
Reactive Oxygen Species Induced Pathways in Heart Failure Pathogenesis and Potential Therapeutic Strategies

 ,
,
Abstract
:1. Introduction
2. ROS Sources, Importance and Danger in Human Cells
3. ROS in the Pathogenesis of CHF Development
3.1. Enzymes Involved in ROS Production
3.2. Mitochondria in ROS Production and Enzyme-Antioxidants
4. Differences in ROS-Induced Pathways between HFrEF and HFpEF
4.1. ROS in HFrEF
4.2. ROS in HFpEF
5. ROS-Induced Pathways as a Treatment Target in HFrEF and HFpEF
5.1. AMPK Activators
5.2. Renin-Angiotensin System Inhibitors
5.3. Inhibitors of ROS-Producing Enzymes
5.4. Antioxidants
5.5. MAO Inhibitors
5.6. Mitochondrial Function Improvement
5.7. Chemicals Increasing cGMP-PKG Signaling
6. Conclusions
7. Perspectives
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbrevations
| ALDH2 | aldehyde dehydrogenase |
| AMP | adenosine monophosphate |
| AMPK | adenosine monophosphate activated protein kinase |
| ANGII | angiotensin II |
| AO | antioxidant |
| AOx | vitamin C, vitamin E, α-lipoic acid (antioxidants) |
| AT1R | angiotensin II receptor |
| ATP | adenosine triphosphate |
| BH4 | dihydrobiopterin-4 |
| BNIP3 | mitochondrial mitophagy marker |
| BNP | brain natriuretic peptide |
| Bax | bcl-2-like protein 4 |
| CAT | chloramphenicol acetyltransferase |
| CHF– | chronic heart failure |
| CI | ETC complex I |
| CIII | ETC complex III |
| CK | creatine kinase |
| CRP– | C-reactive protein |
| Cluh | clustered mitochondria protein homolog |
| CoenzymeQ-10– | ubiquinone |
| DM– | diabetes mellitus |
| DROM | reactive oxidative metabolites |
| DRP1 | dynamin related protein1 |
| ERK | extracellular signal-regulated kinase |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| ETC | electron transport chain |
| GTP | guanosine triphosphate |
| GMP | guanosine monophosphate |
| GPX | glutathione peroxidase |
| GSH | glutathione |
| H2O2 | hydrogen peroxide |
| HF | heart failure |
| HFpEF | heart failure with preserved ejection fraction |
| HFrEF | heart failure with reduced ejection fraction |
| HNO | nytroxyl |
| HSC | hematopoietic stem cells |
| IDH | isocitrate dehydrogenase |
| IDH2 | isocitrate dehydrogenase |
| IL-10 | interleukin-10 |
| IL-6 | interleukin-6 |
| IMM | inner mitochondrial membrane |
| IRE1α | inositol-requiring protein 1α |
| ISDN | isosorbide dinitrate |
| JNK-c | Jun N-terminal kinase |
| KFP | Kaempherol |
| LA | left atrium |
| LKB1 | liver kinase B1 |
| LPHs | lipid hydroperoxide |
| LV | left ventricle |
| LVEDV | left ventricle end-diastolic volume |
| LVESV | left ventricle end-systolic volume |
| LVH | left ventricular hypertrophy |
| LVID | left ventricular internal dimension |
| MAO | monoamine oxidase |
| MAO-A | monoamine oxidase A |
| MAPK | mitogen-activated protein kinase |
| MCU | mitochondrial calcium uniporter |
| MMPs | matrix metalloproteinase |
| NAPDH- | nicotinamide adenine dinucleotide phosphate |
| NFκB | nuclear factor kappa-B |
| NHE-1 | Na+/Ca2+ exchanger |
| NO | nitric oxide |
| NO-2 | plasma nitrite |
| NOS | nitric oxide synthase |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| NP | natriuretic peptide |
| NT-proBNP | N-terminal pro b-type natriuretic peptide |
| NYHA- | New York Heart Association |
| OMM | outer mitochondrial membrane |
| ONOO− | peroxynitrite |
| OPA1 | optic atrophy 1 |
| P38 | a focal point of interactions of the serine/threonine kinases |
| PDE | phosphodiesterases |
| PKA | protein kinase A |
| PKB | protein kinase B |
| PKC | protein kinase C |
| PKG | protein kinase G |
| PKS | protein kinase S |
| PRR | pathogen recognition receptor |
| PRX | peroxiredoxin |
| RAAS | renin-angiotensin-aldosterone system |
| ROS | reactive oxygen species |
| Rac-1 | GTP-binding protein |
| SGLT2 | sodium-glucose cotransporter |
| SIRT | NAD+ dependent class III histone deacetylases |
| SOD | superoxide dismutase |
| TGFβ | tumor growth factor β |
| TNF-α | tumor necrosis factor-α |
| TPP | triphenylphosphonium cation |
| TXNIP | thioredoxin-interacting protein |
| UCP2 | uncoupling protein 2 |
| VDAC1 | voltage-dependent anion channel 1 |
| XBP1s | X-box-binding protein1 |
| XO | xanthine oxidase |
| XOR | xanthine oxidoreductase |
| cAMP | cyclic adenosine monophosphate |
| cGMP | cyclic guanosine monophosphate |
| eNOS | endothelial nitric oxide synthase |
| hs-CRP | high-sensitivity C-reactive protein |
| iNOS | inducible nitric oxide synthase |
| nNOS | neuronal nitric oxide synthase |
| pGC | particulate guanylyl cyclase |
| sGC | soluble guanylate cyclase |
| β-OX | beta-oxidation |
References
- Holland, D.; Kumbhani, D.J.; Ahmed, S.H.; Marwick, T.H. Effects of Treatment on Exercise Tolerance, Cardiac Function, and Mortality in Heart Failure with Preserved Ejection Fraction: A Meta-Analysis. J. Am. Coll. Cardiol. 2011, 57, 1676–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henning, R.J. Diagnosis and treatment of heart failure with preserved left ventricular ejection fraction. World J. Cardiol. 2020, 12, 7–25. [Google Scholar] [CrossRef]
- Shah, A.M.; Mann, D. In search of new therapeutic targets and strategies for heart failure: Recent advances in basic science. Lancet 2011, 378, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Kitzman, D.W.; Borlaug, B.; Van Heerebeek, L.; Zile, M.; Kass, D.A.; Paulus, W.J. Phenotype-Specific Treatment of Heart Failure with Preserved Ejection Fraction. Circulation 2016, 134, 73–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Sorescu, D.; Griendling, K.; Sorescu, D.; Griendling, K. Reactive Oxygen Species, Mitochondria, and NAD(P)H Oxidases in the Development and Progression of Heart Failure. Congest. Heart Fail. 2002, 8, 132–140. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef] [Green Version]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef] [Green Version]
- Wardman, P.; Candeias, L.P. Fenton chemistry: An introduction. Radiat. Res. 1996, 145, 523–531. [Google Scholar] [CrossRef]
