
Immunotherapy for SMARCB1-Deficient Sarcomas: Current Evidence and Future Developments


Abstract
:1. Introduction
2. SMARCB1
2.1. SMARCB1 Structure and Functions
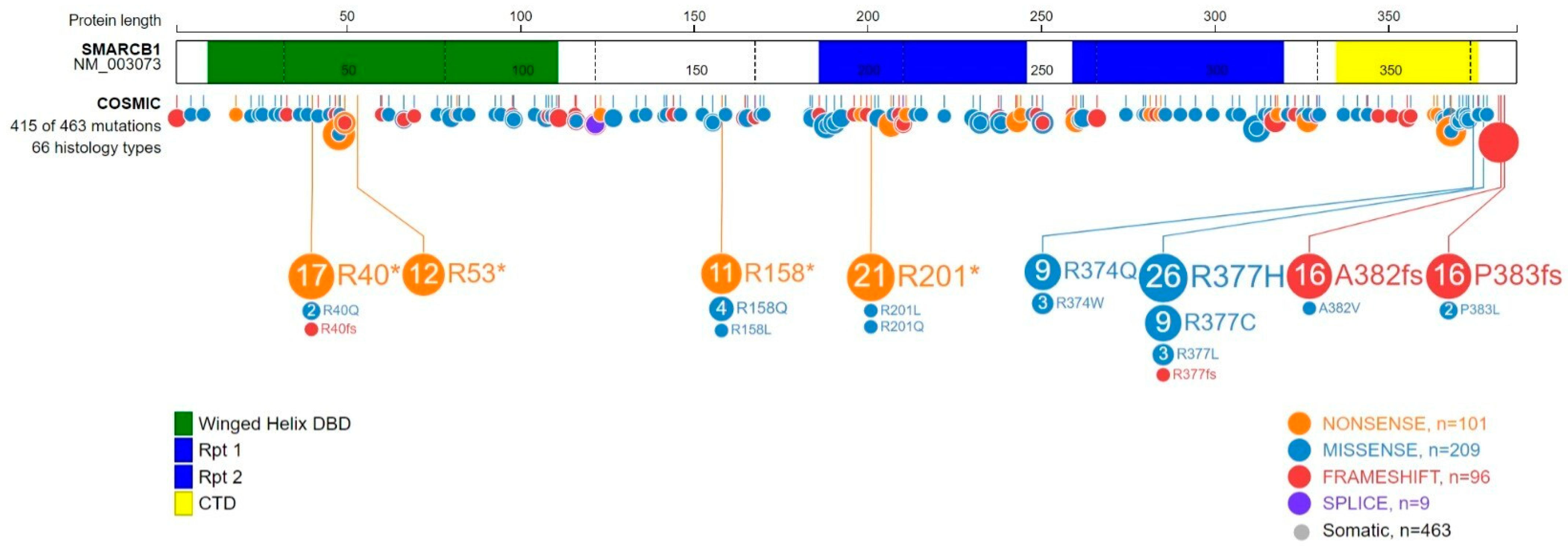
2.2. SMARCB1 Alteration in Cancers
3. SMARCB1 Deficiency and Anti-Tumor Immunity
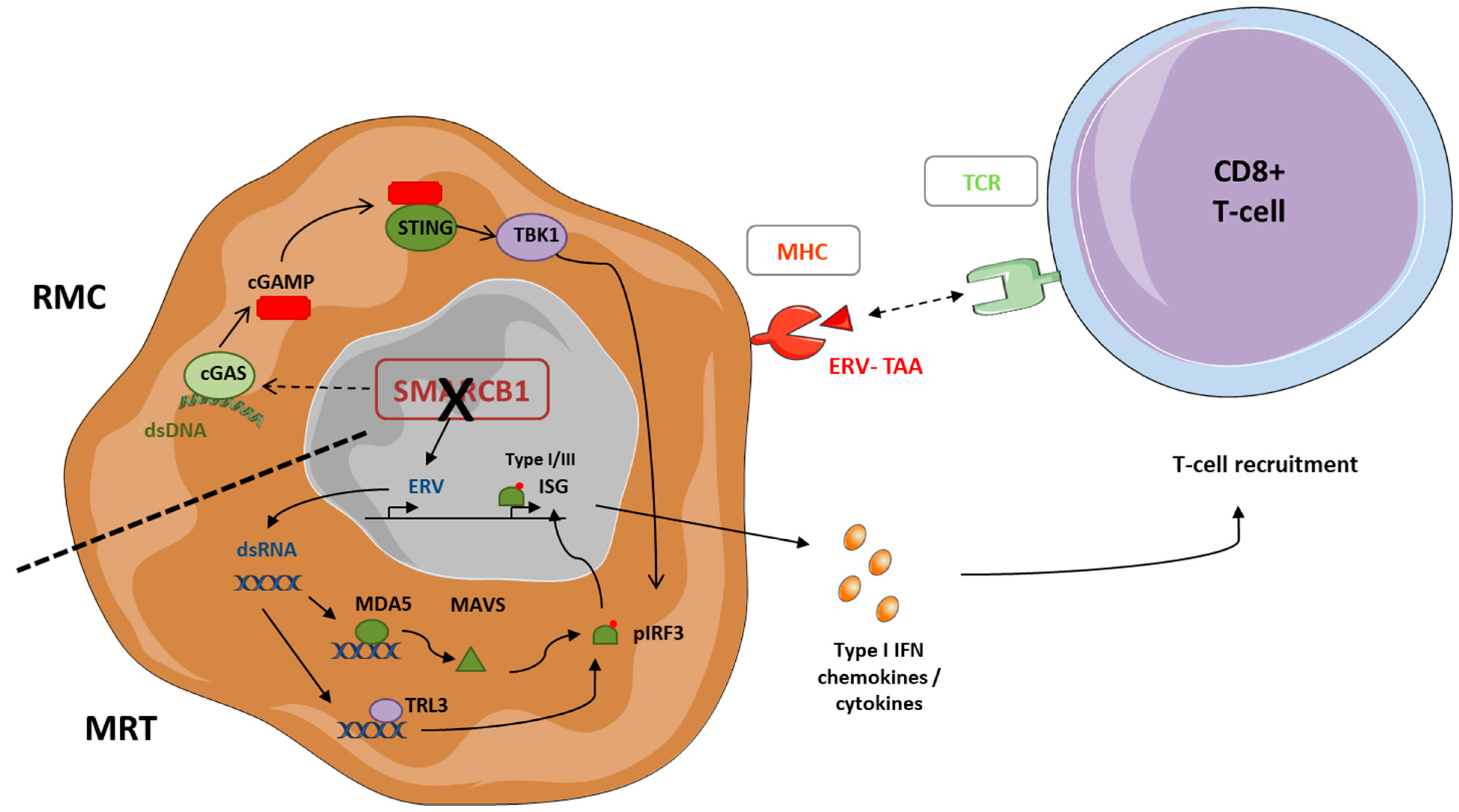
3.1. SMARCB1 Deficiency and Tumor Cell Immunogenicity
3.2. SMARCB1-Deficient Tumors’ Immune Microenvironment
3.2.1. Tumor-Infiltrating Lymphocytes (TILs)
3.2.2. Tertiary Lymphoid Structure (TLS)
3.2.3. Myeloid Populations
3.2.4. Immune Checkpoint Inhibitors
4. Clinical Efficacy of Immune Therapies in SMARCB1-Defective Tumors
4.1. Anti-PD(L)-1 Therapy as a Monotherapy
4.2. Anti-PD-(L)1 Therapy in Combination
4.2.1. Dual ICI Combination
4.2.2. Anti-Angiogenic Agents and ICI
4.2.3. Epigenetic Modulators and ICI Combinations
5. Perspectives and Future Directions
5.1. SMARCB1: A Role Which Is still Unclear in Modulating Tumor Immunogenicity
5.2. Improving Patient Selection for Immune Checkpoint Inhibitors
5.3. Other Immunotherapeutic Approaches
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2020; Volume 3. [Google Scholar]
- Wardelmann, E.; Schildhaus, H.-U.; Merkelbach-Bruse, S.; Hartmann, W.; Reichardt, P.; Hohenberger, P.; Büttner, R. Soft tissue sarcoma: From molecular diagnosis to selection of treatment. Pathological diagnosis of soft tissue sarcoma amid molecular biology and targeted therapies. Ann. Oncol. 2010, 21 (Suppl. 7), vii265–vii269. [Google Scholar] [CrossRef] [PubMed]
- Koelsche, C.; Schrimpf, D.; Stichel, D.; Sill, M.; Sahm, F.; Reuss, D.E.; Blattner, M.; Worst, B.; Heilig, C.E.; Beck, K.; et al. Sarcoma classification by DNA methylation profiling. Nat. Commun. 2021, 12, 498. [Google Scholar] [CrossRef] [PubMed]
- Foersch, S.; Eckstein, M.; Wagner, D.-C.; Gach, F.; Woerl, A.-C.; Geiger, J.; Glasner, C.; Schelbert, S.; Schulz, S.; Porubsky, S.; et al. Deep learning for diagnosis and survival prediction in soft tissue sarcoma. Ann. Oncol. 2021, 32, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, H.C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Morel, D.; Postel-Vinay, S. Exploiting epigenetic vulnerabilities in solid tumors: Novel therapeutic opportunities in the treatment of SWI/SNF-defective cancers. Semin. Cancer Biol. 2020, 61, 180–198. [Google Scholar] [CrossRef]
- Schaefer, I.-M.; Hornick, J.L. SWI/SNF complex-deficient soft tissue neoplasms: An update. Semin. Diagn. Pathol. 2021, 38, 222–231. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF complex in cancer—Biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Leruste, A.; Chauvin, C.; Pouponnot, C.; Bourdeaut, F.; Waterfall, J.J.; Piaggio, E. Immune responses in genomically simple SWI/SNF–deficient cancers. Cancer 2021, 127, 172–180. [Google Scholar] [CrossRef]
- Mashtalir, N.; D’Avino, A.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; Pierre, R.S.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288. [Google Scholar] [CrossRef] [Green Version]
- Mathur, R.; Roberts, C.W. SWI/SNF (BAF) Complexes: Guardians of the Epigenome. Annu. Rev. Cancer Biol. 2018, 2, 413–427. [Google Scholar] [CrossRef]
- Edmonson, M.N.; Patel, A.N.; Hedges, D.J.; Wang, Z.; Rampersaud, E.; Kesserwan, C.A.; Zhou, X.; Liu, Y.; Newman, S.; Rusch, M.C.; et al. Pediatric Cancer Variant Pathogenicity Information Exchange (PeCanPIE): A cloud-based platform for curating and classifying germline variants. Genome Res. 2019, 29, 1555–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalpana, G.V.; Marmon, S.; Wang, W.; Crabtree, G.R.; Goff, S.P. Binding and Stimulation of HIV-1 Integrase by a Human Homolog of Yeast Transcription Factor SNF5. Science 1994, 266, 2002–2006. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.; Yung, E.; Kalpana, G.V. Structure-function analysis of integrase interactor 1/hSNF5L1 reveals differential properties of two repeat motifs present in the highly conserved region. Proc. Natl. Acad. Sci. USA 1998, 95, 1120–1125. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Xie, S.; Du, Y.; Qian, C. Structural Insights into BAF47 and BAF155 Complex Formation. J. Mol. Biol. 2017, 429, 1650–1660. [Google Scholar] [CrossRef]
