; Lahey, L.J.; Kleinschnitz, C.; Mäurer, M.; Schroeter, M.; Braune, S.; Ziemssen, T.; Ho, P.P.; et al. Autoantibodies against central nervous system antigens in a subset of B cell-dominant multiple sclerosis patients. Proc. Natl. Acad. Sci. USA 2020, 117, 21512–21518. [Google Scholar] [CrossRef] [PubMed]
- Chunder, R.; Schropp, V.; Kuerten, S. B cells in multiple sclerosis and virus-induced neuroinflammation. Front. Neurol. 2020, 11, 591894. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Behan, P.O. Multiple sclerosis is not an autoimmune disease. Arch. Neurol. 2004, 61, 1610–1612. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.P.; Barnett, M.H.; Parratt, J.D.E.; Prineas, J.W. Multiple sclerosis: Distribution of inflammatory cells in newly forming lesions. Ann. Neurol. 2009, 66, 739–753. [Google Scholar] [CrossRef]
- Franklin, R.J.M. Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci. 2002, 3, 705–714. [Google Scholar] [CrossRef]
- Dubois-Dalcq, M.; ffrench-Constant, C.; Franklin, R.J.M. Enhancing central nervous system remyelination in multiple sclerosis. Neuron 2005, 48, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Wunsch, M.; Jabari, S.; Voussen, B.; Enders, M.; Srinivasan, S.; Cossais, F.; Wedel, T.; Boettner, M.; Schwarz, A.; Weyer, L.; et al. The enteric nervous system is a potential autoimmune target in multiple sclerosis. Acta Neuropathol. 2017, 134, 281–295. [Google Scholar] [CrossRef]
- McMorris, F.A.; McKinnon, R.D. Regulation of oligodendrocyte development and CNS myelination by growth factors: Prospects for therapy of demyelinating disease. Brain Pathol. 1996, 6, 313–329. [Google Scholar] [CrossRef]
- Lovett-Racke, A.E.; Bittner, P.; Cross, A.H.; Carlino, J.A.; Racke, M.K. Regulation of experimental autoimmune encephalomyelitis with insulin-like growth factor (IGF-1) and IGF-1/IGF-binding protein-3 complex. (IGF-1/IGFBP3). J. Clin. Investig. 1998, 101, 1797–1804. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Hinks, G.L. Understanding CNS remyelination: Clues from developmental and regeneration biology. J. Neurosci. Res. 1999, 58, 207–213. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Gillian, G.L.; Voodruff, R.H.; O’Leary, M.T. What roles do growth factors play in CNS remyelination? Prog. Brain Res. 2001, 132, 185–193. [Google Scholar] [PubMed]
- Armstrong, R.C. Growth factor regulation of remyelination: Behind the growing interest in endogenous cell repair of the CNS. Fut. Neurol. 2007, 2, 689–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudi, V.; Škuljec, J.; Yildiz, O.; Frichert, K.; Skripuletz, T.; Moharregh-Khiabani, D.; Voß, E.; Wissel, K.; Wolter, S.; Stangel, M. Spatial and temporal profiles of growth factor expression during CNS demyelination reveal the dynamics of repair priming. PLoS ONE 2011, 6, e22623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Biancotti, J.C.; Yamaguchi, M.; de Vellis, J. Combination of growth factors enhances remyelination in a cuprizone-induced demyelination mouse model. Neurochem. Res. 2007, 32, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Matthieu, J.-M.; Comte, V.; Tosic, M.; Honegger, P. Myelin gene expression during demyelination and remyelination in aggregating brain cell cultures. J. Neuroimmunol. 1992, 40, 231–234. [Google Scholar] [CrossRef]
- Jarjour, A.A.; Zhang, H.; Bauer, N.; ffrench-Constant, C.; Williams, A. In vitro modelling of central nervous system myelination and remyelination. Glia 2012, 60, 1–12. [Google Scholar] [CrossRef]
- Penderis, J.; Woodruff, R.H.; Lakatos, A.; Li, W.-W.; Dunning, M.D.; Zhao, C.; Marchionni, M.; Franklin, R.J.M. Increasing local levels of neuregulin (glial growth factor-2) by direct infusion into areas of demyelination does not alter remyelination in the rat CNS. Eur. J. Neurosci. 2003, 18, 2253–2264. [Google Scholar] [CrossRef]
- Carlsson, A.; Lindqvist, M.; Magnusson, T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 1957, 180, 1200. [Google Scholar] [CrossRef]
- Nicoletti, F.; Mazzon, E.; Fagone, P.; Mangano, K.; Mammana, S.; Cavalli, E.; Basile, M.S.; Bramanti, P.; Scalabrino, G.; Lange, A.; et al. Prevention of clinical and histological signs of MOG-induced experimental allergic encephalomyelitis by prolonged treatment with recombinant human EGF. J. Neuroimmunol. 2019, 332, 224–232. [Google Scholar] [CrossRef]
- Klein, J.; Hořejší, V. Cytokines and their receptors. In Immunology, 2nd ed.; Klein, J., Hořejší, V., Eds.; Blackwell Science: Oxford, UK, 1997; pp. 291–327. [Google Scholar]