- Münzel, T.; Gori, T.; Keaney, J.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferdinandy, P.; Danial, H.; Ambrus, I.; Rothery, R.; Schulz, R. Peroxynitrite Is a Major Contributor to Cytokine-Induced Myocardial Contractile Failure. Circ. Res. 2000, 87, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, J.M.; Go, Y.-M.; Jones, D.P. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Raza, S.; Shah, A.M. Redox signaling in the cardiomyocyte: From physiology to failure. Int. J. Biochem. Cell Biol. 2016, 74, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Zurlo, G.; Guo, J.; Takada, M.; Wei, W.; Zhang, Q. New Insights into Protein Hydroxylation and Its Important Role in Human Diseases. Biochim. Biophys. Acta 2016, 1866, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Stomberski, C.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [Green Version]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Kotiadis, V.; Duchen, M.R.; Osellame, L.D. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta Gen. Subj. 2013, 1840, 1254–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takimoto, E.; Kass, D.A. Role of Oxidative Stress in Cardiac Hypertrophy and Remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, D.B.; Siwik, D.A.; Xiao, L.; Pimentel, D.R.; Singh, K.; Colucci, W.S. Role of Oxidative Stress in Myocardial Hypertrophy and Failure. J. Mol. Cell. Cardiol. 2002, 34, 379–388. [Google Scholar] [CrossRef]
- Christiansen, L.B.; Dela, F.; Koch, J.; Hansen, C.N.; Leifsson, P.S.; Yokota, T. Impaired cardiac mitochondrial oxidative phosphorylation and enhanced mitochondrial oxidative stress in feline hypertrophic cardiomyopathy. Am. J. Physiol. Circ. Physiol. 2015, 308, H1237–H1247. [Google Scholar] [CrossRef] [Green Version]
- Weissman, D.; Maack, C. Redox signaling in heart failure and therapeutic implications. Free Radic. Biol. Med. 2021, 171, 345–364. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schröder, E. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393–1397. [Google Scholar] [CrossRef]
- Kuster, G.M.; Siwik, D.A.; Pimentel, D.R.; Colucci, W.S. Role of Reversible, Thioredoxin-Sensitive Oxidative Protein Modifications in Cardiac Myocytes. Antioxid. Redox Signal. 2006, 8, 2153–2159. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.-L.A.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A Dynamic Pathway for Calcium-Independent Activation of CaMKII by Methionine Oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [Green Version]
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schröder, E.; Wait, R. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 2006, 281, 21827–21836. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Liu, T.; Zhai, P.; Chen, W.; Li, H.; Molkentin, J.; Vatner, S.F.; Sadoshima, J. A Redox-Dependent Pathway for Regulating Class II HDACs and Cardiac Hypertrophy. Cell 2008, 133, 978–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, H.S.; Cox, M.J.; Tyagi, S.C. Generation of Nitrotyrosine Precedes Activation of Metalloproteinase in Myocardium of Hyperhomocysteinemic Rats. Antioxid. Redox Signal. 2002, 4, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, T.; Tokunaga, E.; Ota, H.; Sugita, H.; Martyn, J.A.J.; Kaneki, M. S-Nitrosylation-dependent Inactivation of Akt/Protein Kinase B in Insulin Resistance. J. Biol. Chem. 2005, 280, 7511–7518. [Google Scholar] [CrossRef] [Green Version]
- Kehat, I.; Molkentin, J.D. Extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in cardiac hypertrophy. Ann. N. Y. Acad. Sci. 2010, 1188, 96–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijles, D.N.; Cull, J.J.; Markou, T.; Cooper, S.T.; Haines, Z.H.; Fuller, S.J.; O’Gara, P.; Sheppard, M.N.; Harding, S.; Sugden, P.H.; et al. Redox Regulation of Cardiac ASK1 (Apoptosis Signal-Regulating Kinase 1) Controls p38-MAPK (Mitogen-Activated Protein Kinase) and Orchestrates Cardiac Remodeling to Hypertension. Hypertension 2020, 76, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C.; Antal, C.E.; Steinberg, S.F. Protein kinase C mechanisms that contribute to cardiac remodelling. Clin. Sci. 2016, 130, 1499–1510. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Dudley, S.C. Redox regulation, NF-kappaB, and atrial fibrillation. Antioxid. Redox Signal 2009, 11, 2265–2277. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Galang, N.; Maulik, N. Redox regulation of NF-kappaB and AP-1 in ischemic reperfused heart. Antioxid. Redox Signal 1999, 1, 317–324. [Google Scholar] [CrossRef]
- Ho, E.; Karimi Galougahi, K.; Liu, C.-C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Fushimi, K.; Kouchi, H.; Mihara, K.; Miyazaki, M.; Ohe, T.; Namba, M. Inhibitory Effects of Antioxidants on Neonatal Rat Cardiac Myocyte Hypertrophy Induced by Tumor Necrosis Factor-α and Angiotensin II. Circulation 1998, 98, 794–799. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, T.; Nakano, M.; Bednarczyk, J.L.; McIntyre, B.W.; Entman, M.; Mann, U.L. Tumor Necrosis Factor-α Provokes a Hypertrophic Growth Response in Adult Cardiac Myocytes. Circulation 1997, 95, 1247–1252. [Google Scholar] [CrossRef]
- Yamazaki, T.; Komuro, I.; Zou, Y.; Kudoh, S.; Shiojima, I.; Hiroi, Y. Norepinephrine induces the raf-1 kinase/mitogen-activated protein kinase cascade through both alpha 1- and beta-adrenoceptors. Circulation 1997, 95, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijnker, P.J.; Sequeira, V.; Kuster, D.W.; Van Der Velden, J. Hypertrophic Cardiomyopathy: A Vicious Cycle Triggered by Sarcomere Mutations and Secondary Disease Hits. Antioxid. Redox Signal. 2019, 31, 318–358. [Google Scholar] [CrossRef]
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Horacio, E.C.; Cingolani, H.E.; Perez, N.G.; Aiello, E.A.; Ennis, I.L.; Garciarena, C.D.; Villa-Abrille, M.C.; Dulce, R.A.; Caldiz, C.I.; Yeves, A.M.; et al. Early signals after stretch leading to cardiac hypertrophy. Key role of NHE-1. Front. Biosci. 2008, 13, 7096–7114. [Google Scholar] [CrossRef] [Green Version]
