
Klotho an Autophagy Stimulator as a Potential Therapeutic Target for Alzheimer’s Disease: A Review
 , , and
, , and
Abstract
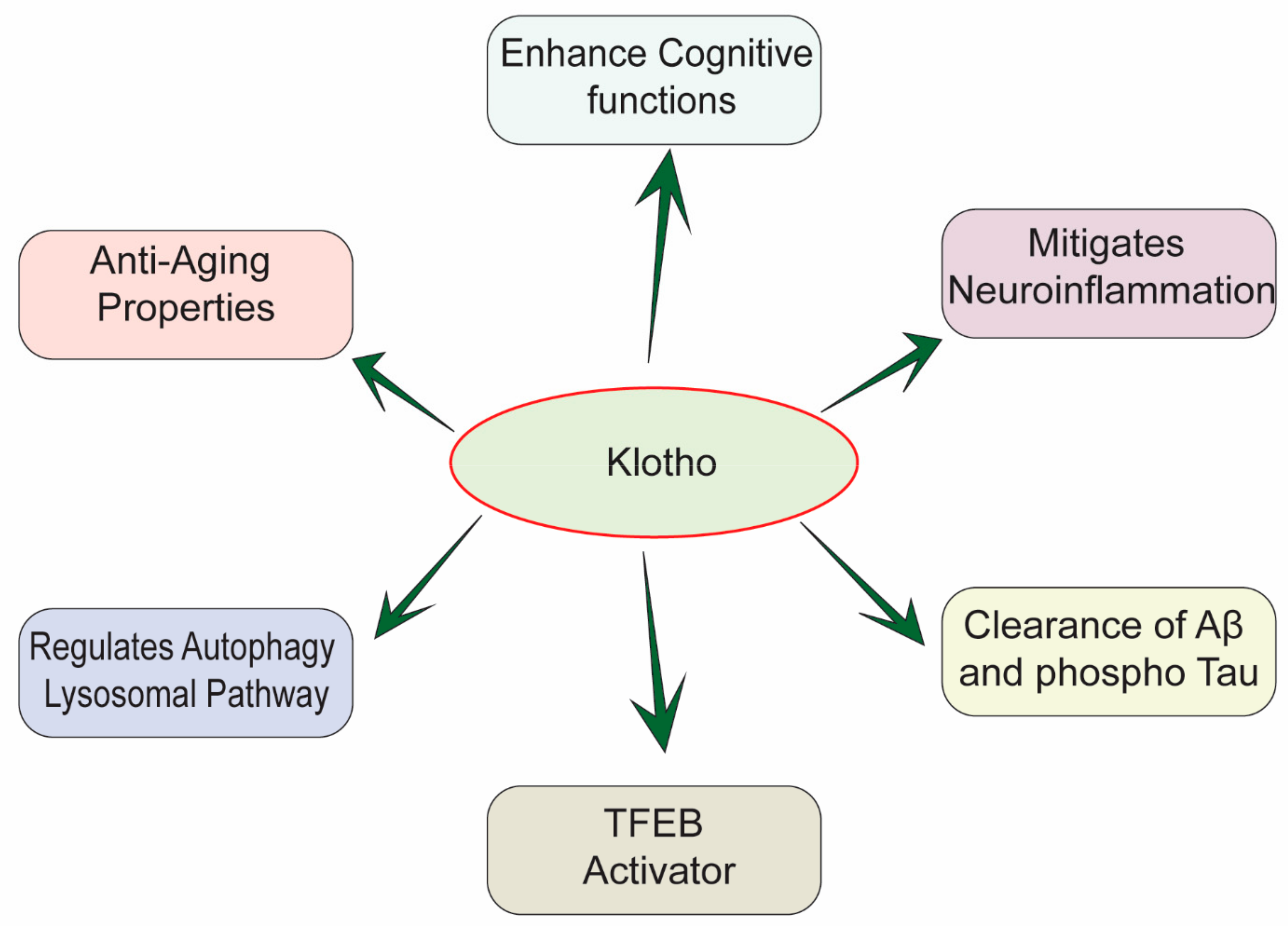
:1. Introduction
2. Expression, Structure and Function of Klotho Protein
3. Klotho Inhibits Neuroinflammation, Promotes Aβ Clearance, and Mitigates Tau Pathology in Alzheimer’s Disease
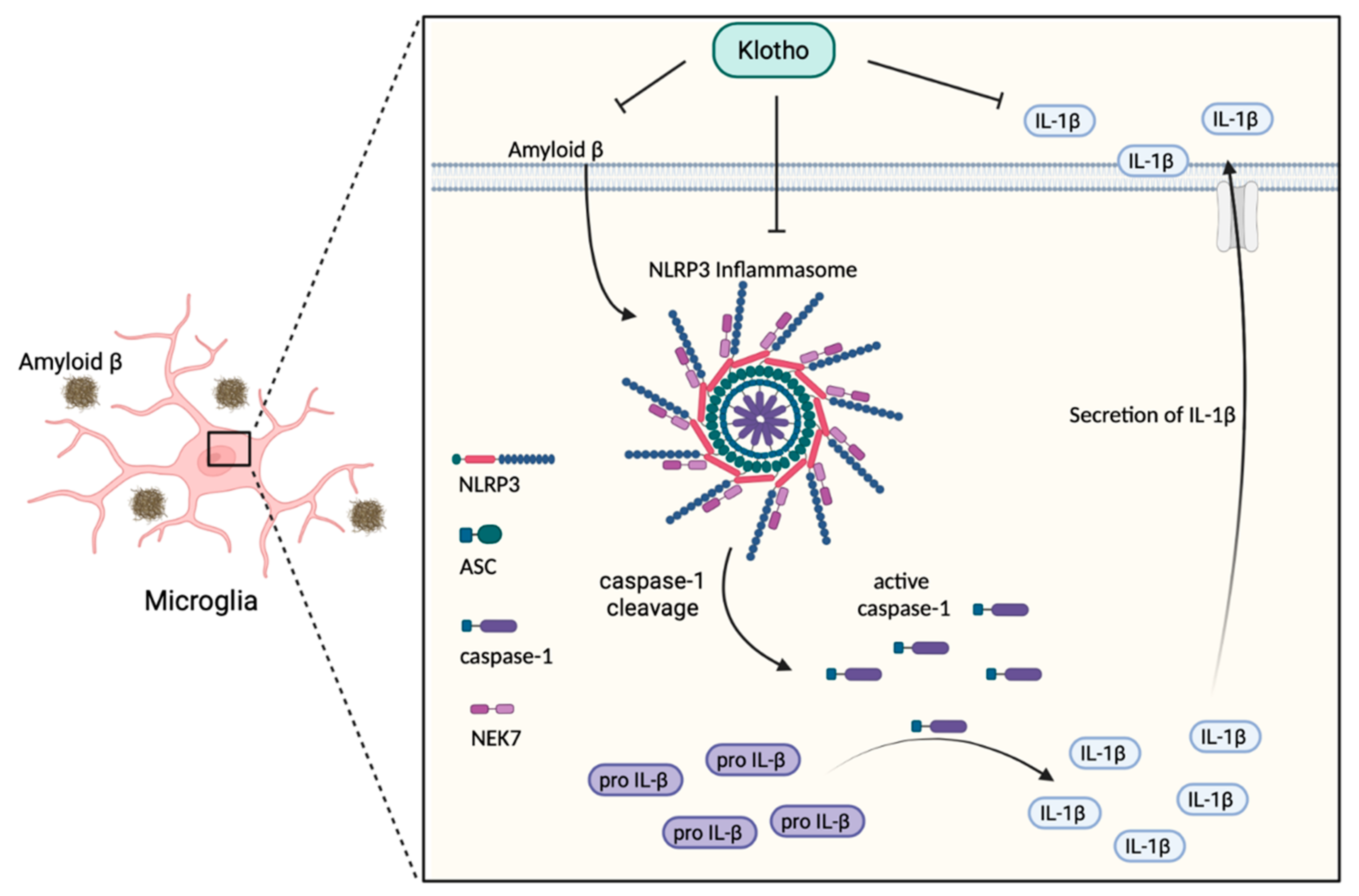
3.1. Klotho Inhibits the NLRP3/Caspase-1 Pathway
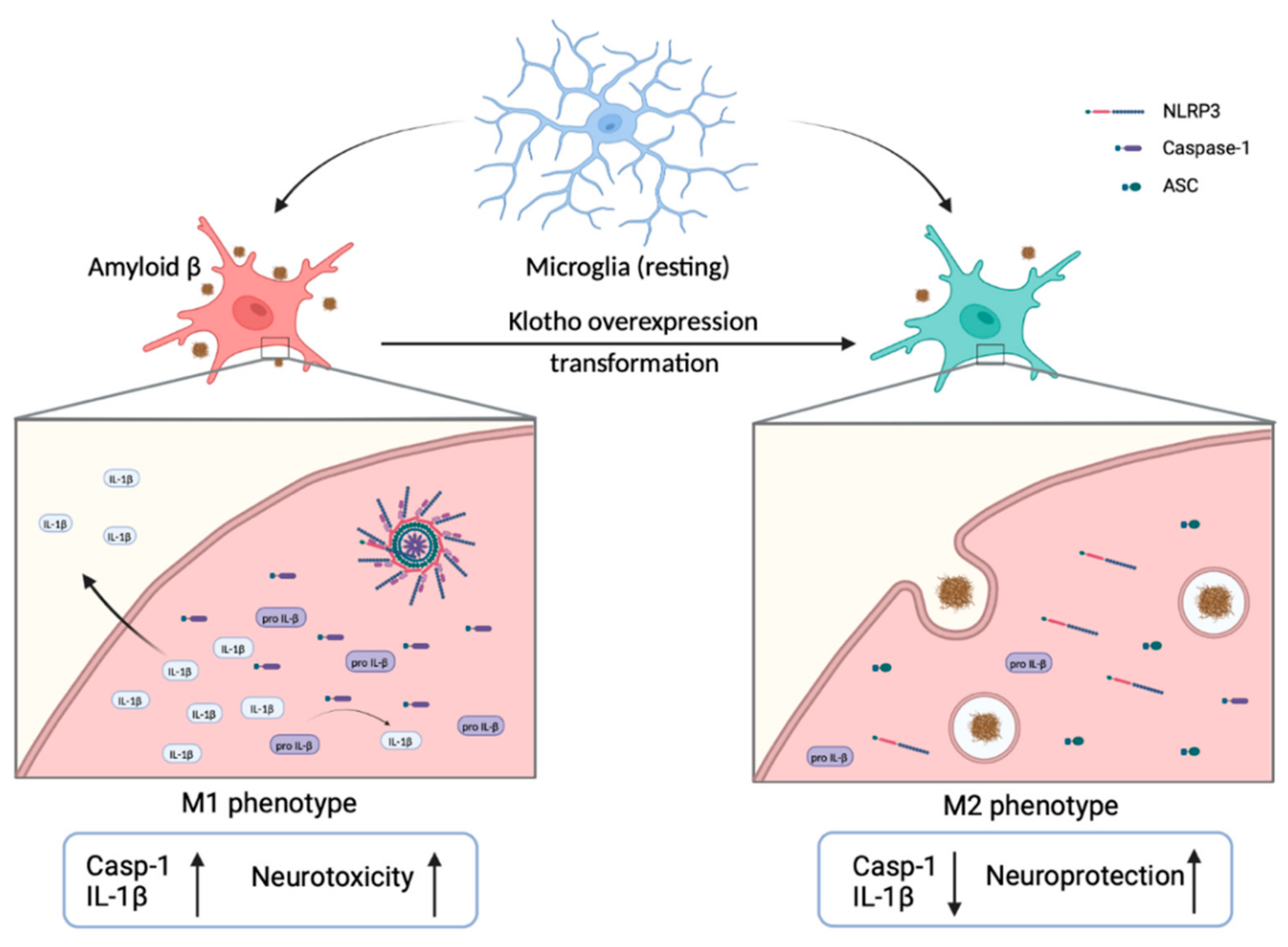
3.2. Klotho Promotes the Transformation of M1 Microglia to M2 Microglia
3.3. Klotho Promote Aβ Transporter-Mediated Aβ Clearance
3.4. Klotho Mitigates Tau Pathology and Enhances Cognitive Function in AD
4. Klotho Acts as an Autophagy Inducer
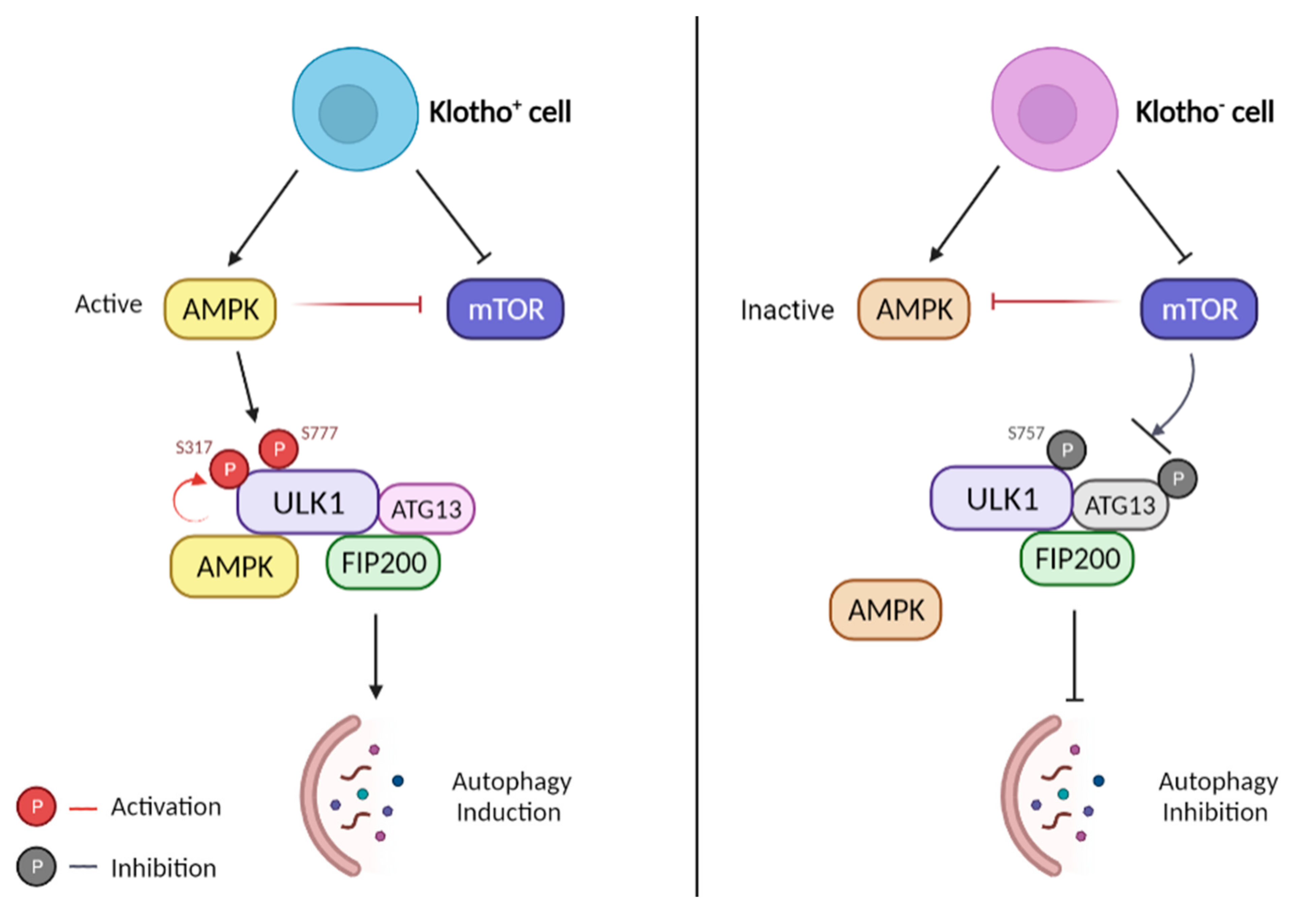
4.1. Klotho Stimulates the Formation of UNC51-like Kinase-1 (ULK1) Complex
4.2. Klotho Inhibits the IGF-1/PI3K/Akt/mTOR Pathway
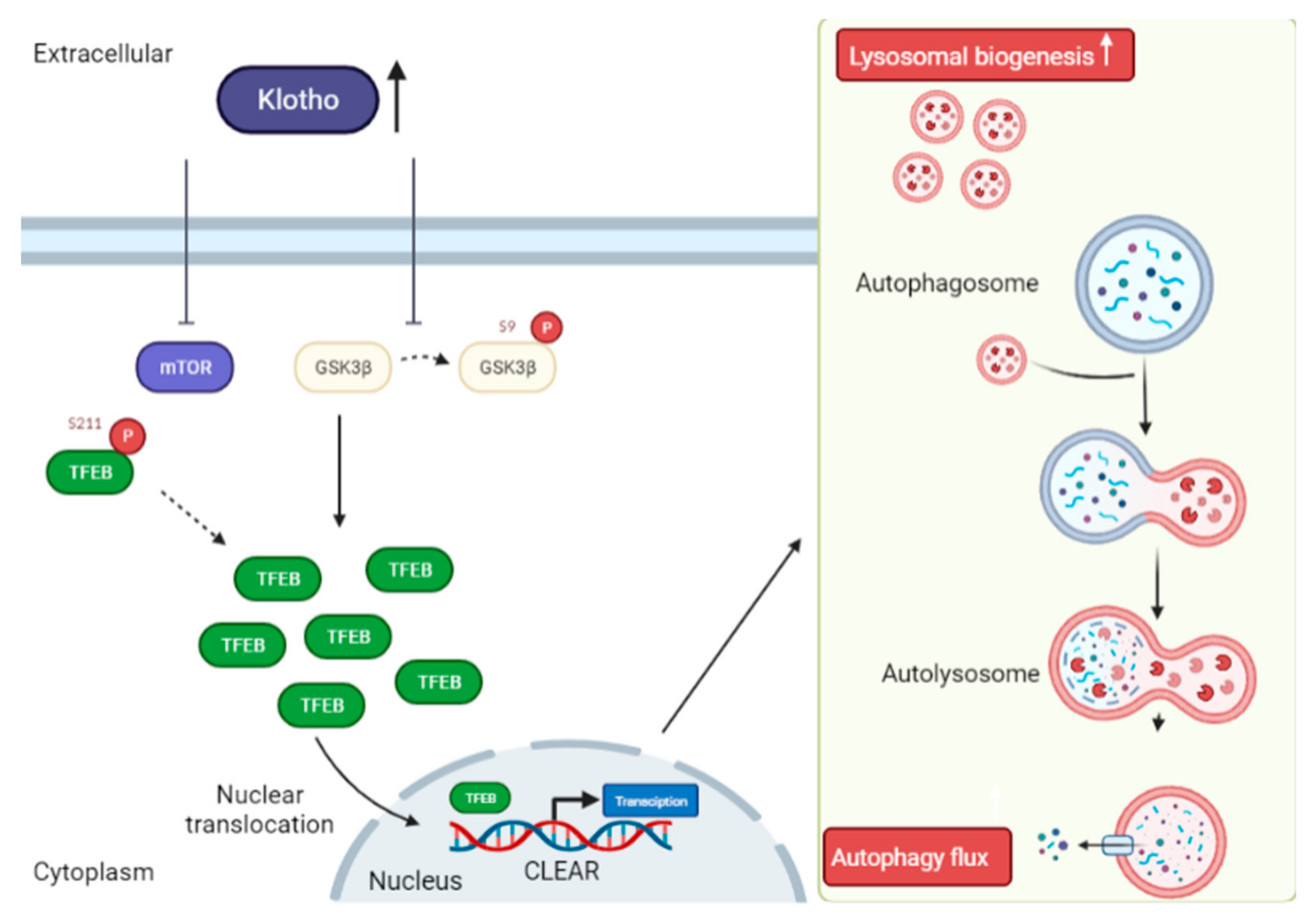
4.3. Klotho Promotes Nuclear Translocation of TFEB
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bolos, M.; Perea, J.R.; Avila, J. Alzheimer’s disease as an inflammatory disease. Biomol. Concepts 2017, 8, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Iyaswamy, A.; Krishnamoorthi, S.K.; Song, J.X.; Yang, C.B.; Kaliyamoorthy, V.; Zhang, H.; Sreenivasmurthy, S.G.; Malampati, S.; Wang, Z.Y.; Zhu, Z.; et al. NeuroDefend, a novel Chinese medicine, attenuates amyloid-beta and tau pathology in experimental Alzheimer’s disease models. J. Food Drug Anal. 2020, 28, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Duijn, C.; Nilsson, P.M.; et al. The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat. Rev. Neurosci. 2022, 23, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients With Early Alzheimer Disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Liu, J.; Zhu, Z.; Su, C.F.; Sreenivasmurthy, S.G.; Iyaswamy, A.; Lu, J.H.; Chen, G.; Song, J.X.; Li, M. Traditional Chinese medicine compounds regulate autophagy for treating neurodegenerative disease: A mechanism review. Biomed. Pharmacother. 2021, 133, 110968. [Google Scholar] [CrossRef] [PubMed]
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2019, 10, 1312. [Google Scholar] [CrossRef] [PubMed]
- Malampati, S.; Song, J.X.; Tong, B.C.-T.; Nalluri, A.; Yang, C.B.; Wang, Z.; Sreenivasmurthy, S.G.; Zhu, Z.; Liu, J.; Su, C.; et al. Targeting Aggrephagy for the Treatment of Alzheimer’s Disease. Cells 2020, 9, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.; Song, J.; Lu, J.; et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanekiyo, T.; Cirrito, J.R.; Liu, C.C.; Shinohara, M.; Li, J.; Schuler, D.R.; Shinohara, M.; Holtzman, D.M.; Bu, G. Neuronal clearance of amyloid-beta by endocytic receptor LRP1. J. Neurosci. 2013, 33, 19276–19283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocahan, S.; Dogan, Z. Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-methyl-D-aspartate Receptors, Tau Protein and Other Risk Factors. Clin. Psychopharmacol. Neurosci. 2017, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Hu, N.; Tan, M.S.; Yu, J.T.; Tan, L. Behavioral and psychological symptoms in Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 927804. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer’s disease. Indian J. Psychiatry 2009, 51, 55–61. [Google Scholar] [CrossRef]
- Yang, C.; Cai, C.Z.; Song, J.X.; Tan, J.Q.; Durairajan, S.S.K.; Iyaswamy, A.; Wu, M.Y.; Chen, L.L.; Yue, Z.; Li, M.; et al. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 2017, 13, 2028–2040. [Google Scholar] [CrossRef]