- Zhornitsky, S.; Yong, V.W.; Weiss, S.; Metz, L.M. Prolactin in multiple sclerosis. Mult. Scler. J. 2012, 19, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, G.H.; Potter, E.; Nicolaisen, A.K.; Evans, R.M.; Rosenfeld, M.G. Epidermal growth factor rapidly stimulates prolactin gene transcription. Nature 1982, 300, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Azar, S.T.; Yamout, B. Prolactin secretion is increased in patients with multiple sclerosis. Endocr. Res. 1999, 25, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Deckx, N.; Lee, W.-P.; Berneman, Z.N.; Cools, N. Neuroendocrine immunoregulation in multiple sclerosis. Clin. Dev. Immunol. 2013, 2013, 705232. [Google Scholar] [CrossRef]
- Van Buul-Offers, S.C.; Kooijman, R. The role of growth hormone and insulin-like growth factors in the immune system. Cell. Mol. Life Sci. 1998, 54, 1083–1094. [Google Scholar] [CrossRef]
- Scafidi, J.; Hammond, T.R.; Scafidi, S.; Ritter, J.; Jablonska, B.; Roncal, M.; Szigeti-Buck, K.; Coman, D.; Huang, Y.; McCarter, R.J., Jr.; et al. Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature 2014, 506, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Knapp, P.E.; Adams, M.H. Epidermal growth factor promotes oligodendrocyte process formation and regrowth after injury. Exp. Cell Res. 2004, 296, 135–144. [Google Scholar] [CrossRef]
- Iwakura, Y.; Piao, Y.-S.; Mizuno, M.; Takei, N.; Kakita, A.; Takahashi, H.; Nawa, H. Influences of dopaminergic lesion on epidermal growth factor-ErbB signals in Parkinson’s disease and its model: Neurotrophic implication in negrostriatal neurons. J. Neurochem. 2005, 93, 974–983. [Google Scholar] [CrossRef]
- Pant, S.S.; Asbury, A.K.; Richardson, E.P., Jr. The myelopathy of pernicious anemia. A neuropathological reappraisal. Acta Neurol Scand. 1968, 44 (Suppl. 35), 1–36. [Google Scholar]
- Agamanolis, D. Metabolic and Toxic Disorders. In Neuropathology; Prayson, R.A., Ed.; Elsevier-Churchill-Livingstone: Philadelphia, PA, USA, 2005; pp. 339–420. [Google Scholar]
- Miller, A.; Korem, M.; Almog, R.; Galboiz, Y. Vitamin B12, demyelination, remyelination, and repair in multiple sclerosis. J. Neurol. Sci. 2005, 233, 93–97. [Google Scholar] [CrossRef]
- Scalabrino, G. Cobalamin (vitamin B12) in subacute combined degeneration and beyond: Traditional interpretations and novel theories. Exp. Neurol. 2005, 192, 463–479. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.R.; Shaygannajad, V.; Mirpourian, M.; Gholamrezaei, A. Vitamin B12 deficiency and multiple sclerosis; is there any association? Int. J. Prev. Med. 2012, 3, 286–289. [Google Scholar] [PubMed]
- Scalabrino, G.; Galimberti, D.; Mutti, E.; Scalabrini, D.; Veber, D.; De Riz, M.; Bamonti, F.; Capello, E.; Mancardi, G.L.; Scarpini, E. Loss of epidermal growth factor regulation by cobalamin in multiple sclerosis. Brain Res. 2010, 1333, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.H. Blood-Brain and Spinal Cord Barriers in Stress. In Blood-Spinal Cord and Brain Barriers in Health and Disease; Sharma, H.S., Westman, J., Eds.; Elsevier-Academic Press: San Diego, CA, USA, 2004; pp. 231–298. [Google Scholar]
- Scalabrino, G.; Veber, D.; De Giuseppe, R.; Roncaroli, F. Low levels of cobalamin, epidermal growth factor, and normal prions in multiple sclerosis spinal cord. Neuroscience 2015, 298, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.L.; Garg, P.; Thin, T.H.; Yoo, S.; Dutta, R.; Trapp, B.D.; Haroutunian, V.; Zhu, J.; Donovan, M.J.; Sharp, A.J.; et al. Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat. Neurosci. 2014, 17, 121–130. [Google Scholar] [CrossRef]
- Huynh, J.L.; Casaccia, P. Epigenetic mechanisms in multiple sclerosis: Implications for pathogenesis and treatment. Lancet Neurol. 2013, 12, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Casaccia-Bonnefil, P.; Pandozy, G.; Mastronardi, F. Evaluating epigenetic landmarks in the brain of multiple sclerosis patients: A contribution to the current debate on disease pathogenesis. Prog. Neurobiol. 2008, 86, 368–378. [Google Scholar]
- Dutta, R.; Trapp, B.D. Gene expression profiling in multiple sclerosis brain. Neurobiol. Dis. 2012, 45, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.W.; Metz, L.M.; Kovalchuk, O. Epigenetic changes in patients with multiple sclerosis. Nat. Rev. Neurol. 2013, 9, 35–43. [Google Scholar] [CrossRef]
- Chomyk, A.M.; Volsko, C.; Tripathi, A.; Deckard, S.A.; Trapp, B.D.; Fox, R.J.; Dutta, R. DNA methylation in demyelinated multiple sclerosis hippocampus. Sci. Rep. 2017, 7, 8696. [Google Scholar] [CrossRef] [Green Version]