- Palomeque, J.; Rueda, O.V.; Sapia, L.; Valverde, C.A.; Salas, M.; Petroff, M.V.; Mattiazzi, A. Angiotensin II–Induced Oxidative Stress Resets the Ca2+ Dependence of Ca2+–Calmodulin Protein Kinase II and Promotes a Death Pathway Conserved Across Different Species. Circ. Res. 2009, 105, 1204–1212. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Marck, P.; Pessoa, M.; Xu, Y.; Kutz, L.; Collins, D.; Yan, Y.; King, C.; Wang, X.; Duan, Q.; Cai, L.; et al. Cardiac Oxidative Signaling and Physiological Hypertrophy in the Na/K-ATPase α1s/sα2s/s Mouse Model of High Affinity for Cardiotonic Steroids. Int. J. Mol. Sci. 2021, 22, 3462. [Google Scholar] [CrossRef]
- Pieske, B.; Maier, L.S.; PiacentinoIII, V.; Weisser, J.; Hasenfuss, G.; Houser, S. Rate Dependence of [Na+] i and Contractility in Nonfailing and Failing Human Myocardium. Circulation 2002, 106, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, H.; Wang, J.; Wei, B.; Zhang, X.; Zhang, M.; Cao, D.; Dai, J.; Wang, Z.; Nyirimigabo, E.; et al. A positive feedback regulation of Heme oxygenase 1 by CELF1 in cardiac myoblast cells. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 209–218. [Google Scholar] [CrossRef]
- AlHayaza, R.; Haque, E.; Karbasiafshar, C.; Sellke, F.W.; Abid, M.R. The Relationship Between Reactive Oxygen Species and Endothelial Cell Metabolism. Front. Chem. 2020, 8, 592688. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Cimmino, G.; Cirillo, P.; Ragni, M.; Conte, S.; Uccello, G.; Golino, P. Reactive oxygen species induce a procoagulant state in endothelial cells by inhibiting tissue factor pathway inhibitor. J. Thromb. Thrombolysis 2015, 40, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH Oxidases in Heart Failure: Poachers or Gamekeepers? Antioxid. Redox Signal 2013, 18, 1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekelund, U.E.G.; Harrison, R.W.; Shokek, O.; Thakkar, R.N.; Tunin, R.S.; Senzaki, H.; Kass, D.A.; Marbaán, E.; Hare, J.M. Intravenous Allopurinol Decreases Myocardial Oxygen Consumption and Increases Mechanical Efficiency in Dogs with Pacing-Induced Heart Failure. Circ. Res. 1999, 85, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovic, A.; Korac, A.; Buzadzic, B.; Stancic, A.; Otasevic, V.; Ferdinandy, P.; Daiber, A.; Korac, B. Targeting the NO/superoxide ratio in adipose tissue: Relevance to obesity and diabetes management. J. Cereb. Blood Flow Metab. 2016, 174, 1570–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Looi, Y.H.; Grieve, D.J.; Siva, A.; Walker, S.J.; Anilkumar, N.; Cave, A.C.; Marber, M.; Monaghan, M.J.; Shah, A.M. Involvement of Nox2 NADPH Oxidase in Adverse Cardiac Remodeling After Myocardial Infarction. Hypertension 2008, 51, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, Physiology, and Pathophysiology of NADPH Oxidases in the Cardiovascular System. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS Signaling: Rapid Mechano-Chemo Transduction in Heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Izu, L.T.; Kohl, P.; Boyden, P.A.; Miura, M.; Banyasz, T.; Chiamvimonvat, N. Mechano-Electric and Mechano-Chemo-Transduction in Cardiomyocytes. J. Physiol. 2020, 598, 1285. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takimoto, E.; Champion, H.C.; Li, M.; Ren, S.; Rodriguez, E.R.; Tavazzi, B.; Lazzarino, G.; Paolocci, N.; Gabrielson, K.L.; Wang, Y.; et al. Oxidant stress from nitric oxide synthase–3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J. Clin. Investig. 2005, 115, 1221–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.; Chen, L.; Tu, W.; Wang, H.; Wang, Q.; Meng, L. Protective effects of valsartan administration on doxorubicin-induced myocardial injury in rats and the role of oxidative stress and NOX2/NOX4 signaling. Mol. Med. Rep. 2020, 22, 4151–4162. [Google Scholar] [CrossRef]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef] [Green Version]
- Nakagami, H.; Takemoto, M.; Liao, J.K. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2003, 35, 851–859. [Google Scholar] [CrossRef]
- Higuchi, Y.; Otsu, K.; Nishida, K.; Hirotani, S.; Nakayama, H.; Yamaguchi, O. The Small GTP-binding Protein Rac1 Induces Cardiac Myocyte Hypertrophy through the Activation of Apoptosis Signal-regulating Kinase 1 and Nuclear Factor-κB*. J. Biol. Chem. 2003, 278, 20770–20777. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Pimental, D.R.; Amin, J.K.; Singh, K.; Sawyer, D.B.; Colucci, W.S. MEK1/2-ERK1/2 mediates alpha1-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J. Mol. Cell Cardiol. 2001, 33, 779–787. [Google Scholar] [CrossRef]
- Byrne, J.A.; Grieve, D.J.; Bendall, J.K.; Li, J.-M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting Roles of NADPH Oxidase Isoforms in Pressure-Overload Versus Angiotensin II–Induced Cardiac Hypertrophy. Circ. Res. 2003, 93, 802–805. [Google Scholar] [CrossRef]
- Bendall, J.K.; Cave, A.C.; Heymes, C.; Gall, N.; Shah, A.M. Pivotal Role of a gp91 phox -Containing NADPH Oxidase in Angiotensin II-Induced Cardiac Hypertrophy in Mice. Circulation 2002, 105, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Grote, K.; Flach, I.; Luchtefeld, M.; Akin, E.; Holland, S.M.; Drexler, H.; Schieffer, B. Mechanical Stretch Enhances mRNA Expression and Proenzyme Release of Matrix Metalloproteinase-2 (MMP-2) via NAD(P)H Oxidase–Derived Reactive Oxygen Species. Circ. Res. 2003, 92, e80–e86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Luchtefeld, M.; Grote, K.; Grothusen, C.; Bley, S.; Bandlow, N.; Selle, T.; Strüber, M.; Haverich, A.; Bavendiek, U.; Drexler, H.; et al. Angiotensin II induces MMP-2 in a p47phox-dependent manner. Biochem. Biophys. Res. Commun. 2005, 328, 183–188. [Google Scholar] [CrossRef]
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006, 20, 1546–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colston, J.T.; De La Rosa, S.D.; Strader, J.R.; Anderson, M.A.; Freeman, G.L. H2O2 activates Nox4 through PLA2-dependent arachidonic acid production in adult cardiac fibroblasts. FEBS Lett. 2005, 579, 2533–2540. [Google Scholar] [CrossRef] [Green Version]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [Green Version]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef]
- Harrison, R. Physiological Roles of Xanthine Oxidoreductase. Drug Metab. Rev. 2004, 36, 363–375. [Google Scholar] [CrossRef]