- Ng, R.C.; Jian, M.; Ma, O.K.; Bunting, M.; Kwan, J.S.; Zhou, G.J.; Senthilkumar, K.; Iyaswamy, A.; Chan, P.K.; Li, M.; et al. Chronic oral administration of adipoRon reverses cognitive impairments and ameliorates neuropathology in an Alzheimer’s disease mouse model. Mol. Psychiatry 2021, 26, 5669–5689. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Chan, H.N.; Ashok, I.; Krishnamoorthi, S.K.; Li, M.; Li, H.W.; Wong, M.S. Amyloid-beta oligomer targeted theranostic probes for in vivo NIR imaging and inhibition of self-aggregation and amyloid-beta induced ROS generation. Talanta 2021, 224, 121830. [Google Scholar] [CrossRef]
- Lehrer, S.; Rheinstein, P.H. Alignment of Alzheimer’s disease amyloid beta-peptide and klotho. World Acad. Sci. J. 2020, 2, 27. [Google Scholar] [CrossRef]
- Tang, B.L. Neuronal protein trafficking associated with Alzheimer disease: From APP and BACE1 to glutamate receptors. Cell Adhes. Migr. 2009, 3, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Sreenivasmurthy, S.G.; Iyaswamy, A.; Krishnamoorthi, S.; Senapati, S.; Malampati, S.; Zhu, Z.; Su, C.F.; Liu, J.; Guan, X.J.; Tong, B.C.; et al. Protopine promotes the proteasomal degradation of pathological tau in Alzheimer’s disease models via HDAC6 inhibition. Phytomedicine 2022, 96, 153887. [Google Scholar] [CrossRef] [PubMed]
- Iyaswamy, A.; Krishnamoorthi, S.K.; Liu, Y.W.; Song, J.X.; Kammala, A.K.; Sreenivasmurthy, S.G.; Malampati, S.; Tong, B.C.K.; Selvarasu, K.; Cheung, K.H.; et al. Yuan-Hu Zhi Tong Prescription Mitigates Tau Pathology and Alleviates Memory Deficiency in the Preclinical Models of Alzheimer’s Disease. Front. Pharmacol. 2020, 11, 584770. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The NLRP3 inflammasome in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Dias, G.P.; Murphy, T.; Stangl, D.; Ahmet, S.; Morisse, B.; Nix, A.; Aimone, L.J.; Aimone, J.B.; Kuro, O.M.; Gage, F.H.; et al. Intermittent fasting enhances long-term memory consolidation, adult hippocampal neurogenesis, and expression of longevity gene Klotho. Mol. Psychiatry 2021, 26, 6365–6379. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Hwang, K.H.; Park, K.S.; Kong, I.D.; Cha, S.K. Biological Role of Anti-aging Protein Klotho. J. Lifestyle Med. 2015, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kuang, X.; Chen, Y.S.; Wang, L.F.; Li, Y.J.; Liu, K.; Zhang, M.X.; Li, L.J.; Chen, C.; He, Q.; Wang, Y.; et al. Klotho upregulation contributes to the neuroprotection of ligustilide in an Alzheimer’s disease mouse model. Neurobiol. Aging 2014, 35, 169–178. [Google Scholar] [CrossRef]
- Mytych, J.; Solek, P.; Bedzinska, A.; Rusinek, K.; Warzybok, A.; Tabecka-Lonczynska, A.; Koziorowski, M. Towards Age-Related Anti-Inflammatory Therapy: Klotho Suppresses Activation of ER and Golgi Stress Response in Senescent Monocytes. Cells 2020, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- Semba, R.D.; Moghekar, A.R.; Hu, J.; Sun, K.; Turner, R.; Ferrucci, L.; O’Brien, R. Klotho in the cerebrospinal fluid of adults with and without Alzheimer’s disease. Neurosci. Lett. 2014, 558, 37–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mytych, J.; Solek, P.; Bedzinska, A.; Rusinek, K.; Warzybok, A.; Tabecka-Lonczynska, A.; Koziorowski, M. Klotho-mediated changes in the expression of Atg13 alter formation of ULK1 complex and thus initiation of ER- and Golgi-stress response mediated autophagy. Apoptosis 2020, 25, 57–72. [Google Scholar] [CrossRef]