- Mastronardi, F.G.; Min, W.; Wang, H.; Winer, S.; Dosch, M.; Boggs, J.M.; Moscarello, M.A. Attenuation of experimental autoimmune encephalomyelitis and nonimmune demyelination by IFN-β plus vitamin B12: Treatment to modify Notch-1/Sonic hedgehog balance. J. Immunol. 2004, 172, 6418–6426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; Ackley, B.D.; Yan, D. Vitamin B12 regulates glial migration and synapse formation through isoform-specific control of PTP-3/LAR PRTP expression. Cell Rep. 2020, 30, 3981–3988. [Google Scholar] [CrossRef] [PubMed]
- Moestrup, S.K.; Birn, H.; Fischer, P.B.; Petersen, C.M.; Verroust, P.J.; Sim, R.B.; Christensen, E.I.; Nexø, E. Megalin-mediated endocytosis of transcobalamin-vitamin-B12 complexes suggests a role of the receptor in vitamin-B12 homeostasis. Proc. Natl. Acad. Sci. USA 1996, 93, 8612–8617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birn, H.; Willnow, T.E.; Nielsen, R.; Norden, A.G.W.; Bönsch, C.; Moestrup, S.K.; Nexø, E.; Christensen, E.I. Megalin is essential for renal proximal tubule reabsorption and accumulation of transcobalamin-B12. Am. J. Physiol. Ren. Physiol. 2002, 282, F408–F416. [Google Scholar] [CrossRef] [Green Version]
- Kozyraki, R.; Cases, O. Cubilin, the intrinsic factor-vitamin B12 receptor in development and disease. Curr. Med. Chem. 2020, 27, 3123–3150. [Google Scholar] [CrossRef]
- Ortega, M.C.; Cases, O.; Merchán, P.; Kozyraki, R.; Clemente, D.; De Castro, F. Megalin mediates the influence of Sonic hedgehog on oligodendrocyte precursor cell migration and proliferation during development. Glia 2012, 60, 851–856. [Google Scholar] [CrossRef]
- Winkler, C.C.; Franco, S.J. Loss of Shh signalling in the neocortex reveals heterogeous cell recovery responses from distinct oligodendrocyte populations. Dev. Biol. 2019, 452, 55–65. [Google Scholar] [CrossRef]
- Mastronardi, F.G.; daCruz, L.A.G.; Wang, H.; Boggs, J.; Moscarello, M.A. The amount of sonic hedgehog in mukltiple sclerosis white matter is decreased and cleavage to the signalling peptide is deficient. Mult. Scler. 2003, 9, 362–371. [Google Scholar] [CrossRef]
- Friese, M.A.; Fugger, L. Pathogenic CD8+ T cells in multiple sclerosis. Ann. Neurol. 2009, 66, 132–141. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Tamura, J.; Kubota, K.; Murakami, H.; Sawamura, M.; Matsushima, T.; Tamura, T.; Saitoh, T.; Kurabayshi, H.; Naruse, T. Immunomodulation by vitamin B12: Augmentation of CD8+ T lymphocytes and natural killer (NK) cell activity in vitamin B12-deficient patients by methyl-B12 treatment. Clin. Exp. Immunol. 1999, 116, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Funada, U.; Wada, M.; Kawata, T.; Mori, K.; Tamai, H.; Kawanishi, T.; Kunou, A.; Tanaka, N.; Tadokoro, T.; Maekawa, A. Changes in CD4+CD8−/CD4−CD8+ ratio and humoral immune functions in vitamin B12-deficient rats. Int. J. Vitam. Nutr. Res. 2000, 70, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Paul, S.; Roy, S.; Sutradhar, H.; Emran, T.B.; Nainu, F.; Khandaker, M.U.; Almalki, M.; Wilairatana, P.; Mubarak, M.S. Exploring the immune-boosting functions of vitamins and minerals as nutritional food bioactive compounds: A comprehensive review. Molecules 2022, 27, 555. [Google Scholar] [CrossRef] [PubMed]
- Mars, L.T.; Saikali, P.; Liblau, R.S.; Arbour, N. Contribution of CD8 T lymphocytes to the immuno-pathogenesis of multiple sclerosis and its animal models. Biochim. Biophys. Acta 2011, 1812, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.; Itani, F.R.; Karandikar, N.J. Immune regulation of multiple sclerosis by CD8+ T cells. Immunol. Res. 2014, 59, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Liebert, A.; Bicknell, B.; Adams, R. Prion protein signalling in the nervous system—A review and perspective. Signal Transduct. Insights 2014, 3, 11–32. [Google Scholar] [CrossRef] [Green Version]
- Encalada, S.E.; Szpankowski, L.; Xia, C.-h.; Lawrence, S.B. Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vescicles. Cell 2011, 144, 551–565. [Google Scholar] [CrossRef] [Green Version]
- Wadsworth, J.D.F.; Asante, E.A.; Collinge, J. Contribution of transgenic models to understanding human prion disease. Neuropathol. Appl. Neurobiol. 2010, 36, 576–597. [Google Scholar] [CrossRef] [Green Version]
- Nazor, K.E.; Seward, T.; Telling, G.C. Motor behavioural and neuropathological deficits in mice deficient for normal prion protein expression. Biochim. Biophys. Acta 2007, 1772, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Baumann, F.; Pahnke, J.; Radovanovic, I.; Rülicke, T.; Bremer, J.; Tolnay, M.; Aguzzi, A. Functionally relevant domains of the prion protein identified In Vivo. PLoS ONE 2009, 4, e6707. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, R.; Piccardo, P.; Ghetti, B.; Harris, D.A. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 1998, 21, 1339–1351. [Google Scholar] [CrossRef] [Green Version]