- Han, J.; Kim, I.; Park, J.-H.; Yun, J.-H.; Joo, K.; Kim, T.; Park, G.-Y.; Ryu, K.-S.; Ko, Y.-J.; Mizutani, K.; et al. A Coil-to-Helix Transition Serves as a Binding Motif for hSNF5 and BAF155 Interaction. Int. J. Mol. Sci. 2020, 21, 2452. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.D.; Freund, S.M.; Zinzalla, G.; Bycroft, M. The SWI/SNF Subunit INI1 Contains an N-Terminal Winged Helix DNA Binding Domain that Is a Target for Mutations in Schwannomatosis. Structure 2015, 23, 1344–1349. [Google Scholar] [CrossRef] [Green Version]
- Valencia, A.; Collings, C.K.; Dao, H.T.; Pierre, R.S.; Cheng, Y.-C.; Huang, J.; Sun, Z.-Y.; Seo, H.-S.; Mashtalir, N.; Comstock, D.; et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell 2019, 179, 1342–1356.e23. [Google Scholar] [CrossRef]
- Wang, X.; Lee, R.S.; Alver, B.H.; Haswell, J.R.; Wang, S.; Mieczkowski, J.; Drier, Y.; Gillespie, S.M.; Archer, T.C.; Wu, J.N.; et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 2017, 49, 289–295. [Google Scholar] [CrossRef]
- Nakayama, R.T.; Pulice, J.L.; Valencia, A.; McBride, M.; McKenzie, Z.M.; Gillespie, M.A.; Ku, W.L.; Teng, M.; Cui, K.; Williams, R.; et al. SMARCB1 is required for widespread BAF complex–mediated activation of enhancers and bivalent promoters. Nat. Genet. 2017, 49, 1613–1623. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet. 2014, 207, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versteege, I.; Sévenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Agaimy, A. The Expanding Family of SMARCB1(INI1)-deficient Neoplasia: Implications of Phenotypic, Biological, and Molecular Heterogeneity. Adv. Anat. Pathol. 2014, 21, 394–410. [Google Scholar] [CrossRef] [PubMed]
- Biegel, J.A.; Kalpana, G.; Knudsen, E.S.; Packer, R.J.; Roberts, C.W.M.; Thiele, C.J.; Weissman, B.; Smith, M. The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: Meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res. 2002, 62, 323–328. [Google Scholar]
- Biegel, J.A.; Tan, L.; Zhang, F.; Wainwright, L.; Russo, P.; Rorke, L.B. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin. Cancer Res. 2002, 8, 3461–3467. [Google Scholar]
- Lee, R.S.; Stewart, C.; Carter, S.L.; Ambrogio, L.; Cibulskis, K.; Sougnez, C.; Lawrence, M.S.; Auclair, D.; Mora, J.; Golub, T.R.; et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J. Clin. Investig. 2012, 122, 2983–2988. [Google Scholar] [CrossRef]
- Guidi, C.J.; Sands, A.T.; Zambrowicz, B.P.; Turner, T.K.; Demers, D.A.; Webster, W.; Smith, T.W.; Imbalzano, A.N.; Jones, S.N. Disruption of Ini1 Leads to Peri-Implantation Lethality and Tumorigenesis in Mice. Mol. Cell. Biol. 2001, 21, 3598–3603. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.; Galusha, S.A.; McMenamin, M.E.; Fletcher, C.D.M.; Orkin, S.H. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 13796–13800. [Google Scholar] [CrossRef] [Green Version]
- Klochendler-Yeivin, A.; Fiette, L.; Barra, J.; Muchardt, C.; Babinet, C.; Yaniv, M. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 2000, 1, 500–506. [Google Scholar] [CrossRef] [Green Version]
- Noujaim, J.; Thway, K.; Bajwa, Z.; Bajwa, A.; Maki, R.G.; Jones, R.L.; Keller, C. Epithelioid Sarcoma: Opportunities for Biology-Driven Targeted Therapy. Front. Oncol. 2015, 5, 186. [Google Scholar] [CrossRef] [Green Version]