- Mytych, J.; Solek, P.; Koziorowski, M. Klotho modulates ER-mediated signaling crosstalk between prosurvival autophagy and apoptotic cell death during LPS challenge. Apoptosis 2019, 24, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Lim, S.W.; Quan, Y.; Cui, S.; Shin, Y.J.; Ko, E.J.; Chung, B.H.; Yang, C.W. Role of Klotho in Chronic Calcineurin Inhibitor Nephropathy. Oxid. Med. Cell. Longev. 2019, 2019, 1825018. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, C.Y.; Yang, T.T.; Zhou, H.J.; Zhao, Y.; Kuang, X.; Duan, W.; Du, J.R. Lentiviral vector-mediated overexpression of Klotho in the brain improves Alzheimer’s disease-like pathology and cognitive deficits in mice. Neurobiol. Aging 2019, 78, 18–28. [Google Scholar] [CrossRef]
- Zhao, Y.; Zeng, C.Y.; Li, X.H.; Yang, T.T.; Kuang, X.; Du, J.R. Klotho overexpression improves amyloid-beta clearance and cognition in the APP/PS1 mouse model of Alzheimer’s disease. Aging Cell 2020, 19, e13239. [Google Scholar] [CrossRef]
- Zhu, L.; Stein, L.R.; Kim, D.; Ho, K.; Yu, G.Q.; Zhan, L.; Larsson, T.E.; Mucke, L. Klotho controls the brain-immune system interface in the choroid plexus. Proc. Natl. Acad. Sci. USA 2018, 115, E11388–E11396. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Sun, Z. Autophagy plays a critical role in Klotho gene deficiency-induced arterial stiffening and hypertension. J. Mol. Med. 2019, 97, 1615–1625. [Google Scholar] [CrossRef]
- Li, D.; Jing, D.; Liu, Z.; Chen, Y.; Huang, F.; Behnisch, T. Enhanced Expression of Secreted alpha-Klotho in the Hippocampus Alters Nesting Behavior and Memory Formation in Mice. Front. Cell. Neurosci. 2019, 13, 133. [Google Scholar] [CrossRef]
- Lin, Y.; Kuro-o, M.; Sun, Z. Genetic deficiency of anti-aging gene klotho exacerbates early nephropathy in STZ-induced diabetes in male mice. Endocrinology 2013, 154, 3855–3863. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rodriguez, M.; Villar-Conde, S.; Astillero-Lopez, V.; Villanueva-Anguita, P.; Ubeda-Banon, I.; Flores-Cuadrado, A.; Martinez-Marcos, A.; Saiz-Sanchez, D. Neurodegeneration and Astrogliosis in the Human CA1 Hippocampal Subfield Are Related to hsp90ab1 and bag3 in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 23, 165. [Google Scholar] [CrossRef]
- Nixon, R.A. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140443. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.; Zhou, H.J.; Thorne, A.H.; Chen, X.N.; Li, L.J.; Du, J.R. Neuroprotective Effect of Ligustilide through Induction of alpha-Secretase Processing of Both APP and Klotho in a Mouse Model of Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 353. [Google Scholar] [CrossRef]
- Lim, S.W.; Shin, Y.J.; Luo, K.; Quan, Y.; Ko, E.J.; Chung, B.H.; Yang, C.W. Effect of Klotho on autophagy clearance in tacrolimus-induced renal injury. FASEB J. 2019, 33, 2694–2706. [Google Scholar] [CrossRef] [Green Version]
- de Mello, N.P.; Orellana, A.M.; Mazucanti, C.H.; de Morais Lima, G.; Scavone, C.; Kawamoto, E.M. Insulin and Autophagy in Neurodegeneration. Front. Neurosci. 2019, 13, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; She, H.; Zhang, T.; Xu, H.; Cheng, L.; Yepes, M.; Zhao, Y.; Mao, Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell Biol. 2018, 217, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Lin, M.G.; Hurley, J.H. Structure and function of the ULK1 complex in autophagy. Curr. Opin. Cell Biol. 2016, 39, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.Z.; Zhuang, X.X.; Zhu, Q.; Wu, M.Y.; Su, H.; Wang, X.J.; Iyaswamy, A.; Yue, Z.; Wang, Q.; Zhang, B.; et al. Enhancing autophagy maturation with CCZ1-MON1A complex alleviates neuropathology and memory defects in Alzheimer disease models. Theranostics 2022, 12, 1738–1755. [Google Scholar] [CrossRef]