- Solomon, I.H.; Schepker, J.A.; Harris, D.A. Prion neurotoxicity: Insights from prion protein mutants. Curr. Issues Mol. Biol. 2010, 12, 51–61. [Google Scholar] [PubMed]
- Moscarello, M.A.; Pritzker, L.; Mastronardi, F.G.; Wood, D.D. Peptidylarginine deaminase: A candidate factor in demyelinating disease. J. Neurochem. 2002, 81, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, B.; Jeon, Y.-C.; Shin, H.-Y.; Lee, Y.-J.; Kim, H.; Kondo, Y.; Ishigami, A.; Kim, Y.-S.; Choi, E.-K. Myelin basic protein citrullination, a hallmark of central nervous system demyelination, assessed by novel monoclonal antibodies in prion diseases. Mol. Neurobiol. 2018, 55, 3172–3184. [Google Scholar] [CrossRef] [PubMed]
- Bribián, A.; Fontana, X.; Llorens, F.; Gavín, R.; Reina, M.; García-Verdugo, M.; Torres, J.M.; de Castro, F.; del Rio, J.A. Role of the cellular prion protein in oligodendrocyte precursor cell proliferation and differentiation in the developing and adult mouse CNS. PLoS ONE 2012, 7, e33872. [Google Scholar] [CrossRef] [Green Version]
- Roucou, X.; LeBlanc, A.C. Cellular prion protein in neuroprotective function: Implications in prion diseases. J. Mol. Med. 2005, 83, 3–11. [Google Scholar] [CrossRef]
- Ishikura, N.; Clever, J.L.; Bouzamondo-Bernstein, E.; Samayoa, E.; Prusiner, S.B.; Huang, E.J.; DeArmond, S.J. Notch-1 activation and dendritic atrophy in prion disease. Proc. Natl. Acad. Sci. USA 2005, 102, 886–891. [Google Scholar] [CrossRef] [Green Version]
- Onodera, T.; Sugiura, K.; Matsuda, S.; Sakudo, A. Function of cellular prion protein. In Prions, 1st ed.; Sakudo, A., Onodera, T., Eds.; Caister Academic Press: Norfolk, UK, 2013; pp. 11–29. [Google Scholar]
- Lazarini, F.; Castelnau, P.; Chermann, J.F.; Deslys, J.P.; Dormont, D. Modulation of prion protein gene expression by growth factors in cultured mouse astrocytes and PC-12 cells. Mol. Brain Res. 1994, 22, 268–274. [Google Scholar] [CrossRef]
- Tsutsui, S.; Hahn, J.N.; Johnson, T.A.; Ali, Z.; Jirik, F.R. Absence if the cellular prion protein exacerbates and prolongs neuroinflammation in experimental autoimmune encephalomyelitis. Am. J. Pathol. 2008, 173, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Gourdain, P.; Ballerini, C.; Nicot, A.B.; Carnaud, C. Exacerbation of experimental autoimmune encephalomyelitis in prion protein protein (PrPc)-null mice: Evidence for a critical role of the central nervous system. J. Neuroinflam. 2012, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Nessler, S.; Hemmer, B.; Eagar, T.N.; Kane, L.P.; Leliveld, S.R.; Müller-Schiffmann, A.; Gocke, A.R.; Lovett-Racke, A.; Ben, L.-H.; et al. Pharmacological prion protein silencing accelerates central nervous system autoimmune disease via T cell receptor signalling. Brain 2010, 133, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, R.; Trapp, B.D. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology 2007, 68 (Suppl. 3), S22–S31. [Google Scholar] [CrossRef] [PubMed]
- Keegan, M.; Noseworthy, J.H. Multiple sclerosis. Annu. Rev. Med. 2002, 53, 285–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassmann, H.; Brück, W.; Lucchinetti, C. Heterogeneity of multiple sclerosis pathogenesis: Implications for diagnosis and therapy. Trends Mol. Med. 2001, 7, 115–121. [Google Scholar] [CrossRef]
- Hemmer, B.; Archelos, J.J.; Hartung, H.-P. New concepts in the immunopathogenesis of multiple sclerosis. Nat. Rev. Neurosci. 2002, 3, 291–301. [Google Scholar] [CrossRef]
- Stys, P.K. Multiple sclerosis: Autoimmune disease or autoimmune reaction? Can. J. Neurol. Sci. 2010, 37, S16–S23. [Google Scholar] [CrossRef] [Green Version]
- Stys, P.K. Pathoetiology of multiple sclerosis: Are we barking up the wrong tree? F1000Prime Rep. 2013, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Barnett, M.H.; Prineas, J.W. Relapsing and remitting multiple sclerosis: Pathology of the newly forming lesion. Ann. Neurol. 2004, 55, 458–468. [Google Scholar] [CrossRef]
- Rahmanzaseh, R.; Brück, W.; Minagar, A.; Sahraian, A. Multiple sclerosis pathogenesis: Missing pieces of an old puzzle. Rev. Neurosci. 2019, 30, 67–83. [Google Scholar] [CrossRef]
