
Novel Biomarkers of Inflammation for the Management of Diabetes: Immunoglobulin-Free Light Chains

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of Virus in the Pathogenesis of Diabetes Mellitus
2.1. SARS-CoV-2
2.2. Hepatitis C Virus
2.3. A New Concept of Pathogenesis of HCV-Induced Diseases
3. Role of Inflammation in the Pathogenesis of Diabetes Mellitus
3.1. Inflammatory Cytokines
3.2. Nuclear Factor-Kappa B (NF-κB)
4. Novel Biomarkers of Inflammation: Immunoglobulin-Free Light Chains (FLCs)
4.1. FLCs as Novel Biomarkers of Chronic Inflammation
4.2. FLCs as Markers of Heart Failure and Myocarditis
4.3. FLCs and COVID-19 and Heart Diseases
4.4. FLCs as Markers of Atrial Fibrillation
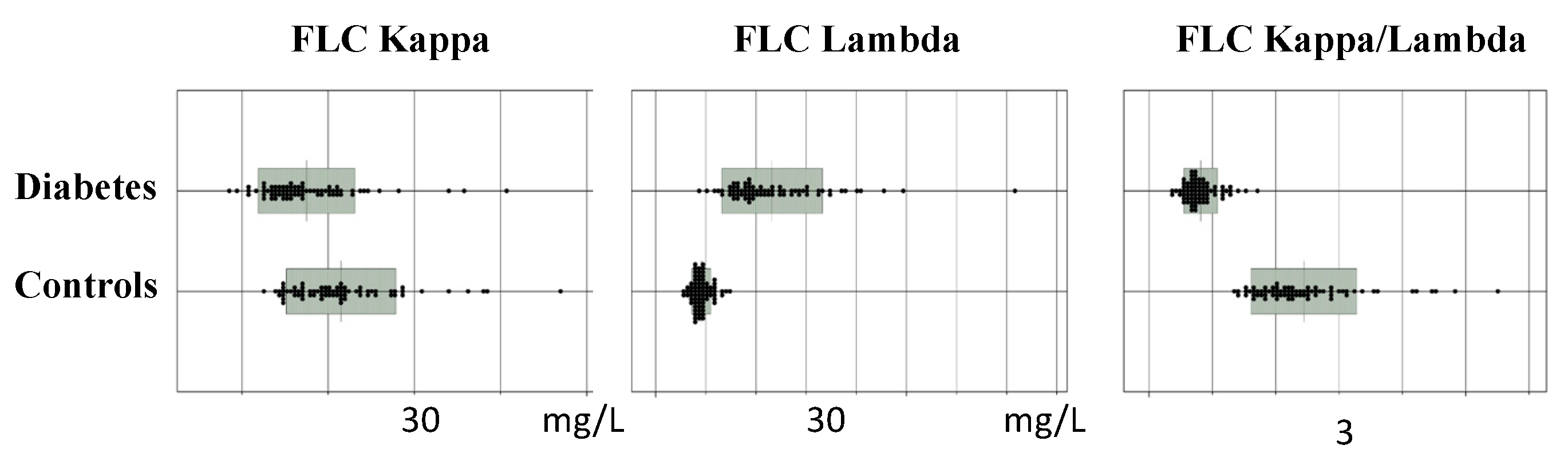
4.5. FLCs as Biomarkers of Diabetes
5. Targeting Inflammation for the Management of Diabetes
5.1. Metformin
5.2. Dipeptidyl Peptidase-4 Inhibitors
5.3. The Glucagon-Like Peptide 1 Receptor Agonists
5.4. SGLT2 Inhibitors
5.5. Anti-IL-1 Agents
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alberti, K.G.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet. Med. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- Yoon, J.W.; Austin, M.; Onodera, T.; Notkins, A.L. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N. Engl. J. Med. 1979, 300, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Sakurami, T.; Nabeya, N.; Nagaoka, K.; Matsumori, A.; Kuno, S.; Honda, A. Antibodies to Coxsackie B viruses and HLA in Japanese with juvenile-onset Type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1982, 22, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Tracy, S.; Drescher, K.M.; Jackson, J.D.; Kim, K.; Kono, K. Enteroviruses, type 1 diabetes and hygiene: A complex relationship. Rev. Med Virol. 2010, 20, 106–116. [Google Scholar] [CrossRef]
- Oever, I.A.M.V.D.; Raterman, H.G.; Nurmohamed, M.T.; Simsek, S. Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediat. Inflamm. 2010, 2010, 792393. [Google Scholar] [CrossRef] [Green Version]
- Goldfine, A.B.; Shoelson, S.E. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Investig. 2017, 127, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donath, M.Y.; Meier, D.T.; Boni-Schnethler, M. Inflammation in the pathophysiology and therapy of cardiometabolic disease. Endocr. Rev. 2019, 40, 1080–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultuybek, G.K.; Soydas, T.; Yenmis, G. NF-κB as the mediator of metformin’s effect on ageing and ageing-related diseases. Clin. Exp. Pharmacol. Physiol. 2019, 46, 413–422. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikk beta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Matsumori, A.; Shimada, T.; Nakatani, E.; Shimada, M.; Tracy, S.; Chapman, N.M.; Drayson, M.T.; Hartz, V.L.; Mason, J.W. Immunoglobulin free light chains as an inflammatory biomarker of heart failure with myocarditis. Clin. Immunol. 2020, 217, 108455. [Google Scholar] [CrossRef]
- Matsumori, A.; Shimada, T.; Shimada, M.; Otani, H.; Drayson, M.T.; Mason, J.W. Immunoglobulin free light chains as inflammatory biomarkers of atrial fibrillation. Circ. Arrhythmia Electrophysiol. 2020, 13, e009017. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, A.; Shimada, T.; Shimada, M.; Drayson, M.T. Immunoglobulin free light chains: An inflammatory biomarker of diabetes. Inflamm. Res. 2020, 69, 715–718, Correction in Inflamm. Res. 2020, 69, 719. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 Diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.M.; Kim, C.; Banerjee, T.; Lee, J.M. Fluctuations in the incidence of type 1 diabetes in the United States from 2001 to 2015: A longitudinal study. BMC Med. 2017, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Rewers, M.; Ludvigsson, J. Environmental risk factors for type 1 diabetes. Lancet 2016, 387, 2340–2348. [Google Scholar] [CrossRef] [Green Version]