- Durairajan, S.S.K.; Selvarasu, K.; Bera, M.R.; Rajaram, K.; Iyaswamy, A.; Li, M. Alzheimer’s Disease and other Tauopathies: Exploring Efficacy of Medicinal Plant-Derived Compounds in Alleviating Tau-Mediated Neurodegeneration. Curr. Mol. Pharmacol. 2021. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Iyaswamy, A.; Krishnamoorthi, S.K.; Zhang, H.; Sreenivasmurthy, S.G.; Zhu, Z.; Liu, J.; Su, C.F.; Guan, X.J.; Wang, Z.Y.; Cheung, K.H.; et al. Qingyangshen mitigates amyloid-beta and Tau aggregate defects involving PPARalpha-TFEB activation in transgenic mice of Alzheimer’s disease. Phytomedicine 2021, 91, 153648. [Google Scholar] [CrossRef] [PubMed]
- Song, J.X.; Malampati, S.; Zeng, Y.; Durairajan, S.S.K.; Yang, C.B.; Tong, B.C.; Iyaswamy, A.; Shang, W.B.; Sreenivasmurthy, S.G.; Zhu, Z.; et al. A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell 2020, 19, e13069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.B.; Liu, J.; Tong, B.C.; Wang, Z.Y.; Zhu, Z.; Su, C.F.; Sreenivasmurthy, S.G.; Wu, J.X.; Iyaswamy, A.; Krishnamoorthi, S.; et al. TFEB, a master regulator of autophagy and biogenesis, unexpectedly promotes apoptosis in response to the cyclopentenone prostaglandin 15d-PGJ2. Acta Pharmacol. Sin. 2021. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Tong, B.C.; Wu, A.J.; Huang, A.S.; Dong, R.; Malampati, S.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Zhu, Z.; Su, C.; et al. Lysosomal TPCN (two pore segment channel) inhibition ameliorates beta-amyloid pathology and mitigates memory impairment in Alzheimer disease. Autophagy 2021. online ahead of print. [Google Scholar] [CrossRef]
- Abraham, C.R.; Mullen, P.C.; Tucker-Zhou, T.; Chen, C.D.; Zeldich, E. Klotho Is a Neuroprotective and Cognition-Enhancing Protein. Vitam. Horm. 2016, 101, 215–238. [Google Scholar] [CrossRef]
- Mytych, J. Klotho and neurons: Mutual crosstalk between autophagy, endoplasmic reticulum, and inflammatory response. Neural Regen. Res. 2021, 16, 1542–1543. [Google Scholar] [CrossRef] [PubMed]
- Mytych, J.; Solek, P.; Tabecka-Lonczynska, A.; Koziorowski, M. Klotho-Mediated Changes in Shelterin Complex Promote Cytotoxic Autophagy and Apoptosis in Amitriptyline-Treated Hippocampal Neuronal Cells. Mol. Neurobiol. 2019, 56, 6952–6963. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Ito, M. Klotho-Related Protein KLrP: Structure and Functions. Vitam. Horm. 2016, 101, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Torbus-Paluszczak, M.; Bartman, W.; Adamczyk-Sowa, M. Klotho protein in neurodegenerative disorders. Neurol. Sci. 2018, 39, 1677–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boksha, I.S.; Prokhorova, T.A.; Savushkina, O.K.; Tereshkina, E.B. Klotho Protein: Its Role in Aging and Central Nervous System Pathology. Biochemistry 2017, 82, 990–1005. [Google Scholar] [CrossRef]
- Dermaku-Sopjani, M.; Kolgeci, S.; Abazi, S.; Sopjani, M. Significance of the anti-aging protein Klotho. Mol. Membr. Biol. 2013, 30, 369–385. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, J.; Sun, Z. Antiaging Gene Klotho Deficiency Promoted High-Fat Diet-Induced Arterial Stiffening via Inactivation of AMP-Activated Protein Kinase. Hypertension 2016, 67, 564–573. [Google Scholar] [CrossRef] [Green Version]