- Lassmann, H. Multiple sclerosis: Lessons from molecular neuropathology. Exp. Neurol. 2014, 262, 2–7. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.K.; Fancy, S.P.J.; Zhan, C.; Rowitch, D.H.; ffrench-Constant, C.; Franklin, R.J.M. Myelin regeneration in multiple sclerosis: Targeting endogenous stem cells. Neurotherapeutics 2011, 8, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, R.J.M.; Goldman, S.A. Glia disease and repair—Remyelination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020594. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; ffrench-Constant, C. Regenerating CNS myelin—From mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Peixoto de Barcelos, I.; Troxell, R.M.; Graves, J.S. Mitochondrial dysfunction and multiple sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in multiple sclerosis: Molecular mechanisms of pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103. [Google Scholar]
- Campbell, G.R.; Mahad, D.J. Mitochondria as crucial players in demyelinated axons: Lessons from neuropathology and experimental demyelination. Autoim. Dis. 2011, 2011, 262847. [Google Scholar] [CrossRef] [Green Version]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [Green Version]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple scelrosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Zambonin, J.L.; Zhao, C.; Ohno, N.; Campbell, G.R.; Engeham, S.; Ziabreva, I.; Schwarz, N.; Lee, S.E.; Frischer, J.M.; Turnbull, D.M.; et al. Increased mitochondrial content in remyelinated axons: Implications for multiple sclerosis. Brain 2011, 134, 1901–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panzica, G.C.; Melcangi, R.C. New perspectives for the action of steroids in the brain. J. Neuroendocrinol. 2018, 30, e12576. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Waller-Evans, H. Biosynthesis and signalling functions of central and peripheral nervous system neurosteroids in health and disease. Essays Biochem. 2020, 64, 591–606. [Google Scholar] [PubMed]
- Giatti, S.; Diviccaro, S.; Serafini, M.M.; Caruso, D.; Garcia-Segura, L.M.; Viviani, B.; Melcangi, R.C. Sex differences in steroid levels and steroidogenesis in the nervous system: Physiopathological role. Front. Neuroendocrinol. 2020, 56, 100804. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Ellestad, K.K.; Maingat, F.; Warren, K.G.; Han, M.H.; Steinman, L.; Baker, G.B.; Power, C. Impaired neurosteroid synthesis in multiple sclerosis. Brain 2011, 134, 2703–2721. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, M.; Hussain, R.; Gago, N.; Oudinet, J.-P.; Mattern, C.; Ghoumari, A.M. Progesterone synthesis in the nervous system: Implications for myelination and myelin repair. Front. Neurosci. 2012, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, C.; Karali, K.; Fodelianaki, G.; Gravanis, A.; Chavakis, T.; Charalampopoulos, I.; Alexaki, V.I. Neurosteroids as regulators of neuroinflammation. Front. Neuroendocrinol. 2019, 55, 100788. [Google Scholar] [CrossRef]
- Stys, P.K.; Zamponi, G.W.; van Minnen, J.; Geurts, J.J.G. Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 2012, 13, 507–514. [Google Scholar] [CrossRef]
- Vohl, K.; Duscha, A.; Gisevius, B.; Kaisler, J.; Gold, R.; Haghikia, A. Predictors for therapy response to intrathecal corticosteroid therapy in multiple sclerosis. Front. Neurol. 2019, 10, 132. [Google Scholar] [CrossRef] [Green Version]
- Morrow, S.A.; McEvan, L.; Alikhani, K.; Hyson, C.; Kremenchutzky, M. MS Patients report excellent compliance with oral prednisone for acute relapses. Can. J. Neurol. Sci. 2012, 39, 352. [Google Scholar] [CrossRef] [Green Version]
- Brambilla, R.; Bracchi-Ricard, V.; Hu, W.-H.; Frydel, B.; Bramwell, A.; Karmally, S.; Green, E.J.; Bethea, J.R. Inhibition of astroglial nuclear factor κB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med. 2005, 202, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambilla, R.; Persuad, T.; Hu, X.; Karmally, S.; Shestopalov, V.I.; Dvoriantchikova, G.; Ivanov, D.; Nathanson, L.; Barnum, S.R.; Bethea, J.R. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J. Immunol. 2009, 182, 2628–2640. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Morton, P.D.; Ashbaugh, J.J.; Karmally, S.; Lambertsen, K.L.; Bethea, J.R. Astrocytes play a key role in EAE pathophysiology by orchestrating in the CNS the inflammatory response of resident and peripheral immune cells and by suppressing remyelination. Glia 2014, 62, 452–467. [Google Scholar] [CrossRef] [PubMed]
