
Vitamin D Counteracts Lipid Accumulation, Augments Free Fatty Acid-Induced ABCA1 and CPT-1A Expression While Reducing CD36 and C/EBPβ Protein Levels in Monocyte-Derived Macrophages

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
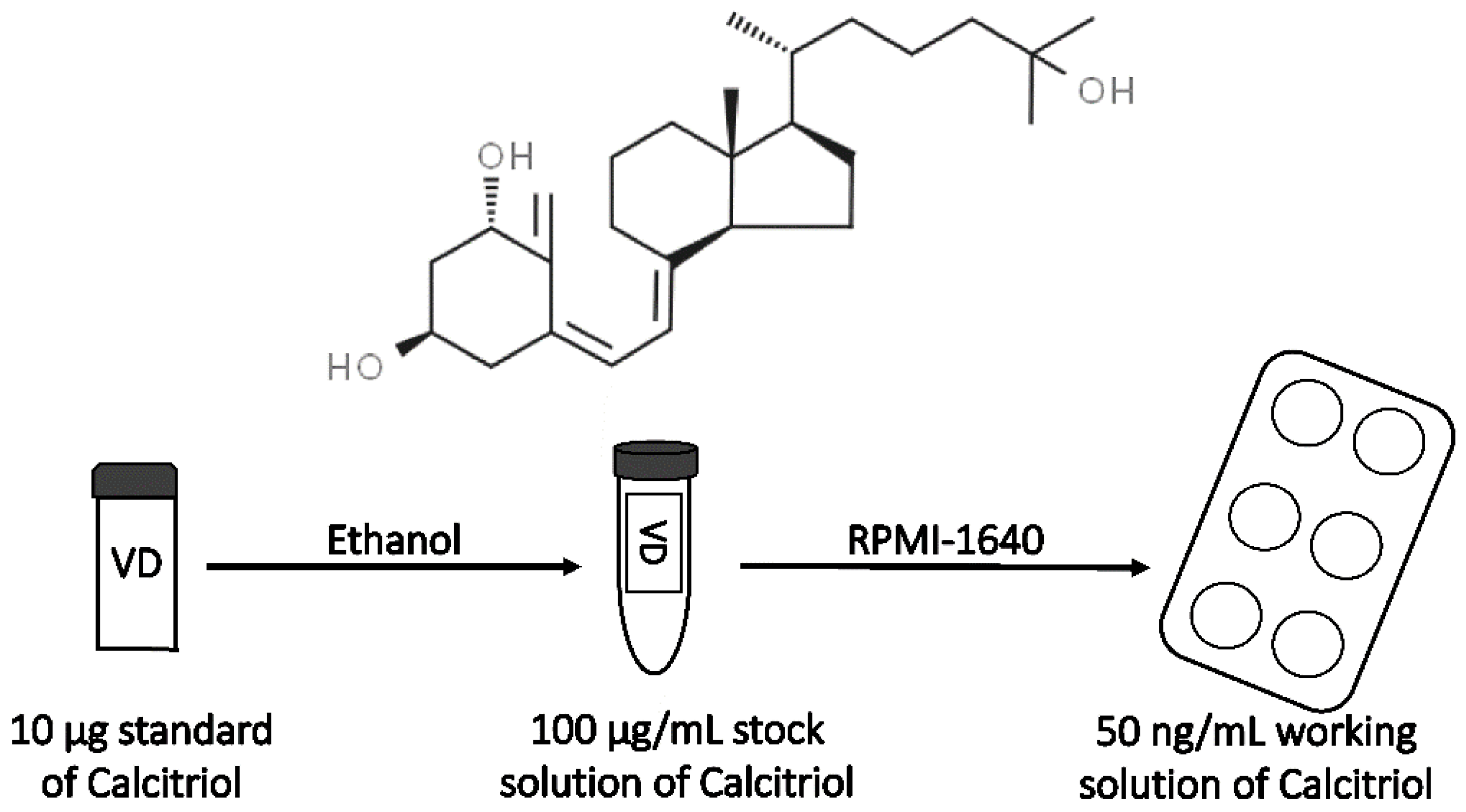
2.2. Preparation of Calcitriol Stock Solution
2.3. Preparation of Fatty Acid Solution and Its Control
2.4. THP-1 Cell Culture
2.5. Viability Assay
2.6. Lipid Accumulation Assay in Macrophages
2.7. Protein Quantification by Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Data Analysis
3. Results
3.1. Effect of VD3 on Cell Viability
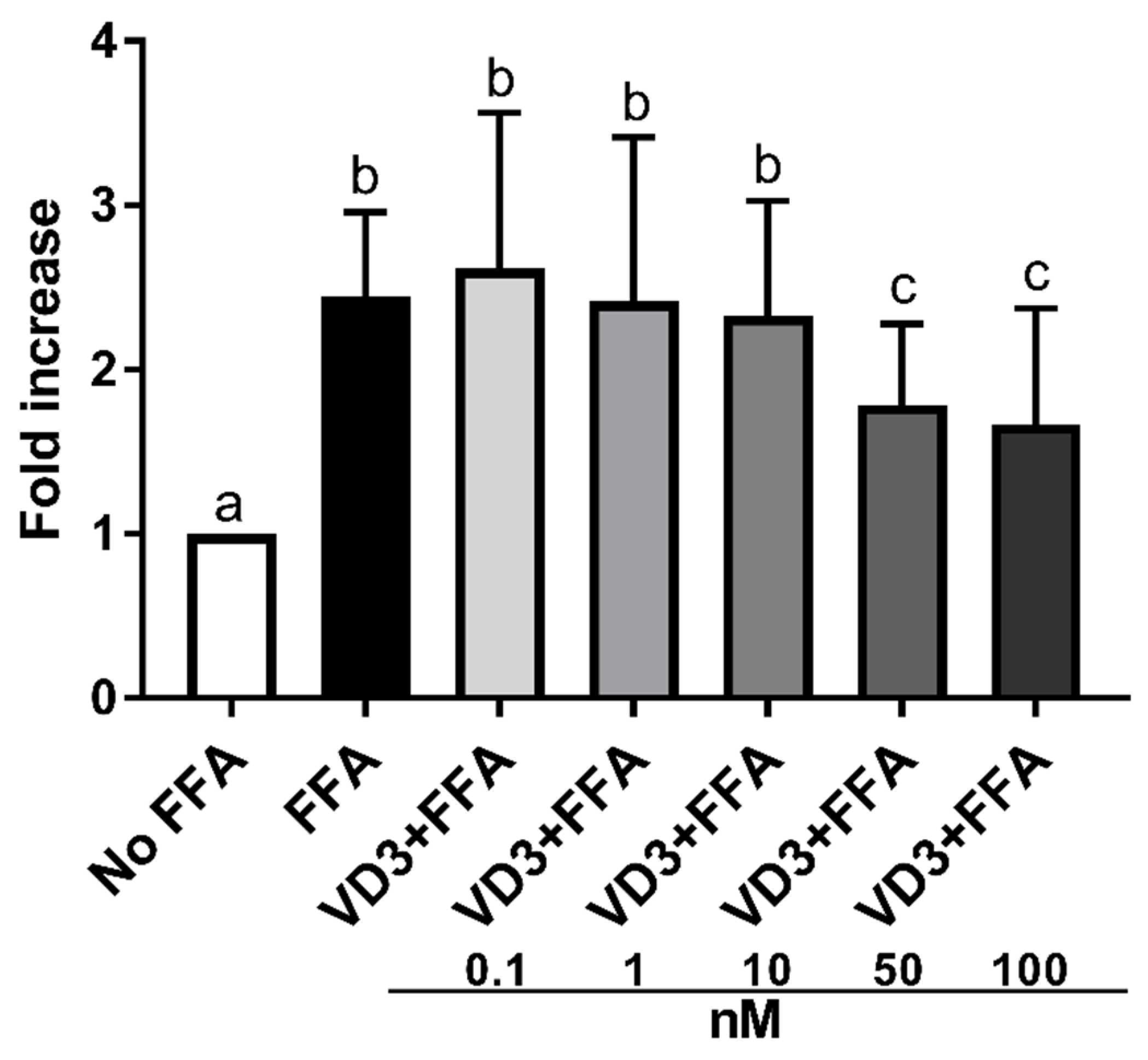
3.2. Effect of VD3 on Lipid Accumulation in THP-1 Derived Macrophages
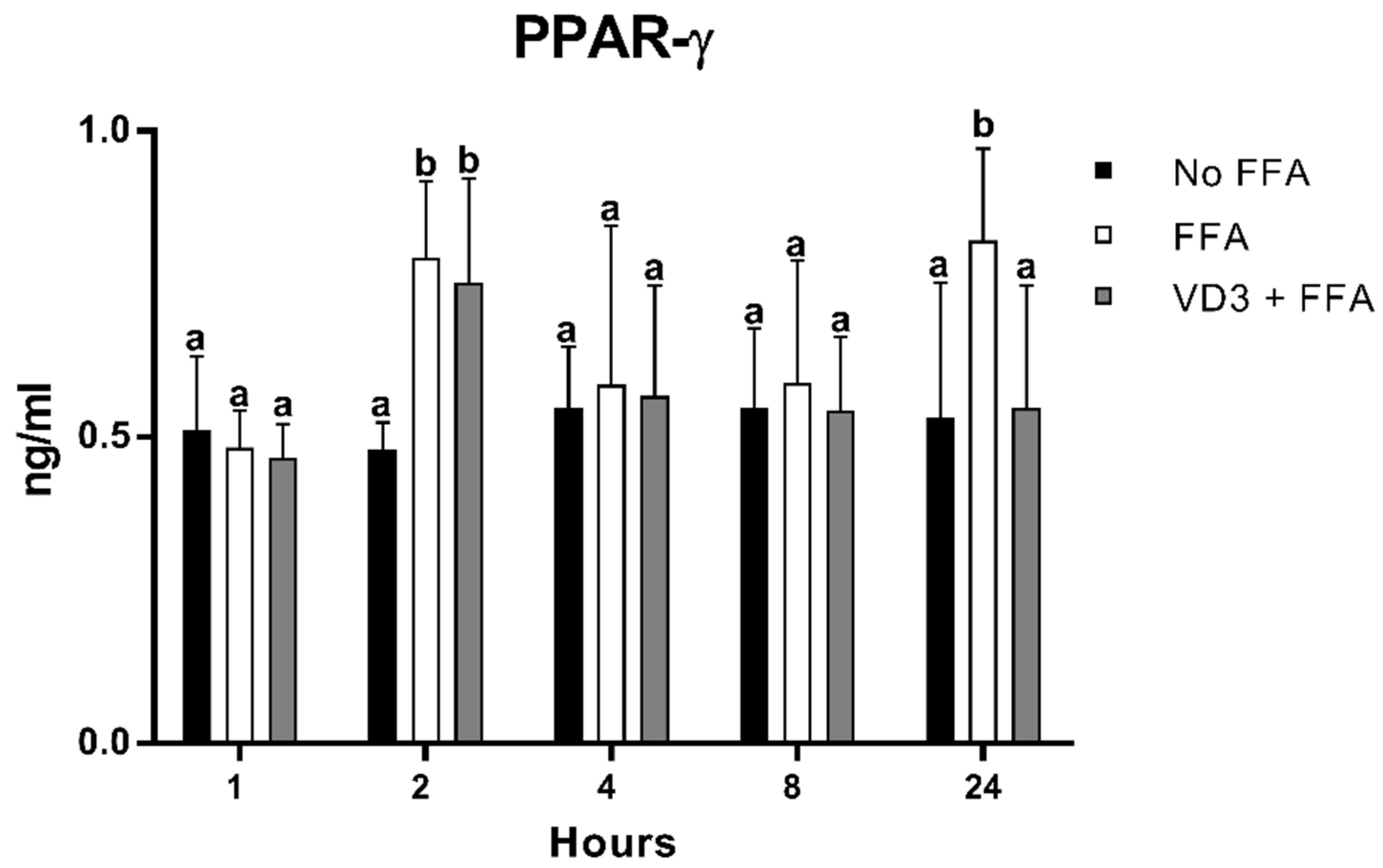
3.3. Effect of VD3 on PPAR-γ1 Protein Levels
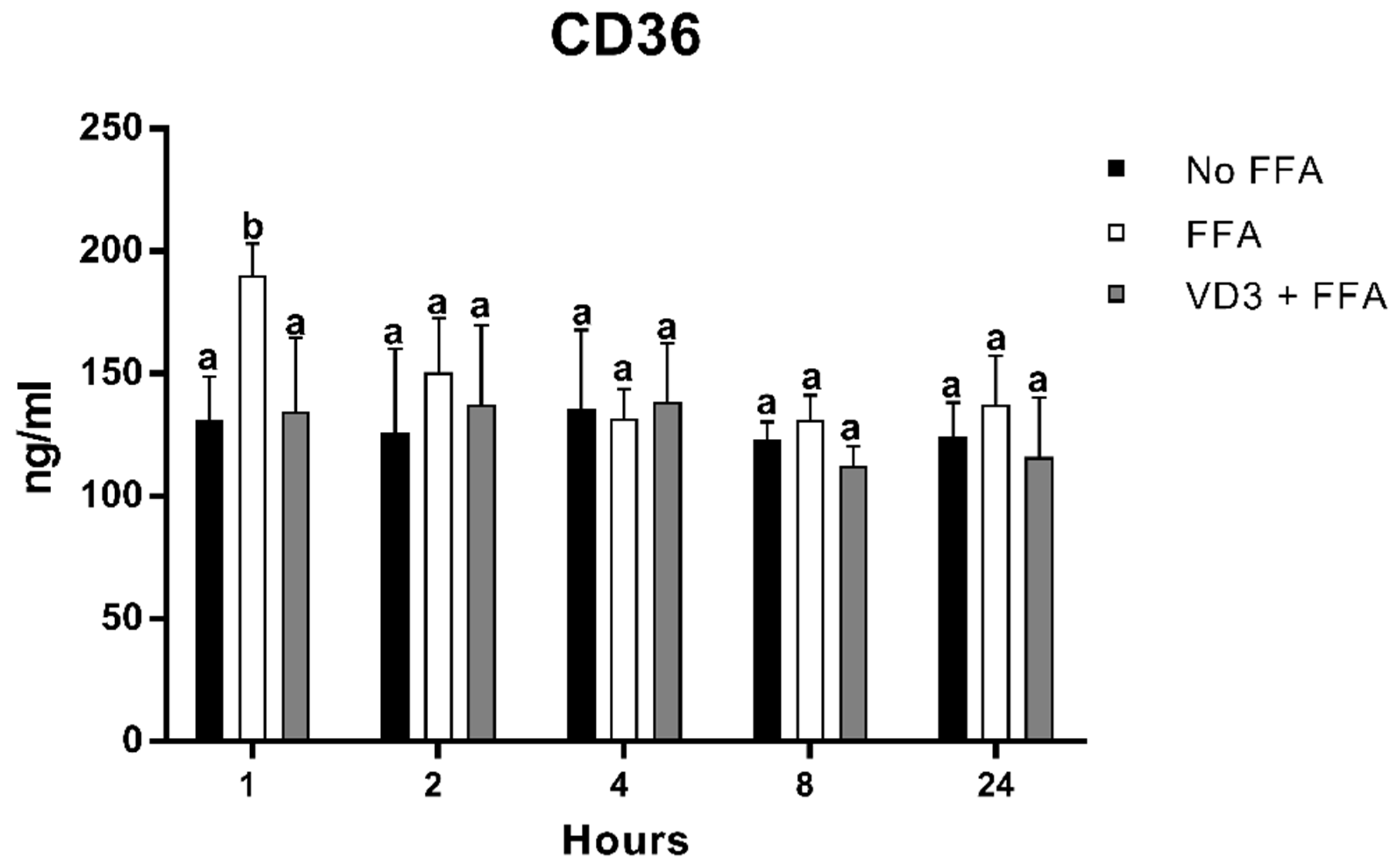
3.4. Effect of VD3 on CD-36 Protein Levels
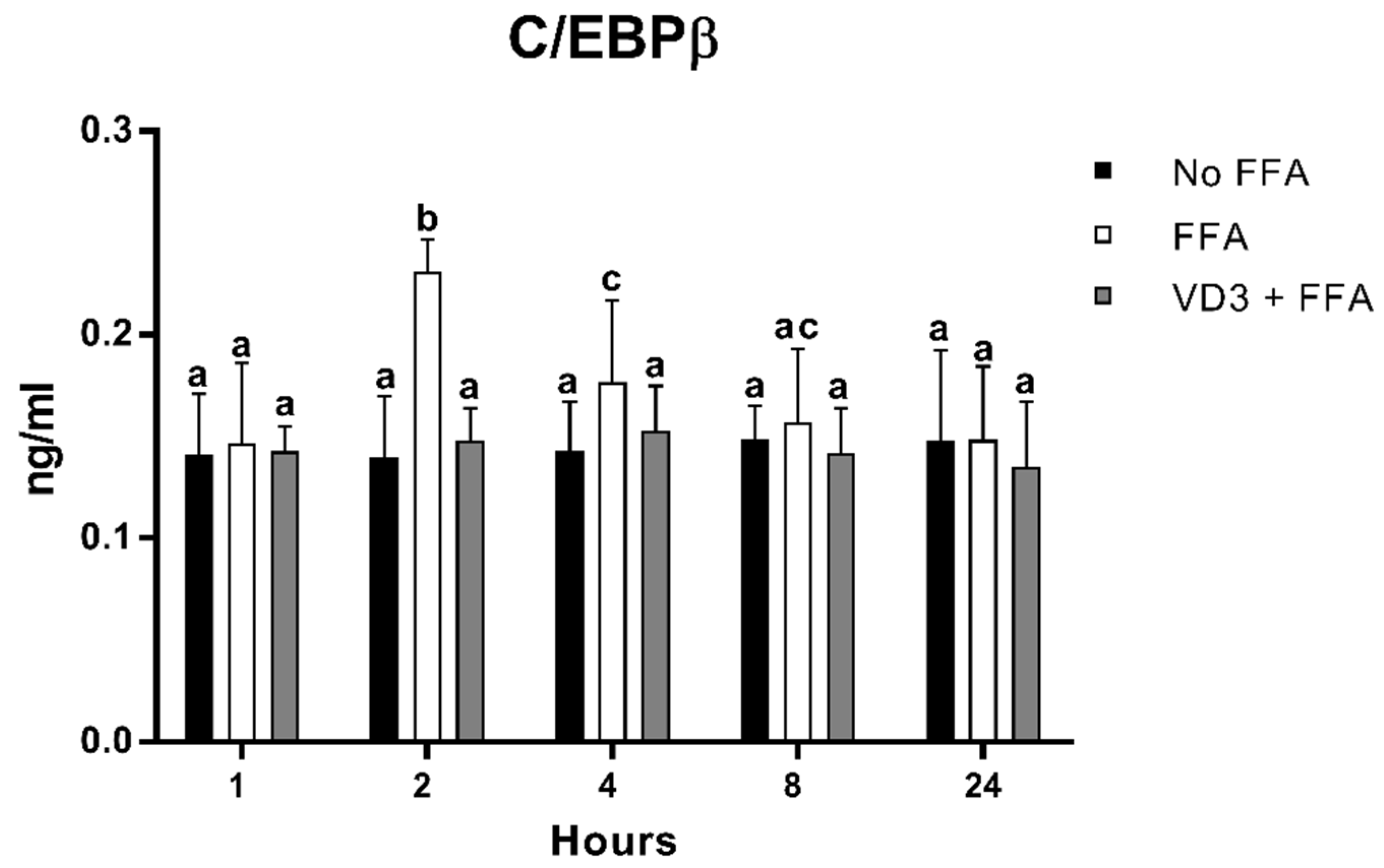
3.5. Effect of VD3 on C/EBPβ Protein Levels
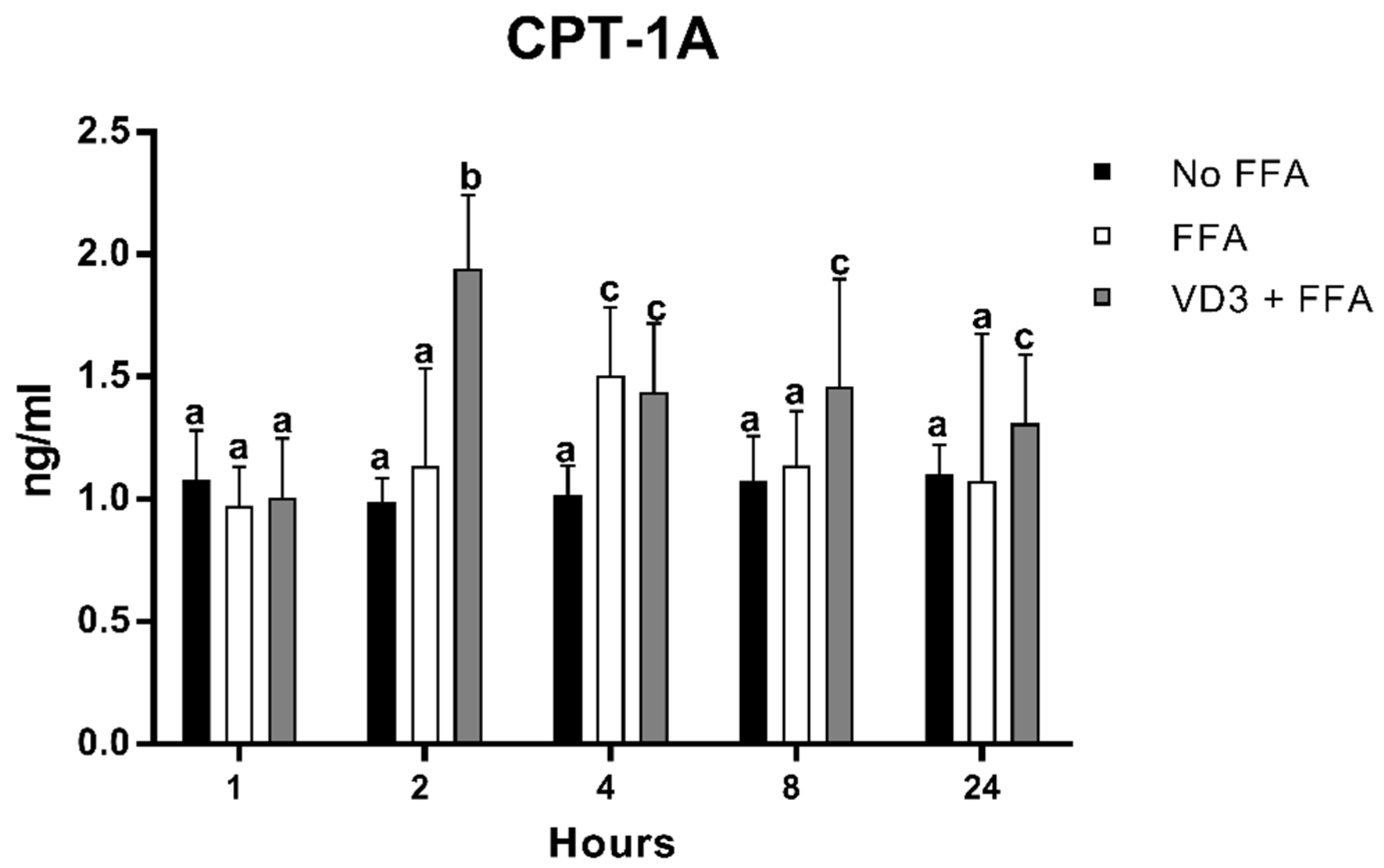
3.6. Effect of VD3 on CPT-1A Protein Levels
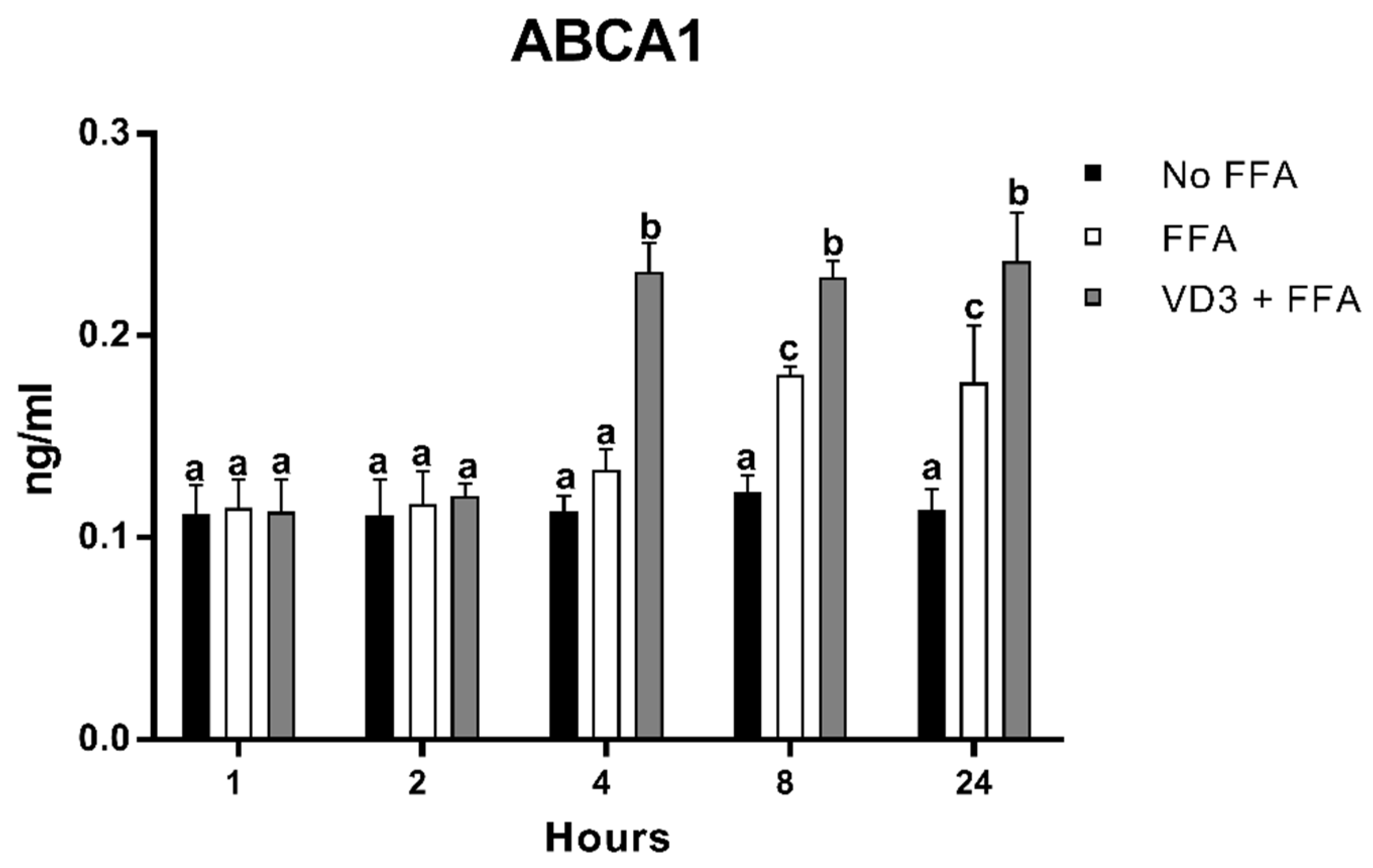
3.7. Effect of VD3 on ABCA1 Protein Levels
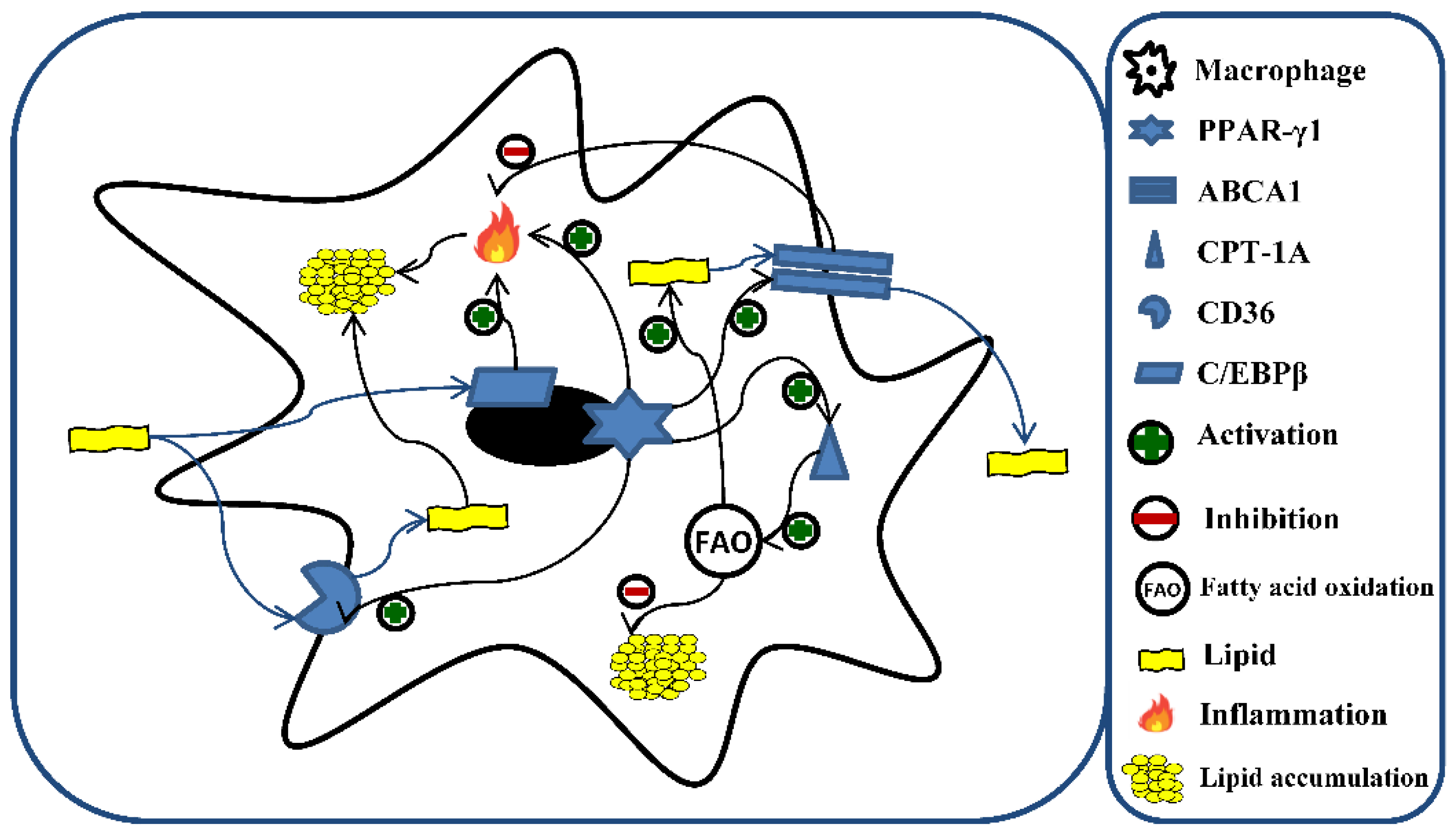
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Norman, A.W. Sunlight, Season, Skin Pigmentation, Vitamin D, and 25-Hydroxyvitamin D: Integral Components of the Vitamin D Endocrine System. Am. J. Clin. Nutr. 1998, 67, 1108–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripkovic, L.; Lambert, H.; Hart, K.; Smith, C.P.; Bucca, G.; Penson, S.; Chope, G.; Hyppönen, E.; Berry, J.; Vieth, R.; et al. Comparison of Vitamin D2 and Vitamin D3 Supplementation in Raising Serum 25-Hydroxyvitamin D Status: A Systematic Review and Meta-Analysis. Am. J. Clin. Nutr. 2012, 95, 1357–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.C.; Chimeh, F.; Lu, Z.; Mathieu, J.; Person, K.S.; Zhang, A.; Kohn, N.; Martinello, S.; Berkowitz, R.; Holick, M.F. Factors That Influence the Cutaneous Synthesis and Dietary Sources of Vitamin D. Arch. Biochem. Biophys. 2007, 460, 213–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinzone, M.R.; Di Rosa, M.; Malaguarnera, M.; Madeddu, G.; Focà, E.; Ceccarelli, G.; D’ettorre, G.; Vullo, V.; Fisichella, R.; Cacopardo, B.; et al. Vitamin D Deficiency in HIV Infection: An Underestimated and Undertreated Epidemic. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 1218–1232. [Google Scholar] [PubMed]