- Olejnik, A.; Franczak, A.; Krzywonos-Zawadzka, A.; Kaluzna-Oleksy, M.; Bil-Lula, I. The Biological Role of Klotho Protein in the Development of Cardiovascular Diseases. Biomed. Res. Int. 2018, 2018, 5171945. [Google Scholar] [CrossRef] [Green Version]
- Paroni, G.; Panza, F.; De Cosmo, S.; Greco, A.; Seripa, D.; Mazzoccoli, G. Klotho at the Edge of Alzheimer’s Disease and Senile Depression. Mol. Neurobiol. 2019, 56, 1908–1920. [Google Scholar] [CrossRef]
- Saar-Kovrov, V.; Donners, M.; van der Vorst, E.P.C. Shedding of Klotho: Functional Implications in Chronic Kidney Disease and Associated Vascular Disease. Front. Cardiovasc. Med. 2020, 7, 617842. [Google Scholar] [CrossRef] [PubMed]
- van Loon, E.P.; Pulskens, W.P.; van der Hagen, E.A.; Lavrijsen, M.; Vervloet, M.G.; van Goor, H.; Bindels, R.J.; Hoenderop, J.G. Shedding of klotho by ADAMs in the kidney. Am. J. Physiol. Renal Physiol. 2015, 309, F359–F368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, S.; Combet, E.; Stenvinkel, P.; Shiels, P.G. Klotho, Aging, and the Failing Kidney. Front. Endocrinol. 2020, 11, 560. [Google Scholar] [CrossRef]
- Shmulevich, R.; Nissim, T.B.; Wolf, I.; Merenbakh-Lamin, K.; Fishman, D.; Sekler, I.; Rubinek, T. Klotho rewires cellular metabolism of breast cancer cells through alteration of calcium shuttling and mitochondrial activity. Oncogene 2020, 39, 4636–4649. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; May, P.; Gu, W.; Mayhaus, M.; Pichler, S.; Spaniol, C.; Glaab, E.; Bobbili, D.R.; Antony, P.; Koegelsberger, S.; et al. A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol. Psychiatry 2020, 25, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Pietri, M.; Dakowski, C.; Hannaoui, S.; Alleaume-Butaux, A.; Hernandez-Rapp, J.; Ragagnin, A.; Mouillet-Richard, S.; Haik, S.; Bailly, Y.; Peyrin, J.M.; et al. PDK1 decreases TACE-mediated alpha-secretase activity and promotes disease progression in prion and Alzheimer’s diseases. Nat. Med. 2013, 19, 1124–1131. [Google Scholar] [CrossRef]
- Lerch, C.; Shroff, R.; Wan, M.; Rees, L.; Aitkenhead, H.; Kaplan Bulut, I.; Thurn, D.; Karabay Bayazit, A.; Niemirska, A.; Canpolat, N.; et al. Effects of nutritional vitamin D supplementation on markers of bone and mineral metabolism in children with chronic kidney disease. Nephrol. Dial. Transplant. 2018, 33, 2208–2217. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Sitara, D.; Sato, T.; Densmore, M.; Saito, H.; Schuler, C.; Erben, R.G.; Lanske, B. PTH ablation ameliorates the anomalies of Fgf23-deficient mice by suppressing the elevated vitamin D and calcium levels. Endocrinology 2011, 152, 4053–4061. [Google Scholar] [CrossRef]
- Kuro-o, M. Klotho and the aging process. Korean J. Intern. Med. 2011, 26, 113–122. [Google Scholar] [CrossRef]
- Razzaque, M.S.; Lanske, B. Hypervitaminosis D and premature aging: Lessons learned from Fgf23 and Klotho mutant mice. Trends Mol. Med. 2006, 12, 298–305. [Google Scholar] [CrossRef]