- Gladden, J.D.; Zelickson, B.R.; Wei, C.-C.; Ulasova, E.; Zheng, J.; Ahmed, M.I.; Chen, Y.; Bamman, M.; Ballinger, S.; Darley-Usmar, V.; et al. Novel insights into interactions between mitochondria and xanthine oxidase in acute cardiac volume overload. Free Radic. Biol. Med. 2011, 51, 1975–1984. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Watanabe, T.; Otaki, Y.; Shishido, T.; Murase, T.; Nakamura, T.; Kato, S.; Tamura, H.; Nishiyama, S.; Takahashi, H.; et al. Impact of plasma xanthine oxidoreductase activity in patients with heart failure with preserved ejection fraction. ESC Heart Fail. 2020, 7, 1735–1743. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factor 1 and Cardiovascular Disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef] [Green Version]
- Massion, P.B.; Balligand, J.-L. Relevance of nitric oxide for myocardial remodeling. Curr. Heart Fail. Rep. 2007, 4, 18–25. [Google Scholar] [CrossRef]
- Soskić, S.S.; Dobutović, B.D.; Sudar, E.M.; Obradović, M.M.; Nikolić, D.M.; Djordjevic, J.D.; Radak, D.J.; Mikhailidis, D.P.; Isenović, E.R. Regulation of Inducible Nitric Oxide Synthase (iNOS) and its Potential Role in Insulin Resistance, Diabetes and Heart Failure. Open Cardiovasc. Med. J. 2011, 5, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Sinclair, D.A. The Intersection Between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Finkel, T.; Deng, C.-X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Cao, Y.; Zheng, Q.; Tu, J.; Zhou, W.; He, J.; Zhong, J.; Chen, Y.; Wang, J.; Cai, R.; et al. SENP1-Sirt3 Signaling Controls Mitochondrial Protein Acetylation and Metabolism. Mol. Cell 2019, 75, 823–834.e5. [Google Scholar] [CrossRef]
- Koentges, C.; Cimolai, M.C.; Pfeil, K.; Wolf, D.; Marchini, T.O.; Tarkhnishvili, A.; Hoffmann, M.M.; Odening, K.E.; Diehl, P.; Mühlen, C.V.Z.; et al. Impaired SIRT3 activity mediates cardiac dysfunction in endotoxemia by calpain-dependent disruption of ATP synthesis. J. Mol. Cell. Cardiol. 2019, 133, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Luo, K.; Huang, W.; Tang, S. Sirt3 enhances glioma cell viability by stabilizing Ku70–BAX interaction. OncoTargets Ther. 2018, 11, 7559–7567. [Google Scholar] [CrossRef] [Green Version]
- Tseng, A.H.; Shieh, S.-S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Bergaggio, E.; Riganti, C.; Garaffo, G.; Vitale, N.; Mereu, E.; Bandini, C.; Pellegrino, E.; Pullano, V.; Omedè, P.; Todoerti, K.; et al. IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood 2019, 133, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Rodriguez, E.; Anderson, C.A.; Thayne, K.; Chitwood, W.R.; Kypson, A.P. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am. J. Physiol. Circ. Physiol. 2011, 300, H118–H124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.G. Reactive oxygen species in TNFalpha-induced signaling and cell death. Mol. Cells 2010, 30, 1–12. [Google Scholar] [CrossRef]
- Anderson, E.J.; Efird, J.T.; Davies, S.W.; O’Neal, W.T.; Darden, T.M.; Thayne, K.A.; Katunga, L.A.; Kindell, L.C.; Ferguson, T.B.; Anderson, C.A.; et al. Monoamine Oxidase is a Major Determinant of Redox Balance in Human Atrial Myocardium and is Associated with Postoperative Atrial Fibrillation. J. Am. Heart Assoc. 2014, 3, e000713. [Google Scholar] [CrossRef] [Green Version]
- Santin, Y.; Sicard, P.; Vigneron, F.; Guilbeau-Frugier, C.; Dutaur, M.; Lairez, O.; Couderc, B.; Manni, D.; Korolchuk, V.I.; Lezoualc’H, F.; et al. Oxidative Stress by Monoamine Oxidase-A Impairs Transcription Factor EB Activation and Autophagosome Clearance, Leading to Cardiomyocyte Necrosis and Heart Failure. Antioxid. Redox Signal. 2016, 25, 10–27. [Google Scholar] [CrossRef]
- Deshwal, S.; Di Sante, M.; Di Lisa, F.; Kaludercic, N. Emerging role of monoamine oxidase as a therapeutic target for cardiovascular disease. Curr. Opin. Pharmacol. 2017, 33, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.-H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative Stress–Dependent Sphingosine Kinase-1 Inhibition Mediates Monoamine Oxidase A–Associated Cardiac Cell Apoptosis. Circ. Res. 2007, 100, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Menazza, S.; Blaauw, B.; Tiepolo, T.; Toniolo, L.; Braghetta, P.; Spolaore, B.; Reggiani, C.; Di Lisa, F.; Bonaldo, P.; Canton, M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet. 2010, 19, 4207–4215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molavian, H.; Tonekaboni, A.M.; Kohandel, M.; Sivaloganathan, S. The Synergetic Coupling among the Cellular Antioxidants Glutathione Peroxidase/Peroxiredoxin and Other Antioxidants and its Effect on the Concentration of H2O2. Sci. Rep. 2015, 5, srep13620. [Google Scholar] [CrossRef]
- Aon, M.A.; Stanley, B.A.; Sivakumaran, V.; Kembro, J.M.; O’Rourke, B.; Paolocci, N.; Cortassa, S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: An experimental-computational study. J. Gen. Physiol. 2012, 139, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal 2008, 10, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Nickel, A.G.; Von Hardenberg, A.; Hohl, M.; Löffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.-C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated Cytosolic Na+ Increases Mitochondrial Formation of Reactive Oxygen Species in Failing Cardiac Myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.; Wenzel, P.; Münzel, T.; Daiber, A. Mitochondrial Redox Signaling: Interaction of Mitochondrial Reactive Oxygen Species with Other Sources of Oxidative Stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef]
- Wenzel, P.; Müller, J.; Zurmeyer, S.; Schuhmacher, S.; Schulz, E.; Oelze, M.; Pautz, A.; Kawamoto, T.; Wojnowski, L.; Kleinert, H.; et al. ALDH-2 deficiency increases cardiovascular oxidative stress—Evidence for indirect antioxidative properties. Biochem. Biophys. Res. Commun. 2008, 367, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Bahoran, T.; Soobrattee, M.A.; Luximon-Ramma, V.; Aruoma, O.I. Free Radicals and Antioxidants in Cardiovascular Health and Disease. Internet J. Med. Updat. 2006, 1, 24–40. [Google Scholar] [CrossRef] [Green Version]