- Norman, P.E.; Powell, J.T. Vitamin D and Cardiovascular Disease. Circ. Res. 2014, 114, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Dix, C.F.; Barcley, J.L.; Wright, O.R.L. The Role of Vitamin D in Adipogenesis. Nutr. Rev. 2018, 76, 47–59. [Google Scholar] [CrossRef]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Evaluation, Treatment, and Prevention of Vitamin D Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, L.J.; Farruggia, M.; Veronese, N.; Barbagallo, M. Vitamin D Sources, Metabolism, and Deficiency: Available Compounds and Guidelines for Its Treatment. Metabolites 2021, 11, 255. [Google Scholar] [CrossRef]
- Lamberg-Allardt, C. Vitamin D in Foods and as Supplements. Prog. Biophys. Mol. Biol. 2006, 92, 33–38. [Google Scholar] [CrossRef]
- Hansen, L.; Tjønneland, A.; Køster, B.; Brot, C.; Andersen, R.; Cohen, A.S.; Frederiksen, K.; Olsen, A. Vitamin D Status and Seasonal Variation among Danish Children and Adults: A Descriptive Study. Nutrients 2018, 10, 1801. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; O’Keefe, J.H.; Bell, D.; Hensrud, D.D.; Holick, M.F. Vitamin D Deficiency. An Important, Common, and Easily Treatable Cardiovascular Risk Factor? J. Am. Coll. Cardiol. 2008, 52, 1949–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holick, M.F. Vitamin D Deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Pencina, M.J.; Booth, S.L.; Jacques, P.F.; Ingelsson, E.; Lanier, K.; Benjamin, E.J.; D’Agostino, R.B.; Wolf, M.; Vasan, R.S. Vitamin D Deficiency and Risk of Cardiovascular Disease. Circulation 2008, 117, 503–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockau, L.; Atkinson, S.A. Vitamin D’s Role in Health and Disease: How Does the Present Inform Our Understanding of the Past? Int. J. Paleopathol. 2018, 23, 6–14. [Google Scholar] [CrossRef]
- Rimondi, E.; Marcuuzzi, A.; Casciano, F.; Tornese, G.; Pellati, A.; Toffoli, B.; Secchiero, P.; Melloni, E. Role of Vitamin D in the Pathogenesis of Atheromatosis. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 344–353. [Google Scholar] [CrossRef]
- Gagnon, C.; Lu, Z.X.; Magliano, D.J.; Dunstan, D.W.; Shaw, J.E.; Zimmet, P.Z.; Sikaris, K.; Ebeling, P.R.; Daly, R.M. Low Serum 25-Hydroxyvitamin D Is Associated with Increased Risk of the Development of the Metabolic Syndrome at Five Years: Results from a National, Population-Based Prospective Study (The Australian Diabetes, Obesity and Lifestyle Study: AusDiab). J. Clin. Endocrinol. Metab. 2012, 97, 1953–1961. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Meza, C.A.; Clarke, H.; Kim, J.S.; Hickner, R.C. Vitamin D and Endothelial Function. Nutrients 2020, 12, 575. [Google Scholar] [CrossRef] [Green Version]