- Hornick, J.L.; Cin, P.D.; Fletcher, C.D. Loss of INI1 Expression is Characteristic of Both Conventional and Proximal-type Epithelioid Sarcoma. Am. J. Surg. Pathol. 2009, 33, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.M.; Folpe, A.L.; Pawel, B.R.; Judkins, A.R.; Biegel, J.A. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod. Pathol. 2013, 26, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Kohashi, K.; Izumi, T.; Oda, Y.; Yamamoto, H.; Tamiya, S.; Taguchi, T.; Iwamoto, Y.; Hasegawa, T.; Tsuneyoshi, M. Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: A useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum. Pathol. 2009, 40, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Papp, G.; Changchien, Y.C.; Péterfia, B.; Pecsenka, L.; Krausz, T.; Stricker, T.P.; Khoor, A.; Donner, L.; Sápi, Z. SMARCB1 protein and mRNA loss is not caused by promoter and histone hypermethylation in epithelioid sarcoma. Mod. Pathol. 2013, 26, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Jamshidi, F.; Bashashati, A.; Shumansky, K.; Dickson, B.; Gokgoz, N.; Wunder, J.S.; Andrulis, I.L.; Lazar, A.J.; Shah, S.P.; Huntsman, D.G.; et al. The genomic landscape of epithelioid sarcoma cell lines and tumours. J. Pathol. 2016, 238, 63–73. [Google Scholar] [CrossRef]
- Folpe, A.L.; Schoolmeester, J.K.; McCluggage, W.G.; Sullivan, L.M.; Castagna, K.; Ahrens, W.A.; Oliva, E.; Biegel, J.A.; Nielsen, G.P. SMARCB1-deficient Vulvar Neoplasms: A Clinicopathologic, Immunohistochemical, and Molecular Genetic Study of 14 Cases. Am. J. Surg. Pathol. 2015, 39, 836–849. [Google Scholar] [CrossRef]
- Kohashi, K.; Yamamoto, H.; Kumagai, R.; Yamada, Y.; Hotokebuchi, Y.; Taguchi, T.; Iwamoto, Y.; Oda, Y. Differential microRNA expression profiles between malignant rhabdoid tumor and epithelioid sarcoma: miR193a-5p is suggested to downregulate SMARCB1 mRNA expression. Mod. Pathol. 2013, 27, 832–839. [Google Scholar] [CrossRef]
- Sápi, Z.; Papp, G.; Szendrői, M.; Pápai, Z.; Plótár, V.; Krausz, T.; Fletcher, C.D. Epigenetic regulation ofSMARCB1By miR-206, -381 and -671-5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer 2016, 55, 786–802. [Google Scholar] [CrossRef]
- Papp, G.; Krausz, T.; Stricker, T.P.; Szendrői, M.; Sápi, Z. SMARCB1expression in epithelioid sarcoma is regulated by miR-206, miR-381, and miR-671-5p on Both mRNA and protein levels. Genes Chromosomes Cancer 2014, 53, 168–176. [Google Scholar] [CrossRef]
- Brenca, M.; Rossi, S.; Lorenzetto, E.; Piccinin, E.; Piccinin, S.; Rossi, F.M.; Giuliano, A.; Dei Tos, A.P.; Maestro, R.; Modena, P. SMARCB1/INI1 genetic inactivation is responsible for tumorigenic properties of epithelioid sarcoma cell line VAESBJ. Mol. Cancer Ther. 2013, 12, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka, A.M.; Sobczuk, P.; Kostrzanowski, M.; Spalek, M.; Chojnacka, M.; Szumera-Ciećkiewicz, A.; Rutkowski, P. Epithelioid Sarcoma—From Genetics to Clinical Practice. Cancers 2020, 12, 2112. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.; Schöffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.W.; Jahan, T.; et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: An international, open-label, phase 2 basket study. Lancet Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef]
- Hasselblatt, M.; Thomas, C.; Hovestadt, V.; Schrimpf, D.; Johann, P.; Bens, S.; Oyen, F.; Peetz-Dienhart, S.; Crede, Y.; Wefers, A.; et al. Poorly differentiated chordoma with SMARCB1/INI1 loss: A distinct molecular entity with dismal prognosis. Acta Neuropathol. 2016, 132, 149–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owosho, A.A.; Zhang, L.; Rosenblum, M.K.; Antonescu, C.R. High sensitivity of FISH analysis in detecting homozygous SMARCB1 deletions in poorly differentiated chordoma: A clinicopathologic and molecular study of nine cases. Genes Chromosomes Cancer 2018, 57, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.R.; Chebib, I.; Deshpande, V.; Dickson, B.C.; Iafrate, A.J.; Nielsen, G.P. Molecular characteristics of poorly differentiated chordoma. Genes Chromosomes Cancer 2019, 58, 804–808. [Google Scholar] [CrossRef]
- Renard, C.; Pissaloux, D.; Decouvelaere, A.V.; Bourdeaut, F.; Ranchère, D. Non-rhabdoid pediatric SMARCB1-deficient tumors: Overlap between chordomas and malignant rhabdoid tumors? Cancer Genet. 2014, 207, 384–389. [Google Scholar] [CrossRef]