- Sverdlov, A.L.; Ngo, D.T.M.; Colucci, W.S. 8—Oxidative Stress in Heart Failure. In Heart Failure: A Companion to Braunwald’s Heart Disease; Elsevier: Philadelphia, PA, USA, 2020; pp. 115–126.e6. [Google Scholar]
- De Keulenaer, G.; Brutsaert, D.L. Diastolic and systolic heart failure are distinct phenotypes within the heart failure spectrum. Circulation 2011, 123, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, T.; Tokitsu, T.; Sueta, D.; Oike, F.; Takae, M.; Fujisue, K.; Usuku, H.; Ito, M.; Kanazawa, H.; Araki, S.; et al. Clinical significance of reactive oxidative metabolites in patients with heart failure with reduced left ventricular ejection fraction. J. Card. Fail. 2020, 27, 57–66. [Google Scholar] [CrossRef]
- Hummel, S.L.; Seymour, E.M.; Brook, R.D.; Kolias, T.J.; Sheth, S.S.; Rosenblum, H.R.; Wells, J.M.; Weder, A.B. Low-Sodium Dietary Approaches to Stop Hypertension Diet Reduces Blood Pressure, Arterial Stiffness, and Oxidative Stress in Hypertensive Heart Failure with Preserved Ejection Fraction. Hypertension 2012, 60, 1200–1206. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H. Morphological Stages of Mitochondrial Vacuolar Degeneration in Phenylephrine-Stressed Cardiac Myocytes and in Animal Models and Human Heart Failure. Medicina 2019, 55, 239. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease with Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Gustafsson, B.; Brunk, U.T. The Involvement of Lysosomes in Myocardial Aging and Disease. Curr. Cardiol. Rev. 2008, 4, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-Induced Phosphorylation and Proteasomal Degradation of Mitofusin 2 Facilitates Mitochondrial Fragmentation and Apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.; LeJemtel, T.; Delafontaine, P. Mitochondrial Pathobiology and Metabolic Remodeling in Progression to Overt Systolic Heart Failure. J. Clin. Med. 2020, 9, 3582. [Google Scholar] [CrossRef]
- Chen, H.C.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; De Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabò, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The Opa1-Dependent Mitochondrial Cristae Remodeling Pathway Controls Atrophic, Apoptotic, and Ischemic Tissue Damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [Green Version]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnauné-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Nan, J.; Zhu, W.; Rahman, M.; Liu, M.; Li, D.; Su, S.; Zhang, N.; Hu, X.; Yu, H.; Gupta, M.P.; et al. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1260–1273. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012, 3, e265. [Google Scholar] [CrossRef] [Green Version]
- Aoki, H.; Kang, P.M.; Hampe, J.; Yoshimura, K.; Noma, T.; Matsuzaki, M.; Izumo, S. Direct Activation of Mitochondrial Apoptosis Machinery by c-Jun N-terminal Kinase in Adult Cardiac Myocytes. J. Biol. Chem. 2002, 277, 10244–10250. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-R.; Blackstone, C. Cyclic AMP-dependent Protein Kinase Phosphorylation of Drp1 Regulates Its GTPase Activity and Mitochondrial Morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter (MCU). Nat. Cell Biol. 2013, 15, 1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Rasmussen, T.P.; Koval, O.M.; Joiner, M.L.A.; Hall, D.D.; Chen, B. The mitochondrial uniporter controls fight or flight heart rate increases. Nat. Commun. 2015, 6, 6081. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Menazza, S.; Holmström, K.M.; Parks, R.J.; Liu, J.; Sun, J. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: Mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ. Heart Fail. 2013, 6, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C., Jr.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 1, e84897. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Li, J. Mouse SIRT3 Attenuates Hypertrophy-Related Lipid Accumulation in the Heart through the Deacetylation of LCAD. PLoS ONE 2015, 10, e0118909. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Juhaszova, M.; Aon, M.A.; Zorov, D.B.; Sollott, S.J. Mitochondrial Ca2+, redox environment and ROS emission in heart failure: Two sides of the same coin? J. Mol. Cell Cardiol. 2021, 151, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Anzai, T.; Kaneko, H.; Mano, Y.; Anzai, A.; Maekawa, Y. C-reactive protein overexpression exacerbates pressure overload-induced cardiac remodeling through enhanced inflammatory response. Hypertension 2011, 57, 208–215. [Google Scholar] [CrossRef]
- Kobayashi, S.; Inoue, N.; Ohashi, Y.; Terashima, M.; Matsui, K.; Mori, T. Interaction of oxidative stress and inflammatory response in coronary plaque instability: Important role of C-reactive protein. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells 2020, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Katayama, Y.; Battista, M.; Kao, W.-M.; Hidalgo, A.; Peired, A.; Thomas, S.A.; Frenette, P.S. Signals from the Sympathetic Nervous System Regulate Hematopoietic Stem Cell Egress from Bone Marrow. Cell 2006, 124, 407–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, P.; Nahrendorf, M. Monocytes in myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1066–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noll, N.A.; Lal, H.; Merryman, W.D. Mouse Models of Heart Failure with Preserved or Reduced Ejection Fraction. Am. J. Pathol. 2020, 190, 1596–1608. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, J.; Lu, Q.; Ren, D.; Sun, X.; Rousselle, T.; Tan, Y.; Li, J. AMPK: A therapeutic target of heart failure—Not only metabolism regulation. Biosci. Rep. 2019, 39, BSR20181767. [Google Scholar] [CrossRef]
- Kanwar, M.; Walter, C.; Clarke, M.; Aponte, M. Targeting heart failure with preserved ejection fraction: Current status and future prospects. Vasc. Health Risk Manag. 2016, 12, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Hamdani, N.; Franssen, C.; Lourenço, A.; Falca o-Pires, I.; Fontoura, D.; Leite, S. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ. Heart Fail. 2013, 6, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Ben-Nun, D.; Buja, L.M.; Fuentes, F. Prevention of heart failure with preserved ejection fraction (HFpEF): Reexamining microRNA-21 inhibition in the era of oligonucleotide-based therapeutics. Cardiovasc. Pathol. 2020, 49, 107243. [Google Scholar] [CrossRef]