- Kassi, E.; Adamopoulos, C.; Basdra, E.K.; Papavassiliou, A.G. Role of Vitamin D in Atherosclerosis. Circulation 2013, 128, 2517–2531. [Google Scholar] [CrossRef] [Green Version]
- Schöttker, B.; Jorde, R.; Peasey, A.; Thorand, B.; Jansen, E.H.J.M.; De Groot, L.; Streppel, M.; Gardiner, J.; Ordóñez-Mena, J.M.; Perna, L.; et al. Vitamin D and Mortality: Meta-Analysis of Individual Participant Data from a Large Consortium of Cohort Studies from Europe and the United States. BMJ 2014, 348, g3656. [Google Scholar] [CrossRef] [Green Version]
- Martins, D.; Wolf, M.; Pan, D.; Zadshir, A.; Tareen, N.; Thadhani, R.; Felsenfeld, A.; Levine, B.; Mehrotra, R.; Norris, K. Prevalence of Cardiovascular Risk Factors and the Serum Levels of 25-Hydroxyvitamin D in the United States. Data From the Third National Health and Nutrition Examination Survey. Arch. Intern. Med. 2007, 167, 1159–1165. [Google Scholar] [CrossRef] [Green Version]
- Wilson, L.R.; Tripkovic, L.; Hart, K.H.; Lanham-New, S.A. Vitamin D Deficiency as a Public Health Issue: Using Vitamin D2 or Vitamin D3 in Future Fortification Strategies. Proc. Nut. Soc. 2017, 76, 392–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlberg, C. Vitamin D Genomics: From in Vitro to in Vivo. Front. Endocrinol. 2018, 9, 250. [Google Scholar] [CrossRef] [PubMed]
- Hewison, M. Vitamin D and Immune Function: An Overview. Proc. Nut. Soc. 2012, 71, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziedzic, E.A.; Przychodzen, S.; Dabrowski, M. The Effects of Vitamin D on Severity of Coronary Artery Atherosclerosis and Lipid Profile of Cardiac Patients. Arch. Med. Sci. 2016, 12, 1199–1206. [Google Scholar] [CrossRef] [Green Version]
- Muscogiuri, G.; Annweiler, C.; Duval, G.; Karras, S.; Tirabassi, G.; Salvio, G.; Balercia, G.; Kimball, S.; Kotsa, K.; Mascitelli, L.; et al. Vitamin D and Cardiovascular Disease: From Atherosclerosis to Myocardial Infarction and Stroke. Int. J. Cardiol. 2017, 230, 577–584. [Google Scholar] [CrossRef]
- Izzo, M.; Carrizzo, A.; Izzo, C.; Cappello, E.; Cecere, D.; Ciccarelli, M.; Iannece, P.; Damato, A.; Vecchione, C.; Pompeo, F. Vitamin D: Not Just Bone Metabolism but a Key Player in Cardiovascular Diseases. Life 2021, 11, 452. [Google Scholar] [CrossRef]
- Latic, N.; Erben, R.G. Vitamin D and Cardiovascular Disease, with Emphasis on Hypertension, Atherosclerosis, and Heart Failure. Int. J. Mol. Sci. 2020, 21, 6483. [Google Scholar] [CrossRef]