- Mobley, B.C.; McKenney, J.K.; Bangs, C.D.; Callahan, K.; Yeom, K.W.; Schneppenheim, R.; Hayden, M.G.; Cherry, A.M.; Gokden, M.; Edwards, M.S.; et al. Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol. 2010, 120, 745–753. [Google Scholar] [CrossRef]
- Shih, A.; Cote, G.M.; Chebib, I.; Choy, E.; Delaney, T.; Deshpande, V.; Hornicek, F.J.; Miao, R.; Schwab, J.H.; Nielsen, G.P.; et al. Clinicopathologic characteristics of poorly differentiated chordoma. Mod. Pathol. 2018, 31, 1237–1245. [Google Scholar] [CrossRef]
- Jo, V.Y.; Fletcher, C.D.M. Epithelioid Malignant Peripheral Nerve Sheath Tumor: Clinicopathologic Analysis of 63 Cases. Am. J. Surg. Pathol. 2015, 39, 673–682. [Google Scholar] [CrossRef]
- Schaefer, I.-M.; Dong, F.; Garcia, E.P.; Fletcher, C.D.M.; Jo, V.Y. Recurrent SMARCB1 Inactivation in Epithelioid Malignant Peripheral Nerve Sheath Tumors. Am. J. Surg. Pathol. 2019, 43, 835–843. [Google Scholar] [CrossRef]
- Gleason, B.C.; Fletcher, C.D.M. Myoepithelial Carcinoma of Soft Tissue in Children: An Aggressive Neoplasm Analyzed in a Series of 29 Cases. Am. J. Surg. Pathol. 2007, 31, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Le Loarer, F.; Zhang, L.; Fletcher, C.D.; Ribeiro, A.; Singer, S.; Italiano, A.; Neuville, A.; Houlier, A.; Chibon, F.; Coindre, J.M.; et al. Consistent SMARCB1 homozygous deletions in epithelioid sarcoma and in a subset of myoepithelial carcinomas can be reliably detected by FISH in archival material. Genes Chromosomes Cancer 2014, 53, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohashi, K.; Oda, Y.; Yamamoto, H.; Tamiya, S.; Oshiro, Y.; Izumi, T.; Taguchi, T.; Tsuneyoshi, M. SMARCB1/INI1 protein expression in round cell soft tissue sarcomas associated with chromosomal translocations involving EWS: A special reference to SMARCB1/INI1 negative variant extraskeletal myxoid chondrosarcoma. Am. J. Surg. Pathol. 2008, 32, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- McBride, M.J.; Pulice, J.L.; Beird, H.C.; Ingram, D.R.; D’Avino, A.R.; Shern, J.F.; Charville, G.W.; Hornick, J.L.; Nakayama, R.T.; Garcia-Rivera, E.M.; et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 2018, 33, 1128–1141.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.; Harview, C.; Yearly, J.; Shintaku, I.; Taylor, E.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Casey, S.C.; Baylot, V.; Felsher, D.W. The MYC oncogene is a global regulator of the immune response. Blood 2018, 131, 2007–2015. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabanon, R.M.; Morel, D.; Eychenne, T.; Colmet-Daage, L.; Bajrami, I.; Dorvault, N.; Garrido, M.; Meisenberg, C.; Lamb, A.; Ngo, C.; et al. PBRM1 Deficiency Confers Synthetic Lethality to DNA Repair Inhibitors in Cancer. Cancer Res. 2021, 81, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Botta, G.P.; Kato, S.; Patel, H.; Fanta, P.; Lee, S.; Okamura, R.; Kurzrock, R. SWI/SNF complex alterations as a biomarker of immunotherapy efficacy in pancreatic cancer. JCI Insight 2021, 6, e150453. [Google Scholar] [CrossRef]
- Leruste, A.; Tosello, J.; Ramos, R.N.; Tauziède-Espariat, A.; Brohard, S.; Han, Z.-Y.; Beccaria, K.; Andrianteranagna, M.; Caudana, P.; Nikolic, J.; et al. Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell 2019, 36, 597–612.e8. [Google Scholar] [CrossRef]
- Larouche, J.-D.; Trofimov, A.; Hesnard, L.; Ehx, G.; Zhao, Q.; Vincent, K.; Durette, C.; Gendron, P.; Laverdure, J.-P.; Bonneil, É.; et al. Widespread and tissue-specific expression of endogenous retroelements in human somatic tissues. Genome Med. 2020, 12, 40. [Google Scholar] [CrossRef]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.-J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Solovyov, A.; Vabret, N.; Arora, K.S.; Snyder, A.; Funt, S.A.; Bajorin, D.F.; Rosenberg, J.E.; Bhardwaj, N.; Ting, D.T.; Greenbaum, B.D. Global Cancer Transcriptome Quantifies Repeat Element Polarization between Immunotherapy Responsive and T Cell Suppressive Classes. Cell Rep. 2018, 23, 512–521. [Google Scholar] [CrossRef]