- Filippi, C.M.; von Herrath, M.G. Viral trigger for type 1 diabetes: Pros and cons. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Calvo, T.; Sabouri, S.; Anquetil, F.; von Herrath, M.G. The viral paradigm in type 1 diabetes: Who are the main suspects? Autoimmun. Rev. 2016, 15, 964–969. [Google Scholar] [CrossRef]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A review on the potential interaction and molecular mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef] [Green Version]
- Burke, R.M.; Tate, J.E.; Jiang, B.; Parashar, U.D. Rotavirus and type 1 diabetes—Is there a connection? A synthesis of the evidence. J. Infect. Dis. 2020, 222, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Haykal, M.; Matsumori, A.; Saleh, A.; Fayez, M.; Negm, H.; Shalaby, M.; Bassuony, S. Diagnosis and treatment of HCV heart diseases. Expert Rev. Cardiovasc. Ther. 2021, 19, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Lontchi-Yimagou, E.; Feutseu, C.; Kenmoe, S.; Zune, A.L.D.; Ekali, S.F.K.; Nguewa, J.L.; Choukem, S.P.; Mbanya, J.C.; Gautier, J.F.; Sobngwi, E. Non-autoimmune diabetes mellitus and the risk of virus infections: A systematic review and meta-analysis of case-control and cohort studies. Sci. Rep. 2021, 11, 8968. [Google Scholar] [CrossRef] [PubMed]
- Khunti, K.; Prato, D.P.; Mathieu, C.; Kahn, S.E.; Gabbay, R.A.; Buse, J.B. COVID-19, hyperglycemia, and new-onset diabetes. Diabetes Care 2021, 44, 2645–2655. [Google Scholar] [CrossRef] [PubMed]
- Bode, B.; Garrett, V.; Messler, J.; McFarland, R.; Crowe, J.; Booth, R.; Klonoff, D.C. Glycemic characteristics and clinical out-comes of COVID-19 patients hospitalized in the United States. J. Diabetes Sci. Technol. 2020, 14, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tian, S.; Chen, T.; Cui, Z.; Shi, N.; Zhong, X.; Qiu, K.; Zhang, J.; Zeng, T.; Chen, L.; et al. Newly diagnosed diabetes is associated with a higher risk of mortality than known diabetes in hospitalized patients with COVID-19. Diabetes Obes. Metab. 2020, 22, 1897–1906. [Google Scholar] [CrossRef]
- Accili, D. Can COVID-19 cause diabetes? Nat. Metab. 2021, 3, 123–125. [Google Scholar] [CrossRef]
- Coppelli, A.; Giannarelli, R.; Aragona, M.; Penno, G.; Falcone, M.; Tiseo, G.; Ghiadoni, L.; Barbieri, G.; Monzani, F.; Virdis, A.; et al. Hyperglycemia at hospital admission is associated with severity of the prognosis in patients hospitalized for COVID-19: The Pisa COVID-19 study. Diabetes Care 2020, 43, 2345–2348. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, E.; Storm, P.; Käräjämäki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Aly, D.M.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Shaharuddin, S.H.; Wang, V.; Santos, R.S.; Gross, A.; Wang, Y.; Jawanda, H.; Zhang, Y.; Hasan, W.; Garcia, G.J.; Arumugaswami, V.; et al. Deleterious effects of SARS-CoV-2 infection on human pancreatic cells. Front. Cell. Infect. Microbiol. 2021, 11, 678482. [Google Scholar] [CrossRef]