- Collier, P.; Watson, C.J.; Voon, V.; Phelan, D.; Jan, A.; Mak, G. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur. J. Heart Fail. 2011, 13, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Willis, M.S.; Patterson, C. Proteotoxicity and cardiac dysfunction—Alzheimer’s disease of the heart? N. Engl. J. Med. 2013, 368, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taube, A.; Schlich, R.; Sell, H.; Eckardt, K.; Eckel, J. Inflammation and metabolic dysfunction: Links to cardiovascular diseases. Am. J. Physiol. Circ. Physiol. 2012, 302, H2148–H2165. [Google Scholar] [CrossRef]
- Jelic, S.; Lederer, D.J.; Adams, T.; Padeletti, M.; Colombo, P.C.; Factor, P.H.; Le Jemtel, T.H. Vascular Inflammation in Obesity and Sleep Apnea. Circulation 2010, 121, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- MacDougall, I.C.; Canaud, B.; De Francisco, A.L.; Filippatos, G.; Ponikowski, P.; Silverberg, D.; Van Veldhuisen, D.J.; Anker, S.D. Beyond the cardiorenal anaemia syndrome: Recognizing the role of iron deficiency. Eur. J. Heart Fail. 2012, 14, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, J.; Sugiyama, S.; Nozaki, T.; Sugamura, K.; Konishi, M.; Ohba, K.; Matsuzawa, Y.; Akiyama, E.; Yamamoto, E.; Sakamoto, K.; et al. Pentraxin 3 Is a New Inflammatory Marker Correlated with Left Ventricular Diastolic Dysfunction and Heart Failure with Normal Ejection Fraction. J. Am. Coll. Cardiol. 2011, 57, 861–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, K.B.; Kop, W.J.; Christenson, R.H.; Diercks, D.B.; Henderson, S.; Hanson, K.; Li, S.-Y.; Defilippi, C.R. Prognostic Utility of ST2 in Patients with Acute Dyspnea and Preserved Left Ventricular Ejection Fraction. Clin. Chem. 2011, 57, 874–882. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.V.; Bronzwaer, J.G.F.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac Inflammation Contributes to Changes in the Extracellular Matrix in Patients with Heart Failure and Normal Ejection Fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Dorsch, M.F.; Lawrance, R.A.; Sapsford, R.J.; Durham, N.; Oldham, J.; Greenwood, D.C.; Jackson, B.M.; Morrell, C.; Robinson, M.B.; Hall, A.S. Poor prognosis of patients presenting with symptomatic myocardial infarction but without chest pain. Heart 2001, 86, 494–498. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Shah, A.M. ROS signalling between endothelial cells and cardiac cells. Cardiovasc. Res. 2014, 102, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulus, W.J.; Vantrimpont, P.J.; Shah, A.M. Paracrine Coronary Endothelial Control of Left Ventricular Function in Humans. Circulation 1995, 92, 2119–2126. [Google Scholar] [CrossRef]
- Denninger, J.W.; Marletta, M.A. Guanylate cyclase and the .NO/cGMP signaling pathway. Biochim. Biophys. Acta 1999, 1411, 334–350. [Google Scholar] [CrossRef] [Green Version]
- Cerra, M.; Pellegrino, D. Cardiovascular cGMP-generating systems in physiological and pathological conditions. Curr. Med. Chem. 2007, 14, 585–599. [Google Scholar] [CrossRef]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, A.J.; Stasch, J.P. Soluble Guanylate Cyclase: Allosteric Activation and Redox Regulation. Nitric Oxide 2010, 1, 301–326. [Google Scholar]
- Hamdani, N.; Bishu, K.G.; Von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2012, 97, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Bishu, K.G.; Hamdani, N.; Mohammed, S.F.; Kruger, M.; Ohtani, T.; Ogut, O.; Brozovich, F.V.; Burnett, J.C., Jr.; Linke, W.A.; Redfield, M.M. Sildenafil and B-Type Natriuretic Peptide Acutely Phosphorylate Titin and Improve Diastolic Distensibility In Vivo. Circulation 2011, 124, 2882–2891. [Google Scholar] [CrossRef] [Green Version]
- Lyle, M.A.; Brozovich, F.V. HFpEF, a Disease of the Vasculature: A Closer Look at the Other Half. Mayo Clin. Proc. 2018, 93, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Lyle, M.A.; Alabdaljabar, M.S.; Han, Y.S.; Brozovich, F.V. The vasculature in HFpEF vs HFrEF: Differences in contractile protein expression produce distinct phenotypes. Heliyon 2019, 6, e03129. [Google Scholar] [CrossRef] [Green Version]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Niwa, M.; Koong, A.C. Targeting the IRE1α–XBP1 branch of the unfolded protein response in human diseases. Semin. Cancer Biol. 2015, 33, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V.; Joubert, F. Bioenergetics of the failing heart. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1360–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Fillmore, N.; Levasseur, J.L.; Fukushima, A.; Wagg, C.S.; Wang, W.; Dyck, J.R.B.; Lopaschuk, G.D. Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol. Med. 2018, 24, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mori, J.; Basu, R.; McLean, B.A.; Das, S.K.; Zhang, L.; Patel, V.B. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: A metabolic contribution to heart failure with normal ejection fraction. Circ. Heart Fail. 2012, 5, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Briston, T.; Roberts, M.; Lewis, S.; Powney, B.; Staddon, J.M.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 2017, 7, 10492. [Google Scholar] [CrossRef]
- Miranda-Silva, D.; Wüst, R.C.I.; Conceição, G.; Gonçalves-Rodrigues, P.; Gonçalves, N.; Gonçalves, A.; Kuster, D.W.D.; Leite-Moreira, A.F.; van der Velden, J.; Beleza, J.M.D.S.; et al. Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk-related rat model of heart failure with preserved ejection fraction. Acta Physiol. 2019, 228, e13378. [Google Scholar] [CrossRef] [Green Version]