- Surdu, A.M.; Pînzariu, O.; Ciobanu, D.M.; Negru, A.G.; Căinap, S.S.; Lazea, C.; Iacob, D.; Săraci, G.; Tirinescu, D.; Borda, I.M.; et al. Vitamin D and Its Role in the Lipid Metabolism and the Development of Atherosclerosis. Biomedicines 2021, 9, 172. [Google Scholar] [CrossRef]
- Autier, P.; Boniol, M.; Pizot, C.; Mullie, P. Vitamin D Status and Ill Health: A Systematic Review. Lancet Diabetes Endocrinol. 2014, 2, 76–89. [Google Scholar] [CrossRef]
- Takeda, M.; Yamashita, T.; Sasaki, N.; Nakajima, K.; Kita, T.; Shinohara, M.; Ishida, T.; Hirata, K.I. Oral Administration of an Active Form of Vitamin D3 (Calcitriol) Decreases Atherosclerosis in Mice by Inducing Regulatory T Cells and Immature Dendritic Cells with Tolerogenic Functions. Arter. Thromb. Vasc. Biol. 2010, 30, 2495–2503. [Google Scholar] [CrossRef] [Green Version]
- Pownall, H.J.; Gotto, A.M. Lipids and Cardiovascular Disease: Putting It All Together. Methodist DeBakey Cardiovasc. J. 2019, 15, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Soppert, J.; Lehrke, M.; Marx, N.; Jankowski, J.; Noels, H. Lipoproteins and Lipids in Cardiovascular Disease: From Mechanistic Insights to Therapeutic Targeting. Adv. Drug Deliv. Rev. 2020, 159, 4–33. [Google Scholar] [CrossRef] [PubMed]
- Ruuth, M.; Nguyen, S.D.; Vihervaara, T.; Hilvo, M.; Laajala, T.D.; Kondadi, P.K.; Gistera, A.; Lahteenmaki, H.; Kiittila, T.; Huusko, J.; et al. Susceptibility of Low-Density Lipoprotein particles to Aggregate Depends on Particle lipidome, Is Modifiable, and Associates With future Cardiovascular Deaths. Eur. Heart J. 2018, 39, 2574–2576. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Freeman, M.W. Scavenger Receptors in Atherosclerosis: Beyond Lipid Uptake. Arter. Thromb. Vasc. Biol. 2006, 26, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nũez, G.; Schnurr, M.; et al. NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Camejo, G. Lysophospholipids: Effectors Mediating the Contribution of Dyslipidemia to Calcification Associated with Atherosclerosis. Atherosclerosis 2010, 211, 36–37. [Google Scholar] [CrossRef]
- Feng, L.; Gu, C.; Li, Y.; Huang, J. High Glucose Promotes CD36 Expression by Upregulating Peroxisome Proliferator-Activated Receptor γ Levels to Exacerbate Lipid Deposition in Renal Tubular Cells. Biomed. Res. Int. 2017, 2017, 1414070. [Google Scholar] [CrossRef] [Green Version]
- Baillie, A.G.S.; Coburn, C.T.; Abumrad, N.A. Reversible Binding of Long-Chain Fatty Acids to Purified FAT, the Adipose CD36 Homolog Membrane Biology. J. Membr. Biol. 1996, 153, 75–81. [Google Scholar] [CrossRef]