- Kolek, O.I.; Hines, E.R.; Jones, M.D.; LeSueur, L.K.; Lipko, M.A.; Kiela, P.R.; Collins, J.F.; Haussler, M.R.; Ghishan, F.K. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: The final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1036–G1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.L. Regulation of ion channels by secreted Klotho: Mechanisms and implications. Kidney Int. 2010, 77, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shardell, M.; Drew, D.A.; Semba, R.D.; Harris, T.B.; Cawthon, P.M.; Simonsick, E.M.; Kalyani, R.R.; Schwartz, A.V.; Kritchevsky, S.B.; Newman, A.B. Plasma Soluble alphaKlotho, Serum Fibroblast Growth Factor 23, and Mobility Disability in Community-Dwelling Older Adults. J. Endocr. Soc. 2020, 4, bvz032. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhou, M.; Peng, J.; Yang, Q.; Chu, J.; Li, R.; Jiang, Y. Mechanism of the Fibroblast Growth Factor 23/alpha-Klotho Axis in Peripheral Blood Mononuclear Cell Inflammation in Alzheimer’s Disease. Immunol. Investig. 2021. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Mencke, R.; Olauson, H.; Hillebrands, J.L. Effects of Klotho on fibrosis and cancer: A renal focus on mechanisms and therapeutic strategies. Adv. Drug Deliv. Rev. 2017, 121, 85–100. [Google Scholar] [CrossRef]
- Chateau, M.T.; Araiz, C.; Descamps, S.; Galas, S. Klotho interferes with a novel FGF-signalling pathway and insulin/Igf-like signalling to improve longevity and stress resistance in Caenorhabditis elegans. Aging 2010, 2, 567–581. [Google Scholar] [CrossRef] [Green Version]
- Bartke, A. Long-lived Klotho mice: New insights into the roles of IGF-1 and insulin in aging. Trends Endocrinol. Metab. 2006, 17, 33–35. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [Green Version]
- Altintas, O.; Park, S.; Lee, S.J. The role of insulin/IGF-1 signaling in the longevity of model invertebrates, C. elegans and D. melanogaster. BMB Rep. 2016, 49, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Kopitar-Jerala, N. The Role of Interferons in Inflammation and Inflammasome Activation. Front. Immunol. 2017, 8, 873. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-alpha signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Typiak, M.; Piwkowska, A. Antiinflammatory Actions of Klotho: Implications for Therapy of Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 956. [Google Scholar] [CrossRef] [PubMed]
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Mankan, A.K.; Dau, T.; Jenne, D.; Hornung, V. The NLRP3/ASC/Caspase-1 axis regulates IL-1beta processing in neutrophils. Eur. J. Immunol. 2012, 42, 710–715. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Tay, T.L.; Prinz, M. Love and death: Microglia, NLRP3 and the Alzheimer’s brain. Cell Res. 2013, 23, 595–596. [Google Scholar] [CrossRef] [Green Version]
- Griffin, W.S.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflammation 2006, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ising, C.; Heneka, M.T. Functional and structural damage of neurons by innate immune mechanisms during neurodegeneration. Cell Death Dis. 2018, 9, 120. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.; Fisher, K.; Hooper, N.M. Exploiting the neuroprotective effects of alpha-klotho to tackle ageing- and neurodegeneration-related cognitive dysfunction. Neuronal Signal. 2021, 5, NS20200101. [Google Scholar] [CrossRef]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Kim, J.; Li, A.; Knoten, A.; Jain, S.; Hirsch-Reinshagen, V.; Wellington, C.L.; Bales, K.R.; et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Investig. 2008, 118, 671–682. [Google Scholar] [CrossRef]
- Zhou, H.; Pu, S.; Zhou, H.; Guo, Y. Klotho as Potential Autophagy Regulator and Therapeutic Target. Front. Pharmacol. 2021, 12, 755366. [Google Scholar] [CrossRef]