- Mazzaro, C.; Quartuccio, L.; Adinolfi, L.E.; Roccatello, D.; Pozzato, G.; Nevola, R.; Tonizzo, M.; Gitto, S.; Andreone, P.; Gattei, V. A review on extrahepatic manifestations of chronic hepatitis C virus infection and the impact of direct-acting antiviral therapy. Viruses 2021, 13, 2249. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jin, M.; Yang, M.; Liu, K.; Li, J.-W. Type 2 diabetes mellitus and the risk of hepatitis C virus infection: A systematic review. Sci. Rep. 2013, 3, srep02981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, D.; Ratziu, V.; El-Serag, H.B. Hepatitis C infection and risk of diabetes: A systematic review and meta-analysis. J. Hepatol. 2008, 49, 831–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naing, C.; Mak, J.W.; Ahmed, S.I.; Maung, M. Relationship between hepatitis C virus infection and type 2 diabetes mellitus: Meta-analysis. World J. Gastroenterol. 2012, 18, 1642–1651. [Google Scholar] [CrossRef]
- Lim, T.R.; Hazlehurst, J.M.; Oprescu, A.I.; Armstrong, M.J.; Abdullah, S.F.; Davies, N.; Flintham, R.; Balfe, P.; Mutimer, D.J.; McKeating, J.A.; et al. Hepatitis C virus infection is associated with hepatic and adipose tissue insulin resistance that improves after viral cure. Clin. Endocrinol. 2018, 90, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Nevola, R.; Acierno, C.; Pafundi, P.C.; Adinolfi, L.E. Chronic hepatitis C infection induces cardiovascular disease and type 2 diabetes: Mechanisms and management. Minerva Med. 2020, 112, 118–200. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Restivo, L.; Guerrera, B.; Sellitto, A.; Ciervo, A.; Iuliano, N.; Rinaldi, L.; Santoro, A.; Vigni, G.L.; Marrone, A. Chronic HCV infection is a risk factor of ischemic stroke. Atherosclerosis 2013, 231, 22–26. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Petta, S.; Fracanzani, A.L.; Coppola, C.; Narciso, V.; Nevola, R.; Rinaldi, L.; Calvaruso, V.; Staiano, L.; Di Marco, V.; et al. Impact of hepatitis C virus clearance by direct-acting antiviral treatment on the incidence of major cardiovascular events: A prospective multicentre study. Atherosclerosis 2020, 296, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, L.E.; Nevola, R.; Guerrera, B.; D’Alterio, G.; Marrone, A.; Giordano, M.; Rinaldi, L. Hepatitis C virus clearance by direct-acting antiviral treatments and impact on insulin resistance in chronic hepatitis C patients. J. Gastroenterol. Hepatol. 2018, 33, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.A.; Yan, P.; Aslam, S.; Shaikh, O.S.; Abou-Samra, A.B. Hepatitis C virus (HCV) treatment with directly acting agents reduces the risk of incident diabetes: Results from electronically retrieved cohort of HCV infected veterans (ERCHIVES). Clin. Infect. Dis. 2020, 70, 1153–1160. [Google Scholar] [CrossRef]
- Matsumori, A.; Matoba, Y.; Sasayama, S. Dilated cardiomyopathy associated with hepatitis C virus infection. Circulation 1995, 92, 2519–2525. [Google Scholar] [CrossRef]
- Matsumori, A.; Yutani, C.; Ikeda, Y.; Kawai, S.; Sasayama, S. Hepatitis C virus from the hearts of patients with myocarditis and cardiomyopathy. Lab. Investig. 2000, 80, 1137–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumori, A. Hepatitis C virus and cardiomyopathy. Circ. Res. 2005, 9, 144–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumori, A.; Shimada, T.; Chapman, N.M.; Tracy, S.M.; Mason, J.W. Myocarditis and heart Failure associated with hepatitis C virus infection. J. Card. Fail. 2006, 12, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, A.; Shimada, M.; Obata, T. Leukocytes are the major target of hepatitis C virus infection: Possible mechanism of multiorgan involvement including the heart. Glob. Heartl 2010, 5, 51–58. [Google Scholar] [CrossRef]