- Wculek, S.K.; Dunphy, G.; Heras-Murillo, I.; Mastrangelo, A.; Sancho, D. Metabolism of Tissue Macrophages in Homeostasis and Pathology. Cell. Mol. Immunol. 2021, 19, 384–408. [Google Scholar] [CrossRef]
- Song, S.; Attia, R.R.; Connaughton, S.; Niesen, M.I.; Ness, G.C.; Elam, M.B.; Hori, R.T.; Cook, G.A.; Park, E.A. Peroxisome Proliferator Activated Receptor α (PPARα) and PPAR Gamma Coactivator (PGC-1α) Induce Carnitine Palmitoyltransferase IA (CPT-1A) via Independent Gene Elements. Mol. Cell. Endocrinol. 2010, 325, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine Transport and Fatty Acid Oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Boisvert, W.A.; Lee, C.-H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPAR-LXR-ABCA1 Pathway in Macrophages Is Involved in Cholesterol Efflux and Atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Cavelier, C.; Lorenzi, I.; Rohrer, L.; von Eckardstein, A. Lipid Efflux by the ATP-Binding Cassette Transporters ABCA1 and ABCG1. Biochim. Biophys. Acta 2006, 1761, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Out, R.; Hoekstra, M.; Habets, K.; Meurs, I.; de Waard, V.; Hildebrand, R.B.; Wang, Y.; Chimini, G.; Kuiper, J.; Van Berkel, T.J.C.; et al. Combined Deletion of Macrophage ABCA1 and ABCG1 Leads to Massive Lipid Accumulation in Tissue Macrophages and Distinct Atherosclerosis at Relatively Low Plasma Cholesterol Levels. Arter. Thromb. Vasc. Biol. 2008, 28, 258–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, K.; Liao, D.-F.; Tang, C.-K. ATP-Binding Membrane Cassette Transporter A1 (ABCA1): A Possible Link between Inflammation and Reverse Cholesterol Transport. Mol. Med. 2010, 16, 438–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesterdal, L.K.; Danielsen, P.H.; Folkmann, J.K.; Jespersen, L.F.; Aguilar-Pelaez, K.; Roursgaard, M.; Loft, S.; Møller, P. Accumulation of Lipids and Oxidatively Damaged DNA in Hepatocytes Exposed to Particles. Toxicol. Appl. Pharm. 2014, 274, 350–360. [Google Scholar] [CrossRef]
- Araya, J.; Rodrigo, R.; Videla, L.A.; Thielemann, L.; Ornellana, M.; Pettinelli, P.; Pniachik, J. Increase in Long-Chain Polyunsaturated Fatty acid N−6/N−3 Ratio in Relation to Hepatic steatosis in Patients with Non-Alcoholic fatty Liver Disease. Clin. Sci 2004, 106, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef]