- Chun, H.-J.E.; Johann, P.D.; Milne, K.; Zapatka, M.; Buellesbach, A.; Ishaque, N.; Iskar, M.; Erkek, S.; Wei, L.; Tessier-Cloutier, B.; et al. Identification and Analyses of Extra-Cranial and Cranial Rhabdoid Tumor Molecular Subgroups Reveal Tumors with Cytotoxic T Cell Infiltration. Cell Rep. 2019, 29, 2338–2354.e7. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Malouf, G.G.; Su, X.; Yao, H.; Tripathi, D.; Soeung, M.; Gao, J.; Rao, P.; Coarfa, C.; Creighton, C.J.; et al. Comprehensive Molecular Characterization Identifies Distinct Genomic and Immune Hallmarks of Renal Medullary Carcinoma. Cancer Cell 2020, 37, 720–734.e13. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Finetti, M.A.; Grabovska, Y.; Bailey, S.; Williamson, D. Translational genomics of malignant rhabdoid tumours: Current impact and future possibilities. Semin. Cancer Biol. 2020, 61, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Sautès-Fridman, C.; Verneau, J.; Sun, C.-M.; Moreira, M.; Chen, T.W.-W.; Meylan, M.; Petitprez, F.; Fridman, W.H. Tertiary Lymphoid Structures and B cells: Clinical impact and therapeutic modulation in cancer. Semin. Immunol. 2020, 48, 101406. [Google Scholar] [CrossRef]
- Liu, X.; Tsang, J.Y.; Hlaing, T.; Hu, J.; Ni, Y.-B.; Chan, S.K.; Cheung, S.Y.; Tse, G.M. Distinct Tertiary Lymphoid Structure Associations and Their Prognostic Relevance in HER2 Positive and Negative Breast Cancers. Oncologist 2017, 22, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Posch, F.; Silina, K.; Leibl, S.; Mündlein, A.; Moch, H.; Siebenhüner, A.; Samaras, P.; Riedl, J.; Stotz, M.; Szkandera, J.; et al. Maturation of tertiary lymphoid structures and recurrence of stage II and III colorectal cancer. Oncoimmunology 2018, 7, e1378844. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Petitprez, F.; Becht, E.; Laurent, A.; Hirsch, T.Z.; Rousseau, B.; Luciani, A.; Amaddeo, G.; Derman, J.; Charpy, C.; et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J. Hepatol. 2019, 70, 58–65. [Google Scholar] [CrossRef]
- Germain, C.; Gnjatic, S.; Tamzalit, F.; Knockaert, S.; Remark, R.; Goc, J.; Lepelley, A.; Becht, E.; Katsahian, S.; Bizouard, G.; et al. Presence of B Cells in Tertiary Lymphoid Structures Is Associated with a Protective Immunity in Patients with Lung Cancer. Am. J. Respir. Crit. Care Med. 2014, 189, 832–844. [Google Scholar] [CrossRef]
- Lin, Q.; Tao, P.; Wang, J.; Ma, L.; Jiang, Q.; Li, J.; Zhang, G.; Liu, J.; Zhang, Y.; Hou, Y.; et al. Tumor-associated tertiary lymphoid structure predicts postoperative outcomes in patients with primary gastrointestinal stromal tumors. OncoImmunology 2020, 9, 1747339. [Google Scholar] [CrossRef] [Green Version]
- Goc, J.; Germain, C.; Vo-Bourgais, T.K.D.; Lupo, A.; Klein, C.; Knockaert, S.; De Chaisemartin, L.; Ouakrim, H.; Becht, E.; Alifano, M.; et al. Dendritic Cells in Tumor-Associated Tertiary Lymphoid Structures Signal a Th1 Cytotoxic Immune Contexture and License the Positive Prognostic Value of Infiltrating CD8+ T Cells. Cancer Res. 2014, 74, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.-W.; Sun, C.-M.; Calderaro, J.; Jeng, Y.-M.; Hsiao, L.-P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell–induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.-Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. OncoImmunology 2020, 9, 1747340. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Abro, B.; Kaushal, M.; Chen, L.; Wu, R.; Dehner, L.P.; Pfeifer, J.D.; He, M. Tumor mutation burden, DNA mismatch repair status and checkpoint immunotherapy markers in primary and relapsed malignant rhabdoid tumors. Pathol.-Res. Pract. 2019, 215, 152395. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.J.; Al-Ibraheemi, A.; Doan, D.; Ward, A.; Clinton, C.M.; Putra, J.; Pinches, R.S.; Kadoch, C.; Chi, S.N.; Dubois, S.G.; et al. Genomic and Immunologic Characterization of INI1-Deficient Pediatric Cancers. Clin. Cancer Res. 2020, 26, 2882–2890. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Kim, E.K.; Jung, H.; Chon, H.J.; Han, J.W.; Shin, K.-H.; Hu, H.; Kim, K.S.; Choi, Y.D.; Kim, S.; et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer 2016, 16, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boxberg, M.; Steiger, K.; Lenze, U.; Rechl, H.; von Eisenhart-Rothe, R.; Wörtler, K.; Weichert, W.; Langer, R.; Specht, K. PD-L1 and PD-1 and characterization of tumor-infiltrating lymphocytes in high grade sarcomas of soft tissue—Prognostic implications and rationale for immunotherapy. OncoImmunology 2018, 7, e1389366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movva, S.; Wen, W.; Chen, W.; Millis, S.Z.; Gatalica, Z.; Reddy, S.; Von Mehren, M.; Van Tine, B.A. Multi-platform profiling of over 2000 sarcomas: Identification of biomarkers and novel therapeutic targets. Oncotarget 2015, 6, 12234–12247. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Shen, J.; Gao, Y.; Liao, Y.; Côté, G.; Choy, E.; Chebib, I.; Mankin, H.; Hornicek, F.; Duan, Z. Expression of programmed cell death ligand 1 (PD-L1) and prevalence of tumor-infiltrating lymphocytes (TILs) in chordoma. Oncotarget 2015, 6, 11139–11149. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.-X.; Peng, A.-B.; Lv, G.-H.; Wang, X.-B.; Li, J.; She, X.-L.; Jiang, Y. Expression of programmed death-1 ligand (PD-L1) in tumor-infiltrating lymphocytes is associated with favorable spinal chordoma prognosis. Am. J. Transl. Res. 2016, 8, 3274–3287. [Google Scholar]
- Geoerger, B.; Kang, H.J.; Yalon-Oren, M.; Marshall, L.V.; Vezina, C.; Pappo, A.; Laetsch, T.W.; Petrilli, A.S.; Ebinger, M.; Toporski, J.; et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): Interim analysis of an open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 121–133. [Google Scholar] [CrossRef]
- Geoerger, B.; Zwaan, C.M.; Marshall, L.V.; Michon, J.; Bourdeaut, F.; Casanova, M.; Corradini, N.; Rossato, G.; Farid-Kapadia, M.; Shemesh, C.S.; et al. Atezolizumab for children and young adults with previously treated solid tumours, non-Hodgkin lymphoma, and Hodgkin lymphoma (iMATRIX): A multicentre phase 1–2 study. Lancet Oncol. 2020, 21, 134–144. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.; Rosen, G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blay, J.-Y.; Penel, N.; Ray-Coquard, I.; Schott, R.; Saada-Bouzid, E.; Bertucci, F.; Chevreau, C.; Bompas, E.; Coquan, E.; Cousin, S.; et al. High clinical benefit rates of pembrolizumab in very rare sarcoma histotypes: First results of the AcSé pembrolizumab study. Ann. Oncol. 2019, 30, v517. [Google Scholar] [CrossRef]
- Kubli, S.P.; Berger, T.; Araujo, D.V.; Siu, L.L.; Mak, T.W. Beyond immune checkpoint blockade: Emerging immunological strategies. Nat. Rev. Drug Discov. 2021, 20, 899–919. Available online: http://www.nature.com/articles/s41573-021-00155-y (accessed on 19 April 2021). [CrossRef] [PubMed]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Pecora, A.; Halpern, S.; Weber, M.; Paleoudis, E.G.; Panush, D.; Patterson, F.; Toretsky, J. Rapid and Complete Response to Combination Anti-CTLA-4 and Anti-PD-1 Checkpoint Inhibitor Therapy in a Patient with Stage IV Refractory End-stage Epithelioid Sarcoma: A Case Report. J. Immunother. 2020, 43, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; Gabrilovich, D.I.; Sempowski, G.D.; Kisseleva, E.; Parman, K.S.; Nadaf, S.; Carbone, D.P. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003, 101, 4878–4886. [Google Scholar] [CrossRef] [PubMed]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Wieder, E.D.; Kolonias, D.; Rosenberg, A.E.; Kerr, D.A.; et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Hindi, N.; Grignani, G.; Martinez-Trufero, J.; Redondo, A.; Valverde, C.; Stacchiotti, S.; Lopez-Pousa, A.; D’Ambrosio, L.; Gutierrez, A.; et al. Nivolumab and sunitinib combination in advanced soft tissue sarcomas: A multicenter, single-arm, phase Ib/II trial. J. Immunother. Cancer 2020, 8, e001561. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Shoushtari, A.N.; Keohan, M.L.; Dickson, M.A.; Gounder, M.M.; Chi, P.; Loo, J.K.; Gaffney, L.; Schneider, L.; Patel, Z.; et al. Combined KIT and CTLA-4 Blockade in Patients with Refractory GIST and Other Advanced Sarcomas: A Phase Ib Study of Dasatinib plus Ipilimumab. Clin. Cancer Res. 2017, 23, 2972–2980. [Google Scholar] [CrossRef] [Green Version]
- Aspeslagh, S.; Morel, D.; Soria, J.-C.; Postel-Vinay, S. Epigenetic modifiers as new immunomodulatory therapies in solid tumours. Ann. Oncol. 2018, 29, 812–824. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alimova, I.; Birks, D.K.; Harris, P.S.; Knipstein, J.; Venkataraman, S.; Marquez, V.E.; Foreman, N.; Vibhakar, R. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro-Oncol. 2012, 15, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Scott, M.P.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothbart, S.B.; Baylin, S.B. Epigenetic Therapy for Epithelioid Sarcoma. Cell 2020, 181, 211. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.N.; Bourdeaut, F.; Laetsch, T.W.; Fouladi, M.; Macy, M.E.; Makin, G.W.; Shukla, N.N.; Wetmore, C.; Margol, A.S.; Casanova, M.; et al. Phase I study of tazemetostat, an enhancer of zeste homolog-2 inhibitor, in pediatric pts with relapsed/refractory integrase interactor 1-negative tumors. J. Clin. Oncol. 2020, 38, 10525. [Google Scholar] [CrossRef]
- Qiu, J.; Sharma, S.; Rollins, R.A.; Paul, T.A. The complex role of EZH2 in the tumor microenvironment: Opportunities and challenges for immunotherapy combinations. Future. Med. Chem. 2020, 12, 1415–1430. [Google Scholar] [CrossRef]
- Ennishi, D.; Takata, K.; Béguelin, W.; Duns, G.; Mottok, A.; Farinha, P.; Bashashati, A.; Saberi, S.; Boyle, M.; Meissner, B.; et al. Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov. 2019, 9, 546–563. [Google Scholar] [CrossRef] [Green Version]
- Zingg, D.; Arenas-Ramirez, N.; Sahin, D.; Rosalia, R.A.; Antunes, A.T.; Haeusel, J.; Sommer, L.; Boyman, O. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017, 20, 854–867. [Google Scholar] [CrossRef] [Green Version]
- Cañadas, I.; Thummalapalli, R.; Kim, J.W.; Kitajima, S.; Jenkins, R.W.; Christensen, C.L.; Campisi, M.; Kuang, Y.; Zhang, Y.; Gjini, E.; et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat. Med. 2018, 24, 1143–1150. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Goswami, S.; Apostolou, I.; Zhang, J.; Skepner, J.; Anandhan, S.; Zhang, X.; Xiong, L.; Trojer, P.; Aparicio, A.; Subudhi, S.K.; et al. Modulation of EZH2 expression in T cells improves efficacy of anti–CTLA-4 therapy. J. Clin. Investig. 2018, 128, 3813–3818. [Google Scholar] [CrossRef]
- Zhou, L.; Mudianto, T.; Ma, X.; Riley, R.; Uppaluri, R. Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti–PD-1 Resistance in Head and Neck Cancer. Clin. Cancer Res. 2020, 26, 290–300. [Google Scholar] [CrossRef] [Green Version]
- Morel, K.L.; Sheahan, A.V.; Burkhart, D.L.; Baca, S.C.; Boufaied, N.; Liu, Y.; Qiu, X.; Cañadas, I.; Roehle, K.; Heckler, M.; et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat. Cancer 2021, 2, 444–456. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Ott, P.A.; Bang, Y.-J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.-C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell–Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated with Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020, 26, 712–719. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| SMARCB1-Deficient Mesenchymal Malignant Tumors | SMARCB1-Deficient Non-Mesenchymal Malignant Tumors |
|---|---|
| Extrarenal malignant rhabdoid tumor | Atypical teratoid rhabdoid tumor |