- Raasch, J.; Zeller, N.; van Loo, G.; Merkler, D.; Mildner, A.; Erny, D.; Knobeloch, K.-P.; Bethea, J.R.; Waisman, A.; Knust, M.; et al. IκB kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-κB in the central nervous system. Brain 2011, 134, 1184–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Farina, C. Astrocytes: Key regulators of neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Leitner, H. Influence of neurosteroids on the pathogenesis of multiple sclerosis. Med. Hypoth. 2010, 75, 229–234. [Google Scholar] [CrossRef]
- Boghozian, R.; McKenzie, B.A.; Saito, L.B.; Metha, N.; Branton, W.G.; Lu, J.Q.; Baker, F.; Noorbakhsh, F.; Power, C. Suppressed oligodendrocyte steroidogenesis in multiple sclerosis: Implications for regulation of neuroinflammation. Glia 2017, 65, 1590–1606. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Koreman, E.; Sun, X.; Lu, Q.R. Chromatin remodeling and epigenetic regulation of oligodendrocyte myelination and myelin repair. Mol. Cell. Neusosci. 2018, 87, 18–26. [Google Scholar] [CrossRef]
- Samudyata; Castelo-Branco, G.; Liu, J. Epigenetic regulation of oligodendrocyte differentiation: From development to demyelinating disorders. Glia 2020, 68, 1619–1630. [Google Scholar] [CrossRef]
- Moyon, S.; Huynh, J.L.; Dutta, D.; Zhang, F.; Ma, D.; Yoo, S.; Lawrence, R.; Wegner, M.; John, G.R.; Emery, B.; et al. Functional characterization of DNA methylation in the oligodendrocyte lineage. Cell Rep. 2016, 15, 748–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyon, S.; Ma, D.; Huynh, J.L.; Coutts, D.J.C.; Zhao, C.; Casaccia, P.; Franklin, R.J.M. Efficient remyelination requires DNA methylation. eNeuro 2017, 4, ENEURO.0336-16.2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedre, X.; Mastronardi, F.; Brück, W.; López-Rodas, G.; Kuhlmann, T.; Casaccia, P. Changed histone acetylation patterns in normal-appearing white matter and early multiple sclerosis lesions. J. Neurosci. 2011, 31, 3435–3445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junker, A.; Hohlfeld, R.; Meinl, E. The emerging role of microRNAs in multiple sclerosis. Nat. Rev. Neurol. 2011, 7, 56–59. [Google Scholar] [CrossRef]
- Teuber-Hanselmann, S.; Meinl, E.; Junker, A. MicroRNAs in gray and white matter multiple sclerosis: Impact on pathophysiology. J. Pathol. 2020, 250, 496–509. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Yeo, G.; Muotri, A.R.; Kuwabara, T.; Gage, F.H. Noncoding RNAs in the mammalian central nervous system. Annu. Rev. Neurosci. 2006, 29, 77–103. [Google Scholar] [CrossRef] [Green Version]
- Martinez, B.; Peplow, P.V. MicroRNAs in blood and cerebrospinal fluid as diagnostic biomarkers of multiple sclerosis and to monitor disease progression. Neural Regen. Res. 2021, 15, 606–619. [Google Scholar]
- De Faria, O., Jr.; Moore, C.S.; Kennedy, T.E.; Antel, J.P.; Bar-Or, A.; Dhaunchak, A.S. MicroRNA dysregulation in multiple sclerosis. Front. Genet. 2013, 3, 311. [Google Scholar]
- Li, J.-S.; Yao, Z.-X. MicroRNAs: Novel regulators of oligodendrocyte differentiation and potential therapeutic targets in demyelination-related diseases. Mol. Neurobiol. 2012, 45, 200–212. [Google Scholar] [CrossRef]
- Dugas, J.C.; Cuellar, T.L.; Scholze, A.; Ason, B.; Ibrahim, A.; Emery, B.; Zamanian, J.L.; Foo, L.C.; McManus, M.T.; Barres, B.A. Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination. Neuron 2010, 65, 597–611. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; He, X.; Han, X.; Yu, Y.; Ye, Y.; Chen, Y.; Hoang, T.N.; Xu, X.; Mi, Q.-S.; Xin, M.; et al. MicroRNA-mediated control of oligodendrocyte differentiation. Neuron 2010, 65, 612–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junker, A.; Krumbholz, M.; Eisele, S.; Mohan, H.; Augstein, F.; Bittner, R.; Lassmann, H.; Wekerle, H.; Hohlfeld, R.; Meinl, E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain 2009, 132, 3342–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marangon, D.; Boda, E.; Parolisi, R.; Negri, C.; Giorgi, C.; Montarolo, F.; Perga, S.; Bertolotto, A.; Buffo, A.; Abbracchio, M.P.; et al. In vivo silencing of miR-125a-3p promotes myelin repair in models of white matter demyelination. Glia 2020, 68, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Marangon, D.; Coppolino, G.T.; Menéndez Méndez, A.; Finardi, A.; Dalla Costa, G.; Martinelli, V.; Furlan, R.; Abbracchio, M.P. MiR-125a-3p timely inhibits oligodendroglial maturation and is pathologically up-regulated in human multiple sclerosis. Sci. Rep. 2016, 6, 34503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghikia, A.; Haghikia, A.; Hellwig, K.; Baraniskin, A.; Holzmann, A.; Décard, B.F.; Thum, T.; Gold, R. Regulated microRNAs in the CSF of patients with multiple sclerosis. Neurology 2012, 79, 2166–2170. [Google Scholar] [CrossRef] [PubMed]