- Shichi, D.; Kikkawa, E.F.; Ota, M.; Katsuyama, Y.; Kimura, A.; Matsumori, A.; Kulski, J.K.; Naruse, T.K.; Inoko, H. The hap-lotype block, NFKBIL1-ATP6V1G2-BAT1-MICB-MICA, within the class III-class I boundary region of the human major his-tocompatibility complex may control susceptibility to hepatitis C virus-associated dilated cardiomyopathy. Tissue Antigens 2005, 66, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Shichi, D.; Matsumori, A.; Naruse, T.K.; Inoko, H.; Kimura, A. HLA-DPbeta chain may confer the susceptibility to hepatitis C virus-associated hypertrophic cardiomyopathy. Int. J. Immunogenet. 2008, 35, 37–43. [Google Scholar] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Pickup, J.C.; Mattock, M.B.; Chusney, G.D.; Burt, D. NIDDM as a disease of the innate immune system: Association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 1997, 40, 1286–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, J.; Kroke, A.; Mohlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Inflammatory cyto-kines and the risk to develop type 2 diabetes: Results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef] [Green Version]
- Herder, C.; Illig, T.; Rathmann, W.; Martin, S.; Haastert, B.; Müller-Scholze, S.; Holle, R.; Thorand, B.; Koenig, W.; Wichmann, H.E.; et al. Inflammation and type 2 diabetes: Results from KORA Augsburg. Das Gesundheitswesen 2005, 67, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Herder, C.; Brunner, E.J.; Rathmann, W.; Strassburger, K.; Tabak, A.G.; Schloot, N.C.; Witte, D.R. Elevated levels of the anti-inflammatory interleukin-1 receptor antagonist precede the onset of type 2 diabetes: The Whitehall II study. Diabetes Care 2009, 32, 421–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, M.; Imhof, A.; Berg, G.; Hutchinson, W.L.; Pepys, M.B.; Boeing, H.; Muche, R.; Brenner, H.; Koenig, W. Association between C-reactive protein and features of the metabolic syndrome: A population-based study. Diabetes Care 2000, 23, 1835–1839. [Google Scholar] [CrossRef] [Green Version]
- Meier, C.A.; Bobbioni, E.; Gabay, C.; Assimacopoulos- Jeannet, F.; Golay, A.; Dayer, J.M. IL-1 receptor antagonist serum levels are increased in human obesity: A possible link to the resistance to leptin? J. Clin. Endocrinol. Metab. 2002, 87, 1184–1188. [Google Scholar] [CrossRef]
- Carstensen, M.; Herder, C.; Kivimaki, M.; Jokela, M.; Roden, M.; Shipley, M.J.; Witte, D.R.; Brunner, E.J.; Tabak, A.G. Accel-erated increase in serum interleukin-1 receptor antagonist starts 6 years before diagnosis of type 2 diabetes: Whitehall II pro-spective cohort study. Diabetes 2010, 59, 1222–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marculescu, R.; Endler, G.; Schillinger, M.; Iordanova, N.; Exner, M.; Hayden, E.; Huber, K.; Wagner, O.; Mannhalter, C. In-terleukin-1 receptor antagonist genotype is associated with coronary atherosclerosis in patients with type 2 diabetes. Diabetes 2002, 51, 3582–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesi-ty-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Mathis, D. Immunological Goings-on in Visceral Adipose Tissue. Cell Metab. 2013, 17, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2002, 110, 851–860. [Google Scholar] [CrossRef]
- Nosadini, R.; Avogaro, A.; Trevisan, A.; Valerio, A.; Tessari, P.; Duner, E.; Tiengo, A.; Velussi, M.M.; Prato, S.D.; Kreutzenberg, S.D.; et al. Effect of metformin on insulin-stimulated glucose turnover and insulin binding to receptors in type II diabetes. Diabetes Care 1987, 10, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Hampson, J.; Turner, A.; Stockley, R. Polyclonal free light chains: Promising new biomarkers in inflammatory disease. Curr. Biomark. Find. 2014, 4, 139. [Google Scholar] [CrossRef] [Green Version]