| Epithelioid sarcoma | Cribriform neuroepithelial tumor |
| Poorly differentiated chordoma | Renal medullary carcinoma |
| Epithelioid MPNST 1 | SMARCB1-deficient sinonasal carcinoma |
| Myoepithelial carcinoma | SMARCB1-deficient carcinoma of the GI tract |
| Myxoid extraskeletal chondrosarcoma |
| Reference | NCT Identifier/ Trial Name | Study Design | Study Description | Number of Patients | Specific Histotype | Best Response | Duration of Best Response |
|---|---|---|---|---|---|---|---|
| Paoluzzi, 2016 | Retrospective series | Nivolumab in relapsed metastatic/unresectable sarcomas | 2 | ES | 1 PR | 3.8 mth | |
| 1 PD | |||||||
| Blay, 2019 | NCT03012620/ AcSé | Phase II | Pembrolizumab for patients with selected rare cancer types | 1 | MRT | PR | NA |
| Georger, 2020 | NCT02541604/ iMATRIX | Phase I/II | Atezolizumab in children and young adults with refractory or relapsed solid tumors, with known or expected PD-L1 expression | 3 | MRT | PR | NA |
| Georger, 2020 | NCT02332668 | Phase I/II | Pembrolizumab in pediatric patients with PD-L1-positive, advanced, relapsed, or refractory solid tumor | 2 | MRT | 1 PR | 17.8 mth |
| 1 PD | |||||||
| 1 | ES | PR | 11.8 mth | ||||
| Forrest, 2020 | Case report | Pembrolizumab | 1 | ES | SD | 12 mth | |
| Nivolumab Pembrolizumab | 1 | PDC | PR | 9 mth | |||
| 1 | MRT | SD | 15 wk |
| Reference | NCT Identifier/Trial Name | Study Design | Study Description | Number of Patients | Specific Histotype | Best Response | Duration of Best Response |
|---|---|---|---|---|---|---|---|
| D’Angelo, 2018 | NCT02500797/ Alliance A091401 | Phase II | Nivolumab with or without ipilimumab treatment for metastatic sarcoma | 1 | ES | 0 | |
| Wilky, 2019 | NCT02636725 | Phase II | Axitinib + pembrolizumab in advanced sarcoma | 1 | ES | PR | 24 wk |
| Martin-Broto, 2020 | NCT03277924 | Phase Ib/II | Nivolumab and sunitinib combination in advanced soft tissue sarcomas | 7 | ES | SD | 17 mth |
| D’Angelo, 2017 | NCT01643278 | Phase Ib | Combined KIT and CTLA-4 Blockade in patients with Refractory GIST and other advanced Sarcomas: Ipilimumab + dasatinib | 1 | ES | SD | 16 wk |
| Pecora, 2020 | Case report | Nivolumab + ipilumab | 1 | ES | Complete response | NA |
| NCT Identifier/ Study Name | Drugs | Clinical Trial Phase | Population | Estimated Enrollment | Primary Completion Date | Location |
|---|---|---|---|---|---|---|
| NCT04741438/ RAR-Immune | Nivolumab + Ipilimumab | III | Metastatic or unresectable advanced sarcoma of rare subtype including ES and chordoma | 96 | February 2025 | Centre Léon Bérard, Lyon, France |
| NCT04416568 | Nivolumab + Ipilimumab | II | Relapsed or refractory INI1-negative cancers in children and young adults from 6 months to 30 years | 45 | October 2023 | Dana-Farber Cancer Institute, United States, Massachusetts |
| NCT04705818/ CAIRE | Durvalumab + Tazemetostat | II | Distinct cohorts of solid tumors including soft-tissue sarcoma and metastatic solid tumor with positive interferon gamma signature and/or presence of TLS | 173 | October 2022 | Multiple French Cancer Institute |
| NCT04095208/ CONGRATS | Nivolumab + Relatlimab | II | Advanced non-resectable/metastatic soft tissue sarcoma with high-level of tertiary lymphoid structures | 67 | March 2022 | Multiple French Cancer Institute |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngo, C.; Postel-Vinay, S. Immunotherapy for SMARCB1-Deficient Sarcomas: Current Evidence and Future Developments. Biomedicines 2022, 10, 650. https://doi.org/10.3390/biomedicines10030650
Ngo C, Postel-Vinay S. Immunotherapy for SMARCB1-Deficient Sarcomas: Current Evidence and Future Developments. Biomedicines. 2022; 10(3):650. https://doi.org/10.3390/biomedicines10030650
Chicago/Turabian StyleNgo, Carine, and Sophie Postel-Vinay. 2022. "Immunotherapy for SMARCB1-Deficient Sarcomas: Current Evidence and Future Developments" Biomedicines 10, no. 3: 650. https://doi.org/10.3390/biomedicines10030650