- Kim, W.S.; Bhatia, S.; Elliott, D.A.; Agholme, L.; Kagedal, K.; McCann, H.; Halliday, G.M.; Barnham, K.J.; Garner, B. Increased ATP-binding cassette transporter A1 expression in Alzheimer’s disease hippocampal neurons. J. Alzheimers Dis. 2010, 21, 193–205. [Google Scholar] [CrossRef]
- Dubal, D.B.; Zhu, L.; Sanchez, P.E.; Worden, K.; Broestl, L.; Johnson, E.; Ho, K.; Yu, G.Q.; Kim, D.; Betourne, A.; et al. Life extension factor klotho prevents mortality and enhances cognition in hAPP transgenic mice. J. Neurosci. 2015, 35, 2358–2371. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, J.; Franzmeier, N.; Rubinski, A.; Dichgans, M.; Brendel, M.; Alzheimer’s Disease Neuroimaging Initiative (ADNI); Malik, R.; Ewers, M. KL-VS heterozygosity is associated with lower amyloid-dependent tau accumulation and memory impairment in Alzheimer’s disease. Nat. Commun. 2021, 12, 3825. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Perez-Rojas, J.M.; Hernandez-Damian, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, H.; Wang, Z.L.; Li, W.H.; Liu, H.; Zhao, Y.X. The PI3K/AKT/mTOR pathway regulates autophagy to induce apoptosis of alveolar epithelial cells in chronic obstructive pulmonary disease caused by PM2.5 particulate matter. J. Int. Med. Res. 2020, 48, 300060520927919. [Google Scholar] [CrossRef] [PubMed]
- Mytych, J. Actions of Klotho on hippocampal neuronal cells. Vitam. Horm. 2022, 118, 223–246. [Google Scholar] [CrossRef]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef]
- Liu, Q.; Guan, J.Z.; Sun, Y.; Le, Z.; Zhang, P.; Yu, D.; Liu, Y. Insulin-like growth factor 1 receptor-mediated cell survival in hypoxia depends on the promotion of autophagy via suppression of the PI3K/Akt/mTOR signaling pathway. Mol. Med. Rep. 2017, 15, 2136–2142. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.W.; Yuan, L.J.; Yang, Y.; Zhang, M.; Chen, W.F. IGF-1 inhibits MPTP/MPP(+)-induced autophagy on dopaminergic neurons through the IGF-1R/PI3K-Akt-mTOR pathway and GPER. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E734–E743. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Liu, L.; Xie, A. The Role of Insulin/IGF-1/PI3K/Akt/GSK3beta Signaling in Parkinson’s Disease Dementia. Front. Neurosci. 2018, 12, 73. [Google Scholar] [CrossRef] [Green Version]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhang, X.Q.; Zhang, Z. Transcription factor EB agonists from natural products for treating human diseases with impaired autophagy-lysosome pathway. Chin. Med. 2020, 15, 123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, X.; Wang, S.; Chen, Y.; Liu, H. Regulation of TFEB activity and its potential as a therapeutic target against kidney diseases. Cell Death Discov. 2020, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Su, C.F.; Jiang, L.; Zhang, X.W.; Iyaswamy, A.; Li, M. Resveratrol in Rodent Models of Parkinson’s Disease: A Systematic Review of Experimental Studies. Front. Pharmacol. 2021, 12, 644219. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, J.; Yang, C. Celastrol, a TFEB (transcription factor EB) agonist, is a promising drug candidate for Alzheimer disease. Autophagy 2022. online ahead of print. [Google Scholar] [CrossRef]
- Yang, C.; Zhu, Z.; Tong, B.C.; Iyaswamy, A.; Xie, W.J.; Zhu, Y.; Sreenivasmurthy, S.G.; Senthilkumar, K.; Cheung, K.H.; Song, J.X.; et al. A stress response p38 MAP kinase inhibitor SB202190 promoted TFEB/TFE3-dependent autophagy and lysosomal biogenesis independent of p38. Redox Biol. 2020, 32, 101445. [Google Scholar] [CrossRef]