- Dispenzieri, A.; Katzmann, J.A.; Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Colby, C.L.; Clark, R.J.; Mead, G.P.; Kumar, S.; Melton, L.J., 3rd; et al. Use of nonclonal serum immunoglobulin free light chains to predict overall survival in the general population. Mayo Clin. Proc. 2012, 87, 517–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulli, F.; Napodano, C.; Marino, M.; Ciasca, G.; Pocino, K.; Basile, V.; Visentini, M.; Stefanile, A.; Todi, L.; De Spirito, M.; et al. Serum immunoglobulin free light chain levels in systemic autoimmune rheumatic diseases. Clin. Exp. Immunol. 2019, 199, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Terrier, B.; Sène, D.; Saadoun, D.; Ghillani-Dalbin, P.; Thibault, V.; Delluc, A.; Piette, J.-C.; Cacoub, P. Serum-free light chain assessment in hepatitis C virus-related lymphoproliferative disorders. Ann. Rheum. Dis. 2009, 68, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Basile, U.; Gragnani, L.; Piluso, A.; Gulli, F.; Urraro, T.; Dell’Abate, M.T.; Torti, E.; Stasi, C.; Monti, M.; Rapaccini, G.L.; et al. Assessment of free light chains in HCV-positive patients with mixed cryoglobulinaemia vasculitis undergoing rituximab treatment. Liver Int. 2015, 35, 2100–2107. [Google Scholar] [CrossRef]
- Matsumori, A.; Shimada, M.; Jie, X.; Higuchi, H.; Kormelink, T.G.; Redegeld, F.A. Effects of Free Immunoglobulin Light Chains on Viral Myocarditis. Circ. Res. 2010, 106, 1533–1540. [Google Scholar] [CrossRef]
- Matsumori, A. Viral myocarditis from animal models to human diseases. In Advances in Medicine and Biology; Berhardt, L.V., Ed.; Nova Medicine & Health: New York, NY, USA, 2022; pp. 40–74. [Google Scholar]
- Matsumori, A. Novel biomarkers for diagnosis and management of myocarditis and heart Failure: Immunoglobulin free light chains. 21st Century Cardiol. 2022, 2, 114. [Google Scholar]
- Roshdy, A.; Zaher, S.; Fayed, H.; Coghlan, J.G. COVID-19 and the Heart: A systematic review of cardiac autopsies. Front. Cardiovasc. Med. 2020, 7, 626975. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.; Zidar, D.A.; Bristow, M.R.; Cameron, S.J.; Chan, T.; Harding, C.V., III; Kwon, D.H.; Singh, T.; Tilton, J.C.; Tsai, E.J.; et al. COVID-19 and cardiovascular disease. From bench to bedside. Circ. Res. 2021, 128, 1214–1236. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, A.; Mason, J.W. The new FLC biomarker for a novel treatment of myocarditis, COVID-19 disease and other in-flammatory disorders. Intern. Cardiovasc. Forum J. 2022, in press. [Google Scholar]
- Komiyama, M.; Hasegawa, K.; Matsumori, A. Dilated cardiomyopathy risk in patients with coronavirus disease 2019: How to identify and characterise it early? Eur. Cardiol. Rev. 2020, 15, e49. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Matsumori, A.; Abdelrazek, S.; Eltaweel, S.; Salous, A.; Neumann, F.-J.; Antz, M. Myocardial involvement in coro-navirus disease. Herz 2020, 45, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Green, J.B.; Halperin, J.L.; Piccini, J.P. Atrial fibrillation and diabetes mellitus. J. Am. Coll. Cardiol. 2019, 74, 1107–1115. [Google Scholar] [CrossRef]
- Feng, X.; Chen, W.; Ni, X.; Little, P.J.; Xu, S.; Tang, L.; Weng, J. Metformin, macrophage dysfunction and atherosclerosis. Front. Immunol. 2021, 12, 682853. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhu, N.; Sun, R.; Liao, W.; Fan, S.; Shi, F.; Lin, H.; Jiang, S.; Ying, Y. Metformin inhibits chemokine expression through the AMPK/NF-Kappab signaling pathway. J. Interferon Cytokine Res. 2018, 38, 63–369. [Google Scholar] [CrossRef]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.K.; Savinko, T.; Wong, A.K.F.; et al. Anti-Inflammatory effects of metformin irrespective of diabetes status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A. Metformin inhibits the production of reactive oxygen species from NADH: Ubiquinone oxidoreductase to limit induction of interleukin-1β (IL-1β) and boosts interleukin-10 (IL-10) in lipopoly-saccharide (LPS)-activated macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Shang, F.; Hui, L.; Zang, K.; Sun, G. The alleviative effects of metformin for lipopolysaccharide-induced acute lung injury rat model and its underlying mechanism. Saudi Pharm. J. 2017, 25, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.K.; Adya, R.; Shan, X.; Aghilla, M.; Lehnert, H.; Keay, S.D.; Randeva, H.S. The Anti- atherogenic aspect of metformin treatment in insulin resistant women with the polycystic ovary syndrome: Role of the newly established pro-inflammatory adipokine acute-phase serum amyloid A; Evidence of an adipose tissue-monocyte axis. Atherosclerosis 2011, 216, 402–408. [Google Scholar] [CrossRef]
- Dai, Y.; Dai, D.; Wang, X.; Ding, Z.; Mehta, J.L. DPP-4 inhibitors repress NLRP3 inflammasome and interleukin-1 beta via GLP-1 receptor in macrophages through protein kinase C pathway. Cardiovasc. Drugs Ther. 2014, 28, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Ussher, J.R.; Drucker, D.J. Cardiovascular biology of the incretin system. Endocr. Rev. 2012, 33, 187–215. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Ghanim, H.; Vora, M.; Sia, C.L.; Korzeniewski, K.; Dhindsa, S.; Makdissi, A.; Dandona, P. Exenatide exerts a potent antiinflammatory effect. J. Clin. Endocrinol. Metab. 2012, 97, 198–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Zeng, F.; Luo, X.; Lei, Y.; Li, J.; Lu, S.; Huang, X.; Lan, Y.; Liu, R. GLP-1 Receptor: A New Target for Sepsis. Front. Pharmacol. 2021, 12, 706908. [Google Scholar] [CrossRef]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Verma, S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (SGLT2) Inhibitors: A state-of-the-art review. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyani, C.N.; Plastira, I.; Sourij, H.; Hallström, S.; Schmidt, A.; Rainer, P.P.; Bugger, H.; Frank, S.; Malle, E.; von Lewinski, D. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol. Res. 2020, 158, 104870. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleu-kin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. CANTOS Trial Group. Antiinflammatory therapy with canakinumab for athrosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Donath, M.Y.; Pradhan, A.D.; Thuren, T.; Pais, P.; Nicolau, J.C.; Glynn, R.J.; Libby, P.; Ridker, P.M. Anti-in-flammatory therapy with canakinumab for the prevention and management of diabetes. J. Am. Coll. Cardiol. 2018, 71, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Kataria, Y.; Ellervik, C.; Mandrup-Poulsen, T. Treatment of type 2 diabetes by targeting interleukin-1: A meta-analysis of 2921 patients. Semin. Immunopathol. 2019, 41, 413–425. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumori, A. Novel Biomarkers of Inflammation for the Management of Diabetes: Immunoglobulin-Free Light Chains. Biomedicines 2022, 10, 666. https://doi.org/10.3390/biomedicines10030666
Matsumori A. Novel Biomarkers of Inflammation for the Management of Diabetes: Immunoglobulin-Free Light Chains. Biomedicines. 2022; 10(3):666. https://doi.org/10.3390/biomedicines10030666
Chicago/Turabian StyleMatsumori, Akira. 2022. "Novel Biomarkers of Inflammation for the Management of Diabetes: Immunoglobulin-Free Light Chains" Biomedicines 10, no. 3: 666. https://doi.org/10.3390/biomedicines10030666