- Piket, E.; Zheleznyakova, G.Y.; Kular, L.; Jagodic, M. Small non-coding RNAs as important players, biomarkers and therapeutic targets in multiple sclerosis: A comprehensive overview. J. Autoimmun. 2019, 101, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Dolati, S.; Marofi, F.; Babaloo, Z.; Aghebati-Maleki, L.; Roshangar, L.; Ahmadi, M.; Rikhtegar, R.; Yousefi, M. Dysregulated network of miRNAs involved in the pathogenesis of multiple sclerosis. Biomed. Pharmacother. 2018, 104, 280–290. [Google Scholar] [CrossRef]
- Walsh, A.D.; Nguyen, L.T.; Binder, M.D. miRNAs in microglia: Important players in multiple sclerosis pathology. ASN Neuro 2021, 13, 1–23. [Google Scholar] [CrossRef]
- Gao, Y.; Han, D.; Feng, J. MicroRNA in multiple sclerosis. Clin. Chim. Acta 2021, 516, 92–99. [Google Scholar] [CrossRef]
- Meinl, E.; Meister, G. MicroRNAs in the CSF. Neurology 2012, 79, 2162–2163. [Google Scholar] [CrossRef]
- Zhou, Z.; Xiong, H.; Xie, F.; Wu, Z.; Feng, Y. A meta-analytic review of the value of miRNA for multiple sclerosis diagnosis. Front. Neurol. 2020, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.G.; Lu, M.; Wang, X.; Shang, X.; Elias, S.B.; Chopp, M. Mir-146a promotes remyelination in a cuprizone model of demyelinating injury. Neuroscience 2017, 348, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ren, C.; Qu, X.; Wu, X.; Dong, F.; Chand, Y.K.; Fan, H.; Yao, R.; Geng, D. miR-219 attenuates demyelination in cuprizone-induced demyelinated mice by regulating monocarboxylate transporter 1. Eur. J. Neurosci. 2017, 45, 249–259. [Google Scholar] [CrossRef]
- Fan, H.-B.; Chen, L.-X.; Qu, X.-B.; Ren, C.-L.; Wu, X.-X.; Dong, F.-X.; Zhang, B.-L.; Gao, D.-S.; Yao, R.-Q. Transplanted miR-219-overexpressing oligodendrocyte precursor cells promoted remyelination and improved functional recovery in a chronic demyelinated model. Sci. Rep. 2017, 7, 41407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musse, A.A.; Harauz, G. Molecular “negativity” may underlie multiple sclerosis: Role of the myelin basic protein family in the pathogenesis of MS. Int. Rev. Neurobiol. 2007, 79, 149–172. [Google Scholar] [PubMed]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Űber das ausgebreitete Vorkommen einer dem Nervenmark analogen Substanz in den tierischen Geweben. Virchow Arch. Pathol. Anat. 1854, 6, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Snaidero, N.; Simons, M. Myelination at a glance. J. Cell Sci. 2014, 127, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Pöllinger, B.; Krishnamoorthy, G.; Berer, K.; Lassmann, H.; Bösl, M.R.; Dunn, R.; Domingues, H.S.; Holz, A.; Kurschus, F.C.; Wekerle, H. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J. Exp. Med. 2009, 206, 1303–1316. [Google Scholar] [CrossRef]
- Scheikl, T.; Pignolet, B.; Mars, L.T.; Liblau, R.S. Transgenic mouse models of multiple sclerosis. Cell. Mol. Life Sci. 2010, 67, 4011–4034. [Google Scholar] [CrossRef]
- Burrows, D.J.; McGown, A.; Jain, S.A.; De Felice, M.; Ramesh, T.M.; Sharrack, B.; Majid, A. Animal models of multiple sclerosis: From rodents to zebrafish. Mult. Scler. J. 2019, 25, 306–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, H.Y.F.; Yong, V.W. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat. Rev. Neurol. 2022, 18, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Greer, J.M. NF-kB, a potential therapeutic target for the treatment of multiple sclerosis. CNS Neutol. Dis. 2008, 7, 536–557. [Google Scholar] [CrossRef] [Green Version]
- Awad, A.; Hemmer, B.; Hartung, H.-P.; Kieseier, B.; Bennett, J.L.; Stüve, O. Analyses of cerebrospinal fluid in the diagnosis and monitoring of multiple sclerosis. J. Neuroimmunol. 2010, 219, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stangel, M.; Fredrikson, S.; Meinl, E.; Petzold, A.; Stüve, O.; Tumano, H. The utility of cerebrospinal fluid analysis in patients with multiple sclerosis. Nat. Rev. Neurol. 2013, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Tumani, H.; Hartung, H.-P.; Hemmer, B.; Teunissen, C.; Deisenhammer, F.; Giovannoni, G.; Zettl, U.; The BioMS Study Group. Cerebrospinal fluid biomarkers in multiple sclerosis. Neurobiol. Dis. 2009, 35, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Popper, K.R. The Logic of Scientific Discovery; Routledge: Abingdon-on-Thames, UK, 1959; pp. 27–48. [Google Scholar]