- Krzystolik, A.; Dziedziejko, V.; Safranow, K.; Kurzawski, G.; Rać, M.; Sagasz-Tysiewicz, D.; Poncyljusz, W.; Jakubowska, K.; Chlubek, D.; Rać, M.E. Is Plasma Soluble CD36 Associated with Cardiovascular Risk Factors in Early Onset Coronary Artery Disease Patients? Scand. J. Clin. Lab. 2015, 75, 398–406. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, M.; Li, L.; Li, C.; Min, X.; Zheng, M. Expression and Clinical Significance of ATP-Binding Cassette Transporter 1 in Serum and Placental Tissue in Chinese Patients with Preeclampsia. Gynecol. Obstet. Invest. 2014, 78, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Riek, A.E.; Oh, J.; Bernal-Mizrachi, C. 1,25(OH)2 Vitamin D Suppresses Macrophage Migration and Reverses Atherogenic Cholesterol Metabolism in Type 2 Diabetic Patients. J. Steroid. Biochem. Mol. Biol. 2013, 136, 309–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Riek, A.E.; Darwech, I.; Funai, K.; Shao, J.S.; Chin, K.; Sierra, O.L.; Carmeliet, G.; Ostlund, R.E.; Bernal-Mizrachi, C. Deletion of Macrophage Vitamin D Receptor Promotes Insulin Resistance and Monocyte Cholesterol Transport to Accelerate Atherosclerosis in Mice. Cell Rep. 2015, 10, 1872–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, K.; You, Y.; Swier, V.; Tang, L.; Radwan, M.M.; Pandya, A.N.; Agrawal, D.K. Vitamin D Protects Against Atherosclerosis via Regulation of Cholesterol Efflux and Macrophage Polarization in Hypercholesterolemic Swine. Arter. Thromb. Vasc. Biol. 2015, 35, 2432–2442. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Mihalcioiu, M.; Li, L.; Zakikhani, M.; Camirand, A.; Kremer, R. Vitamin D Prevents Lipid Accumulation in Murine Muscle through Regulation of PPARγ and Perilipin-2 Expression. J. Steroid Biochem. Mol. Biol. 2018, 177, 116–124. [Google Scholar] [CrossRef]
- Chang, E.; Kim, Y. Vitamin D Decreases Adipocyte Lipid Storage and Increases NAD-SIRT1 Pathway in 3T3-L1 Adipocytes. Nutrition 2016, 32, 702–708. [Google Scholar] [CrossRef] [Green Version]
- Barbier, O.; Torra, I.P.; Duguay, Y.; Blanquart, C.; Fruchart, J.C.; Glineur, C.; Staels, B. Pleiotropic Actions of Peroxisome Proliferator-Activated Receptors in Lipid Metabolism and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2002, 22, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Marino, M.; del Bo’, C.; Tucci, M.; Venturi, S.; Mantegazza, G.; Taverniti, V.; Møller, P.; Riso, P.; Porrini, M. A Mix of Chlorogenic and Caffeic Acid Reduces C/EBPß and PPAR-Γ1 Levels and Counteracts Lipid Accumulation in Macrophages. Eur. J. Nutr. 2021, 61, 1003–1014. [Google Scholar] [CrossRef]