- Nelson, P.T.; Head, E.; Schmitt, F.A.; Davis, P.R.; Neltner, J.H.; Jicha, G.A.; Abner, E.L.; Smith, C.D.; Van Eldik, L.J.; Kryscio, R.J.; et al. Alzheimer’s disease is not “brain aging”: Neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011, 121, 571–587. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author, Year | Title | Finding | Reference |
|---|---|---|---|
| Kuang et al., 2014 | Klotho upregulation contributes to the neuroprotection of ligustilide in an Alzheimer’s disease mouse model | Ligustilide (LIG)-induced Klotho overexpression is neuroprotective towards AD by downregulating the insulin/IGF-1 signaling pathway, thereby triggering Forkhead-box class O (FoxO) transcription factor to relieve oxidative stress in the brain. Ligustilide increased mitochondrial manganese-superoxide dismutase, catalase expression and activity, and decreased malondialdehyde, protein carbonyl, and 8-hydroxydesoxyguanosine levels in the brain. | [27] |
| Kuang et al., 2017 | Neuroprotective effect of Ligustilide through induction of α-secretase processing of both APP and Klotho in a mouse model of Alzheimer’s disease | Ligustilide (LIG)-induced the expression of both soluble APPα (sAPPα) and soluble Klotho (sKL) protein, thus facilitating the inhibition of IGF-1/Akt/mTOR signaling. The neuroprotective role of LIG against AD is highly associated with an increased level of Klotho, and ADAM10 proteins, eventually, promoting cerebral Aβ clearance and improving cognitive function. | [43] |
| Zeng et al., 2019 | Lentiviral vector mediated overexpression of Klotho in the brain improves Alzheimer’s disease-like pathology and cognitive deficits in mice | Klotho protein is strongly associated with Aβ clearance via AKT/mTOR signaling pathway repression and the activation of the autophagy-lysosome system, resulting in the improvement of cognitive function and beneficial pathological changes in an AD mouse model. | [34] |
| Zhao et al., 2020 | Klotho overexpression improves amyloid-β clearance and cognition in the APP/PS1 mouse model of Alzheimer’s disease | Klotho treatment remarkably improved AD-induced neuropathies in an aged transgenic mice model. It suppressed NLRP3, promoted microglia transformation and modulated the expression of Aβ transporter, eventually enhancing Aβ clearance in the brain. | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fung, T.Y.; Iyaswamy, A.; Sreenivasmurthy, S.G.; Krishnamoorthi, S.; Guan, X.-J.; Zhu, Z.; Su, C.-F.; Liu, J.; Kan, Y.; Zhang, Y.; et al. Klotho an Autophagy Stimulator as a Potential Therapeutic Target for Alzheimer’s Disease: A Review. Biomedicines 2022, 10, 705. https://doi.org/10.3390/biomedicines10030705
Fung TY, Iyaswamy A, Sreenivasmurthy SG, Krishnamoorthi S, Guan X-J, Zhu Z, Su C-F, Liu J, Kan Y, Zhang Y, et al. Klotho an Autophagy Stimulator as a Potential Therapeutic Target for Alzheimer’s Disease: A Review. Biomedicines. 2022; 10(3):705. https://doi.org/10.3390/biomedicines10030705
Chicago/Turabian StyleFung, Tsz Yan, Ashok Iyaswamy, Sravan G. Sreenivasmurthy, Senthilkumar Krishnamoorthi, Xin-Jie Guan, Zhou Zhu, Cheng-Fu Su, Jia Liu, Yuxuan Kan, Yuan Zhang, and et al. 2022. "Klotho an Autophagy Stimulator as a Potential Therapeutic Target for Alzheimer’s Disease: A Review" Biomedicines 10, no. 3: 705. https://doi.org/10.3390/biomedicines10030705