- Marchionni, M.A.; Cannella, B.; Hoban, C.; Gao, Y.L.; Garcia-Arenas, R.; Lawson, D.; Happel, E.; Noel, F.; Tofilon, P.; Gwynne, D.; et al. Neuregulin in neuron/glia interactions in the central nervous system. GGF2 diminishes autoimmune demyelination, promotes oligodendrocyte progenitor expansion, and enhances remyelination. Adv. Exp. Med. Biol. 1999, 468, 283–295. [Google Scholar]
- Chang, A.; Turtelotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Gallo, V. The translational biology of remyelination: Past, present, and future. Glia 2014, 62, 1905–1915. [Google Scholar] [CrossRef]
- De Castro, F.; Bribián, A.; Ortega, M.C. Regulation of oligodendrocyte precursor migration during development, in adulthood and in pathology. Cell. Mol. Life Sci. 2013, 70, 4355–4368. [Google Scholar] [CrossRef]
- Jones, E.V.; Bouvier, D.S. Astrocyte-secreted matricellular proteins in CNS remodelling during development and disease. Neur. Plas. 2014, 2014, 321209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Traiffort, E.; Kassoussi, A.; Zahaf, A.; Laouarem, Y. Astrocytes and microglia as major players of myelin production in normal and pathological conditions. Front. Cell. Neurosci. 2020, 14, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nave, K.-A.; Trapp, B.D. Axon-glial signaling and the glial support of axon function. Annu. Rev. Neurosci. 2008, 31, 535–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, V.; Armstrong, R. Myelin repair strategies: A cellular view. Curr. Opin. Neurol. 2008, 21, 278–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waxman, S.G. Multiple Sclerosis as a Neurodegenerative Disease. In Molecular Neurology; Waxman, S.G., Ed.; Elsevier-Academic Press: Amsterdam, The Netherlands, 2007; pp. 333–346. [Google Scholar]
- Villoslada, P.; Steinman, L. New targets and therapeutics for neuroparotection, remyelination and repair in multiple sclerosis. Expert Opin. Investig. Drugs 2020, 29, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H.; Mi, S. Dissecting demyelination. Nat. Neurosci. 2007, 10, 1351–1354. [Google Scholar] [CrossRef]
- Norgren, N.; Sundström, P.; Svenningsson, A.; Rosengren, L.; Stigbrand, T.; Gunnarsson, M. Neurofilament and glial fibrillary acidic protein in multiple sclerosis. Neurology 2004, 63, 1586–1590. [Google Scholar] [CrossRef]
- Petzold, A. Glial fibrillary acidic protein is a body fluid biomarker for glial pathology in human disease. Brain Res. 2015, 1600, 1–31. [Google Scholar] [CrossRef]
- Mastronardi, F.G.; Moscarello, M.A. Molecules affecting myelin stability: A novel hypothesis regarding the pathogenesis of multiple sclerosis. J. Neurosci. Res. 2005, 80, 301–308. [Google Scholar] [CrossRef]




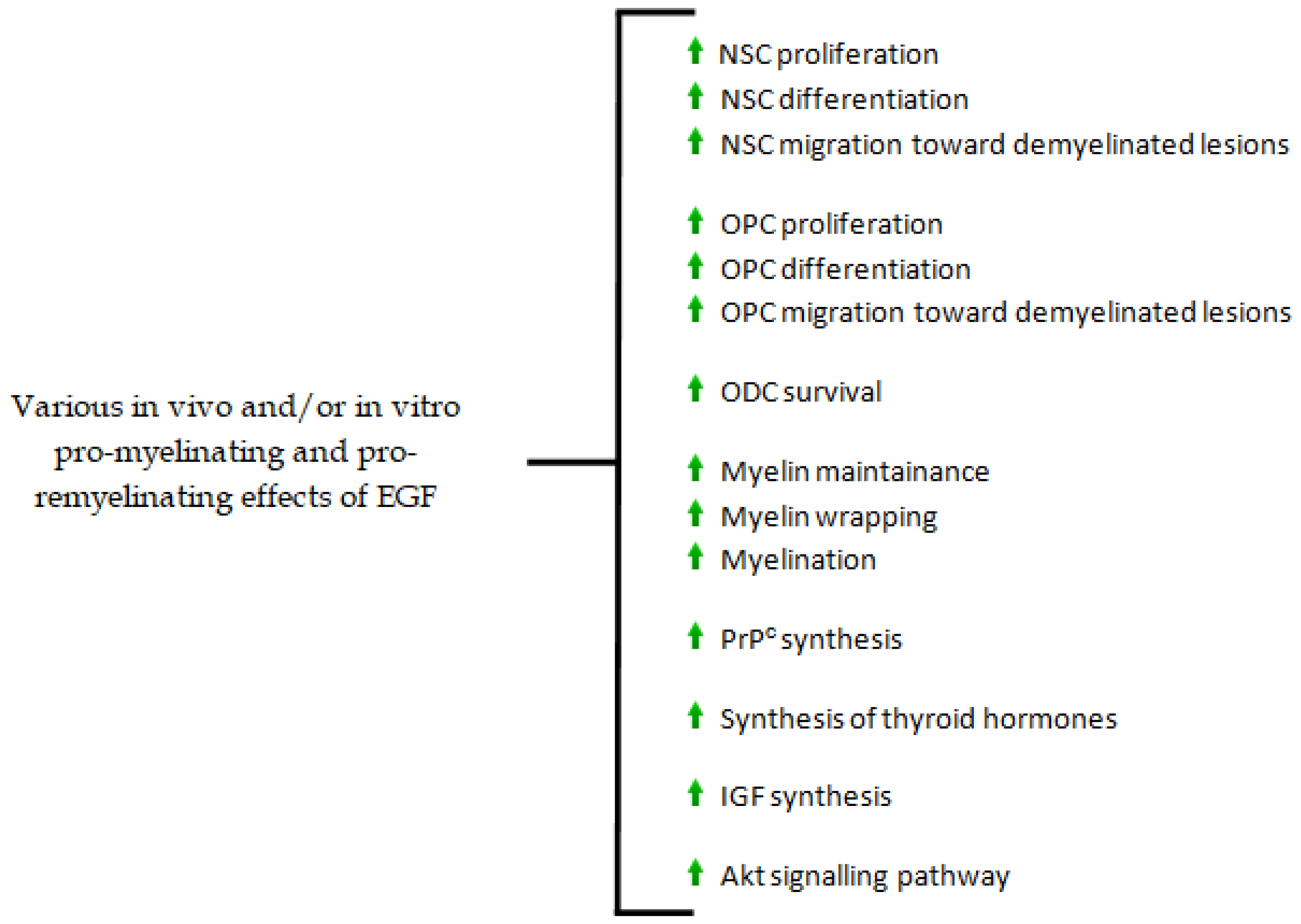{kind=link}
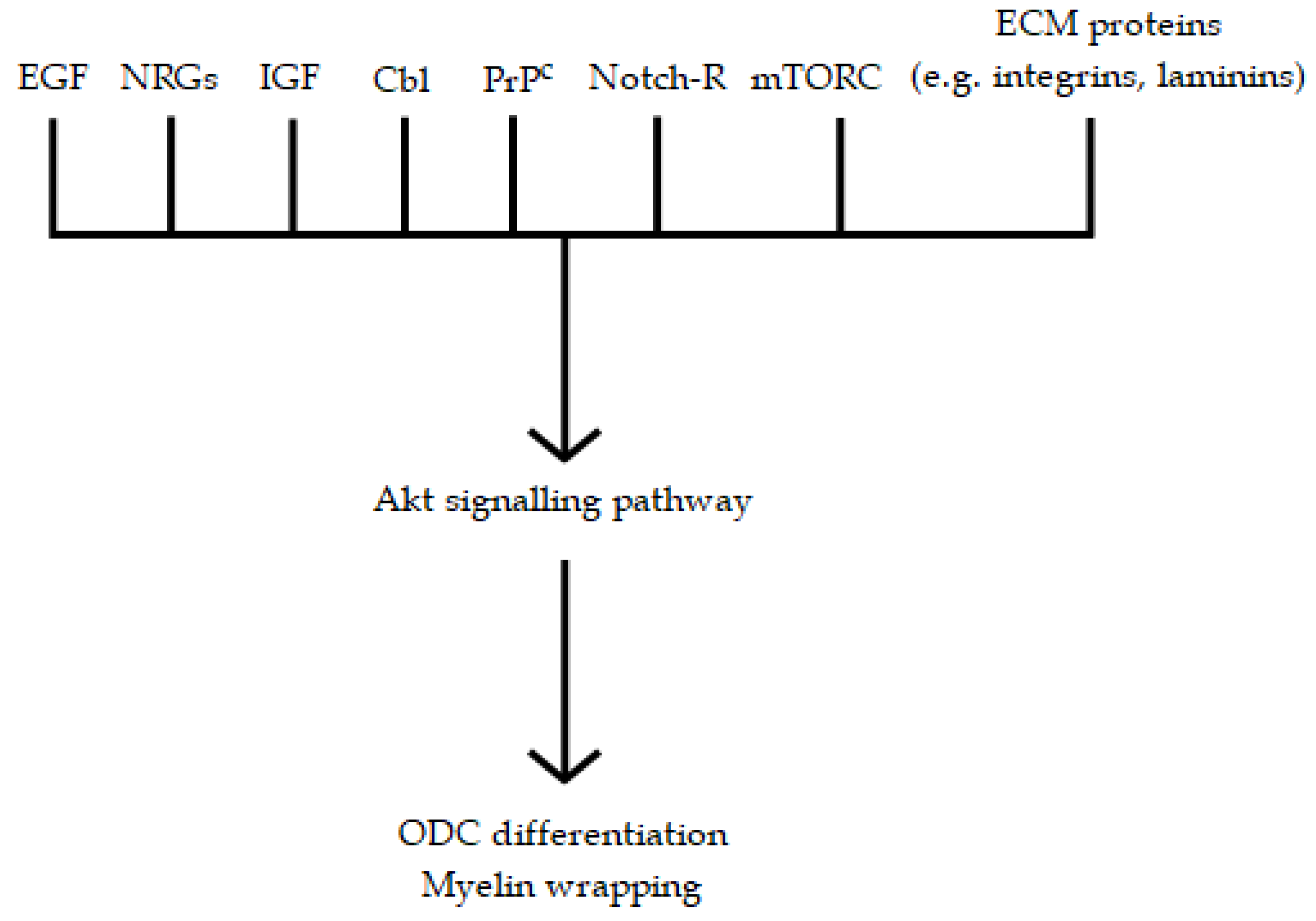{kind=link}
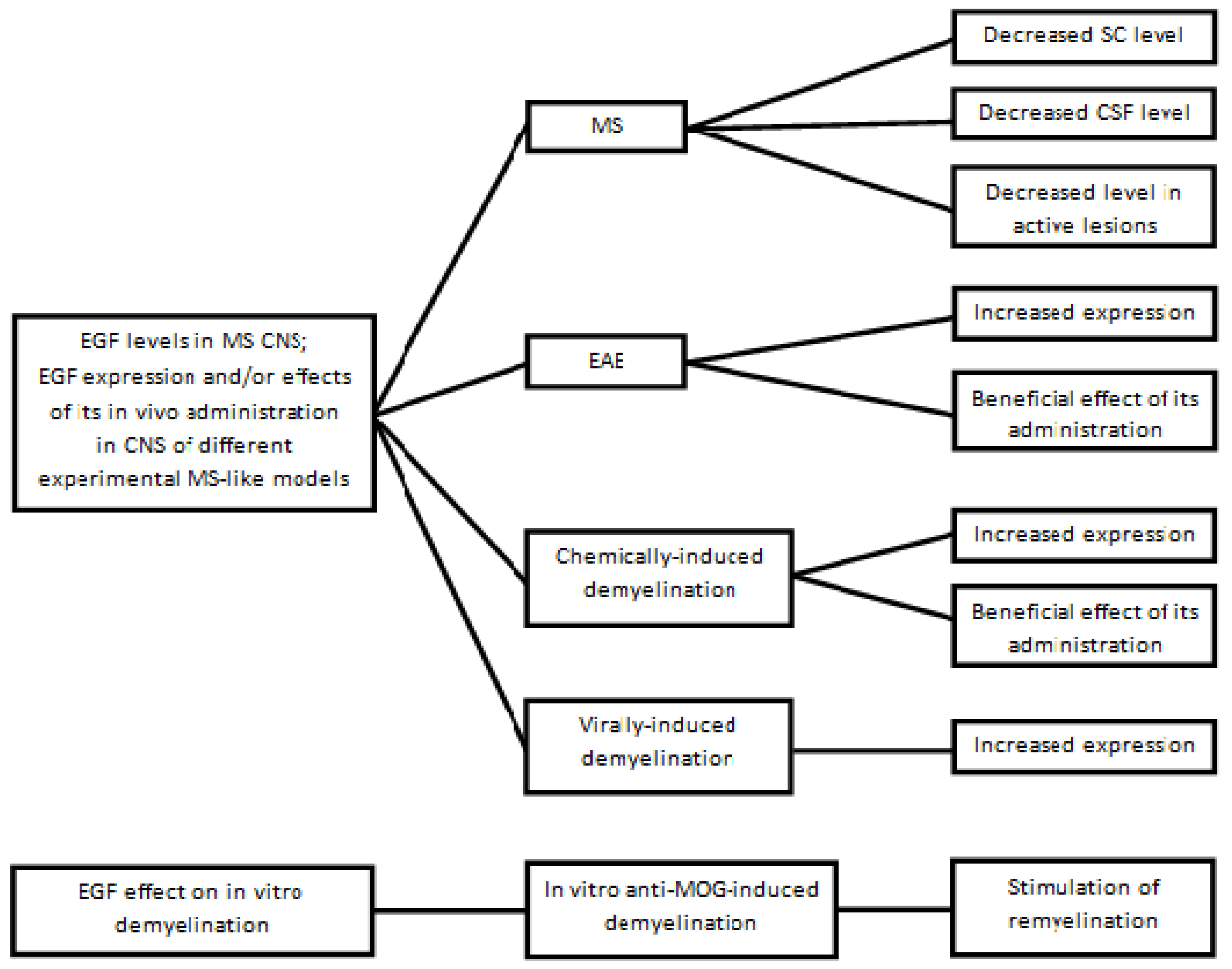{kind=link}
| Change | References |
|---|---|
| Decreased PrPC levels | [46,48] |
| Abnormalities in myelin structure | [88,89,90,91,92,93,94,95,96,97] |
| Changes in ODC transcriptome | [104,105,106,107,280] |
| Changes in acetylation and/or deacetylation levels of histones | [107,439] |
| Changes in ECM composition leading to a predominance of inhibitors of OPC differentiation and proliferation over promoters | [279,288,479,485] |
| Increased levels of inhibitors of differentiation and maturation inside the OPC→ODC cell lineage | [279,288,479,485] |
| Increased glutamate levels | [288] |
| Decreased levels of dome ODC- and myelino-trophic factors (mainly PDGF and EGF) | [288,357,359] |
| Opposite changes in the Cbl levels of SC and CSF | [357,359] |
| Changes in DNA methylation level | [361,362,363,436] |
| Reduced efficiency of mitochondrial respiratory chain | [411,412,416] |
| Decreased levels of some ‘’neurosteroids’’ | [421,432,433] |
| Changes in miRNA profiles | [444,453,454,455] |
| Dysregulated neuronal Na+ channel expression | [483] |
| Increased GFAP levels | [486,487] |
| Abnormalities in myelin composition | [488] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalabrino, G. Newly Identified Deficiencies in the Multiple Sclerosis Central Nervous System and Their Impact on the Remyelination Failure. Biomedicines 2022, 10, 815. https://doi.org/10.3390/biomedicines10040815
Scalabrino G. Newly Identified Deficiencies in the Multiple Sclerosis Central Nervous System and Their Impact on the Remyelination Failure. Biomedicines. 2022; 10(4):815. https://doi.org/10.3390/biomedicines10040815
Chicago/Turabian StyleScalabrino, Giuseppe. 2022. "Newly Identified Deficiencies in the Multiple Sclerosis Central Nervous System and Their Impact on the Remyelination Failure" Biomedicines 10, no. 4: 815. https://doi.org/10.3390/biomedicines10040815