- Scrimieri, R.; Cazzaniga, A.; Castiglioni, S.; Maier, J.A.M. Vitamin d Prevents High Glucose-Induced Lipid Droplets Accumulation in Cultured Endothelial Cells: The Role of Thioredoxin Interacting Protein. Biomedicines 2021, 9, 1874. [Google Scholar] [CrossRef]
- Oh, J.; Weng, S.; Felton, S.K.; Bhandare, S.; Riek, A.; Butler, B.; Proctor, B.M.; Petty, M.; Chen, Z.; Schechtman, K.B.; et al. 1,25(OH) 2 Vitamin D Inhibits Foam Cell Formation and Suppresses Macrophage Cholesterol Uptake in Patients with Type 2 Diabetes. Circulation 2009, 120, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Salamon, H.; Bruiners, N.; Lakehal, K.; Shi, L.; Ravi, J.; Yamaguchi, K.D.; Pine, R.; Gennaro, M.L. Vitamin D Regulates Lipid Metabolism in Mycobacterium Tuberculosis Infection. J. Immunol. 2014, 193, 30–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeto, F.L.; Reardon, C.A.; Yoon, D.; Wang, Y.; Wong, K.E.; Chen, Y.; Kong, J.; Liu, S.Q.; Thadhani, R.; Getz, G.S.; et al. Vitamin D Receptor Signaling Inhibits Atherosclerosis in Mice. Mol. Endocrinol. 2012, 26, 1091–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, S.; Mirshafiey, A.; Djalali, M.; Alvandi, E.; Honarvar, N.M.; Javanbakht, M.H. Vitamin D3 Induces Gene Expression of Ox-LDL Scavenger Receptors in Streptozotocin-Induced Diabetic Rat Aortas: New Insight into the Role of Vitamin D in Diabetic Atherosclerosis. Rep. Biochem. Mol. Biol. 2018, 6, 170–177. [Google Scholar] [PubMed]
- Blumberg, J.M.; Tzameli, I.; Astapova, I.; Lam, F.S.; Flier, J.S.; Hollenberg, A.N. Complex Role of the Vitamin D Receptor and Its Ligand in Adipogenesis in 3T3-L1 Cells. J. Biol. Chem. 2006, 281, 11205–11213. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves-de-Albuquerque, C.F.; Medeiros-de-Moraes, I.M.; de Oliveira, F.M.J.; Burth, P.; Bozza, P.T.; Faria, M.V.C.; Silva, A.R.; de Castro-Faria-Neto, H.C. Omega-9 Oleic Acid Induces Fatty Acid Oxidation and Decreases Organ Dysfunction and Mortality in Experimental Sepsis. PLoS ONE 2016, 11, e0153607. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Li, K.; Hui, X.; Kong, X.; Sweeney, G.; Wang, Y.; Xu, A.; Teng, M.; Liu, P.; Wu, D. Carnitine Palmitoyltransferase 1A Prevents Fatty Acid-Induced Adipocyte Dysfunction through Suppression of c-Jun N-Terminal Kinase. Biochem. J. 2011, 435, 723–732. [Google Scholar] [CrossRef]
- Chang, E.; Kim, Y. Vitamin D Ameliorates Fat Accumulation with AMPK/SIRT1 Activity in C2C12 Skeletal Muscle Cells. Nutrients 2019, 11, 2806. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Yu, Z.; Xia, M.; Luo, X.; Lu, X.; Ling, W. Vitamin D Attenuates High Fat Diet-Induced Hepatic Steatosis in Rats by Modulating Lipid Metabolism. Eur. J. Clin. Investig. 2012, 42, 1189–1196. [Google Scholar] [CrossRef]