- Kraigher-Krainer, E.; Shah, A.M.; Gupta, D.K.; Santos, A.; Claggett, B.; Pieske, B.; Zile, M.; Voors, A.A.; Lefkowitz, M.P.; Packer, M.; et al. Impaired Systolic Function by Strain Imaging in Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2013, 63, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.-S.; Hamdani, N.; Holewinski, R.J.; Jo, S.-H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef]
- Wang, L.; Huang, Z.; Huang, W.; Chen, X.; Shan, P.; Zhong, P.; Khan, Z.; Wang, J.; Fang, Q.; Liang, G.; et al. Inhibition of epidermal growth factor receptor attenuates atherosclerosis via decreasing inflammation and oxidative stress. Sci. Rep. 2017, 7, 45917. [Google Scholar] [CrossRef] [Green Version]
- Zaha, V.; Young, L.H. AMP-Activated Protein Kinase Regulation and Biological Actions in the Heart. Circ. Res. 2012, 111, 800–814. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.-M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Reichert, K.; Pereira do Carmo, H.R.; Galluce Torina, A.; Diógenes de Carvalho, D.; Carvalho Sposito, A.; De Souza Vilarinho, K.A. Atorvastatin Improves Ventricular Remodeling after Myocardial Infarction by Interfering with Collagen Metabolism. PLoS ONE 2016, 11, e0166845. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, P.; Taccardi, A.A.; Barsotti, A. Long term cardioprotective action of trimetazidine and potential effect on the inflammatory process in patients with ischaemic dilated cardiomyopathy. Heart 2005, 91, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Weikel, K.A.; Cacicedo, J.M.; Ido, Y. Resveratrol-Induced AMP-Activated Protein Kinase Activation Is Cell-Type Dependent: Lessons from Basic Research for Clinical Application. Nutrients 2017, 9, 751. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Quan, N.; Sun, W.; Chen, X.; Cates, C.; Rousselle, T.; Zhou, X.; Zhao, X.; Li, J. Cardiomyocyte-specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc. Res. 2018, 114, 805–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Xia, N.; Förstermann, U. Cardiovascular effects and molecular targets of resveratrol. Nitric Oxide 2012, 26, 102–110. [Google Scholar] [CrossRef]
- Senni, M.; Mcmurray, J.; Wachter, R.; McIntyre, H.F.; Reyes, A.; Majercak, I.; Andreka, P.; Shehova-Yankova, N.; Anand, I.; Yilmaz, M.B.; et al. Initiating sacubitril/valsartan (LCZ696) in heart failure: Results of TITRATION, a double-blind, randomized comparison of two uptitration regimens. Eur. J. Heart Fail. 2016, 18, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Baruch, L.; Anand, I.; Cohen, I.S.; Ziesche, S.; Judd, D.; Cohn, J.N. Augmented Short- and Long-Term Hemodynamic and Hormonal Effects of an Angiotensin Receptor Blocker Added to Angiotensin Converting Enzyme Inhibitor Therapy in Patients with Heart Failure. Circulation 1999, 99, 2658–2664. [Google Scholar] [CrossRef] [Green Version]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 132–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazza, A.; Torin, G.; Schiavon, L.; Rossetti, C.; Cuppini, S. Role of sacubitril/valsartan in the treatment of chronic heart failure with reduced ejection fraction in elderly hypertensives with comorbidity: From clinical trials to real world. J. Hypertens. 2021, 39 (Suppl. S1), e279. [Google Scholar] [CrossRef]
- Lund, L.H.; Claggett, B.; Liu, J.; Lam, C.S.; Jhund, P.; Rosano, G.M.; Swedberg, K.; Yusuf, S.; Granger, C.B.; Pfeffer, M.A.; et al. Heart failure with mid-range ejection fraction in CHARM: Characteristics, outcomes and effect of candesartan across the entire ejection fraction spectrum. Eur. J. Heart Fail. 2018, 20, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- D’Elia, E.; Iacovoni, A.; Vaduganathan, M.; Lorini, L.F.; Perlini, S.; Senni, M. Neprilysin inhibition in heart failure: Mechanisms and substrates beyond modulating natriuretic peptides. Eur. J. Heart Fail. 2017, 19, 710–717. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- Gori, M.; Volterrani, M.; Piepoli, M.; Senni, M. Angiotensin receptor–neprilysin inhibitor (ARNi): Clinical studies on a new class of drugs. Int. J. Cardiol. 2016, 226, 136–140. [Google Scholar] [CrossRef]
- Solomon, S.D.; Zile, M.; Pieske, B.; Voors, A.; Shah, A.; Kraigher-Krainer, E.; Shi, V.; Bransford, T.; Takeuchi, M.; Gong, J.; et al. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: A phase 2 double-blind randomised controlled trial. Lancet 2012, 380, 1387–1395. [Google Scholar] [CrossRef]
- Jhund, P.S.; Claggett, B.; Packer, M.; Zile, M.; Voors, A.A.; Pieske, B.; Lefkowitz, M.; Shi, V.; Bransford, T.; McMurray, J.J.V.; et al. Independence of the blood pressure lowering effect and efficacy of the angiotensin receptor neprilysin inhibitor, LCZ696, in patients with heart failure with preserved ejection fraction: An analysis of the PARAMOUNT trial. Eur. J. Heart Fail. 2014, 16, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, 1810–1852. [Google Scholar] [CrossRef]
- Tsao, C.W.; Lyass, A.; Enserro, D.; Larson, M.G.; Ho, J.E.; Kizer, J.R.; Gottdiener, J.S.; Psaty, B.M.; Vasan, R.S. Temporal Trends in the Incidence of and Mortality Associated with Heart Failure with Preserved and Reduced Ejection Fraction. JACC Heart Fail. 2018, 6, 678–685. [Google Scholar] [CrossRef]
- Calderon-Montano, J.M.; Burgos-Moron, E.; Perez-Guerrero, C.; Lopez-Lazaro, M. A review on the dietary flavonoid kaempferol. Mini Rev. Med. Chem. 2011, 11, 298–344. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.K.S.; Martin-Romero, F.J.; Gutiérrez-Merino, C. Kaempferol blocks oxidative stress in cerebellar granule cells and reveals a key role for reactive oxygen species production at the plasma membrane in the commitment to apoptosis. Free Radic. Biol. Med. 2004, 37, 48–61. [Google Scholar] [CrossRef]
- Du, Y.; Han, J.; Zhang, H.; Xu, J.; Jiang, L.; Ge, W. Kaempferol Prevents Against Ang II-induced Cardiac Remodeling Through Attenuating Ang II-induced Inflammation and Oxidative Stress. J. Cardiovasc. Pharmacol. 2019, 74, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M. Understanding and preventing mitochondrial oxidative damage. Biochem. Soc. Trans. 2016, 44, 1219–1226. [Google Scholar] [CrossRef]