- Ning, C.; Liu, L.; Lv, G.; Yang, Y.; Zhang, Y.; Yu, R.; Wang, Y.; Zhu, J. Lipid Metabolism and Inflammation Modulated by Vitamin D in Liver of Diabetic Rats. Lipids Health Dis. 2015, 14, 31. [Google Scholar] [CrossRef] [Green Version]
- He, P.; Gelissen, I.C.; Ammit, A.J. Regulation of ATP Binding Cassette Transporter A1 (ABCA1) Expression: Cholesterol-Dependent and-Independent Signaling Pathways with Relevance to Inflammatory Lung Disease. Respir. Res. 2020, 21, 250. [Google Scholar] [CrossRef]
- Bi, X.; Vitali, C.; Cuchel, M. ABCA1 and Inflammation From Animal Models to Humans. Arter. Thromb. Vasc. Biol. 2015, 35, 1551–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominski, A.T.; Kim, T.K.; Qayyum, S.; Song, Y.; Janjetovic, Z.; Oak, A.S.W.; Slominski, R.M.; Raman, C.; Stefan, J.; Mier-Aguilar, C.A.; et al. Vitamin D and Lumisterol Derivatives Can Act on Liver X Receptors (LXRs). Sci. Rep. 2021, 11, 8002. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | VD3 Concentration (nM) | % of Cell Viability * |
|---|---|---|
| Control | - | 90.7 ± 1.5 |
| Triton X-100 | - | 20 ± 6.2 ** |
| FFA | - | 92.1 ± 2.2 |
| FFA + VD3 | 0.1 | 91.3 ± 2.6 |
| FFA + VD3 | 1 | 90.9 ± 1.8 |
| FFA + VD3 | 10 | 93.4 ± 3.3 |
| FFA + VD3 | 50 | 92.5 ± 2.4 |
| FFA + VD3 | 100 | 90.3 ± 1.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marino, M.; Venturi, S.; Del Bo’, C.; Møller, P.; Riso, P.; Porrini, M. Vitamin D Counteracts Lipid Accumulation, Augments Free Fatty Acid-Induced ABCA1 and CPT-1A Expression While Reducing CD36 and C/EBPβ Protein Levels in Monocyte-Derived Macrophages. Biomedicines 2022, 10, 775. https://doi.org/10.3390/biomedicines10040775
Marino M, Venturi S, Del Bo’ C, Møller P, Riso P, Porrini M. Vitamin D Counteracts Lipid Accumulation, Augments Free Fatty Acid-Induced ABCA1 and CPT-1A Expression While Reducing CD36 and C/EBPβ Protein Levels in Monocyte-Derived Macrophages. Biomedicines. 2022; 10(4):775. https://doi.org/10.3390/biomedicines10040775
Chicago/Turabian StyleMarino, Mirko, Samuele Venturi, Cristian Del Bo’, Peter Møller, Patrizia Riso, and Marisa Porrini. 2022. "Vitamin D Counteracts Lipid Accumulation, Augments Free Fatty Acid-Induced ABCA1 and CPT-1A Expression While Reducing CD36 and C/EBPβ Protein Levels in Monocyte-Derived Macrophages" Biomedicines 10, no. 4: 775. https://doi.org/10.3390/biomedicines10040775