- Ribeiro Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; O’Connell, K.A.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.; Murphy, M.P. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef]
- Tauskela, J.S. MitoQ—A mitochondria-targeted antioxidant. IDrugs 2007, 10, 399–412. [Google Scholar]
- Chen, W.; Guo, C.; Jia, Z.; Wang, J.; Xia, M.; Li, C.; Li, M.; Yin, Y.; Tang, X.; Chen, T.; et al. Inhibition of Mitochondrial ROS by MitoQ Alleviates White Matter Injury and Improves Outcomes after Intracerebral Haemorrhage in Mice. Oxid. Med. Cell. Longev. 2020, 2020, 8285065. [Google Scholar] [CrossRef]
- Fuentes, E.; Araya-Maturana, R.; Urra, F.A. Regulation of mitochondrial function as a promising target in platelet activation-related diseases. Free Radic. Biol. Med. 2019, 136, 172–182. [Google Scholar] [CrossRef]
- James, A.M.; Sharpley, M.S.; Manas, A.-R.B.; Frerman, F.E.; Hirst, J.; Smith, R.A.J.; Murphy, M.P. Interaction of the Mitochondria-targeted Antioxidant MitoQ with Phospholipid Bilayers and Ubiquinone Oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, G.A.; Bottomley, P.A.; Gerstenblith, G.; Weiss, R.G. Allopurinol Acutely Increases Adenosine Triphospate Energy Delivery in Failing Human Hearts. J. Am. Coll. Cardiol. 2012, 59, 802–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, H.E.; Plastino, J.A.; Escudero, E.M.; Mangal, B.; Brown, J.; Pérez, N.G. The Effect of Xanthine Oxidase Inhibition Upon Ejection Fraction in Heart Failure Patients: La Plata Study. J. Card. Fail. 2006, 12, 491–498. [Google Scholar] [CrossRef]
- Doehner, W.; Schoene, N.; Rauchhaus, M.; Leyva-Leon, F.; Pavitt, D.V.; Reaveley, D.A. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: Results from 2 placebo-controlled studies. Circulation 2002, 105, 2619–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratchford, S.M.; Clifton, H.L.; Gifford, J.R.; LaSalle, D.T.; Thurston, T.S.; Bunsawat, K. Impact of acute antioxidant administration on inflammation and vascular function in heart failure with preserved ejection fraction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R607–R614. [Google Scholar] [CrossRef]
- Hare, J.M.; Mangal, B.; Brown, J.; Fisher, C.; Freudenberger, R.; Colucci, W.; Mann, D.; Liu, P.; Givertz, M.M.; Schwarz, R.P. Impact of Oxypurinol in Patients with Symptomatic Heart Failure: Results of the OPT-CHF Study. J. Am. Coll. Cardiol. 2008, 51, 2301–2309. [Google Scholar] [CrossRef] [Green Version]
- Givertz, M.M.; Anstrom, K.J.; Redfield, M.M.; Deswal, A.; Haddad, H.; Butler, J. Effects of Xanthine Oxidase Inhibition in Hyperuricemic Heart Failure Patients: The Xanthine Oxidase Inhibition for Hyperuricemic Heart Failure Patients (EXACT-HF) Study. Circulation 2015, 131, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Yokota, T.; Fukushima, A.; Kinugawa, S.; Okumura, T.; Murohara, T.; Tsutsui, H. Randomized Trial of Effect of Urate-Lowering Agent Febuxostat in Chronic Heart Failure Patients with Hyperuricemia (LEAF-CHF). Int. Heart J. 2018, 59, 976–982. [Google Scholar] [CrossRef] [Green Version]
- Carnicer, R.; Duglan, D.; Ziberna, K.; Recalde, A.; Reilly, S.; Simon, J.N.; Mafrici, S.; Arya, R.; Roselló-Lletí, E.; Chuaiphichai, S.; et al. BH4 Increases nNOS Activity and Preserves Left Ventricular Function in Diabetes. Circ. Res. 2021, 128, 585–601. [Google Scholar] [CrossRef]
- Bunsawat, K.; Ratchford, S.M.; Alpenglow, J.K.; Park, S.H.; Jarrett, C.L.; Stehlik, J.; Drakos, S.G.; Richardson, R.S.; Wray, D.W. Chronic antioxidant administration restores macrovascular function in patients with heart failure with reduced ejection fraction. Exp. Physiol. 2020, 105, 1384–1395. [Google Scholar] [CrossRef]
- Dey, S.; DeMazumder, D.; Sidor, A.; Brian Foster, D.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Hoshino, A.; Okawa, Y.; Ariyoshi, M.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Matoba, S. Oxidative Post-Translational Modifications Develop LONP1 Dysfunction in Pressure Overload Heart Failure. Circ. Heart Fail. 2014, 7, 500–509. [Google Scholar] [CrossRef] [Green Version]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of Glutathione Peroxidase Prevents Left Ventricular Remodeling and Failure After Myocardial Infarction in Mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Chen, H.; Qin, H.; Sankarapandi, S.; Becher, M.W.; Wong, P.C.; Zweier, J.L. Overexpression of human copper, zinc-superoxide dismutase (SOD1) prevents postischemic injury. Proc. Natl. Acad. Sci. USA 1998, 95, 4556–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, F.; Lennon-Edwards, S.; Lancel, S.; Biolo, A.; Siwik, D.A.; Pimentel, D.R. Cardiac-specific overexpression of catalase identifies hydrogen peroxide-dependent and independent-phases of myocardial remodeling, and prevents the progression to overt heart failure in Gαq-overexpressing transgenic mice. Circ. Heart Fail. 2010, 3, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maack, C.; Eschenhagen, T.; Hamdani, N.; Heinze, F.R.; Lyon, A.R.; Manstein, D.J. Treatments targeting inotropy: A position paper of the Committees on Translational Research and Acute Heart Failure of the Heart Failure Association of the European Society of Cardiology. Eur. Heart J. 2019, 40, 3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaludercic, N.; Carpi, A.; Menabò, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1323–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, P.; Pimentel, D.R.; Murphy, M.; Colucci, W.; Parini, A. A new hypertrophic mechanism of serotonin in cardiac myocytes: Receptor-independent ROS generation. FASEB J. 2005, 19, 1–15. [Google Scholar] [CrossRef]
- Kaludercic, N.; Takimoto, E.; Nagayama, T.; Feng, N.; Lai, E.W.; Bedja, D.; Chen, K.; Gabrielson, K.L.; Blakely, R.D.; Shih, J.C.; et al. Monoamine Oxidase A–Mediated Enhanced Catabolism of Norepinephrine Contributes to Adverse Remodeling and Pump Failure in Hearts with Pressure Overload. Circ. Res. 2010, 106, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Kaludercic, N.; Carpi, A.; Menabò, R.; Giorgio, M. Mitochondrial pathways for ROS formation and myocardial injury: The relevance of p66(Shc) and monoamine oxidase. Basic Res. Cardiol. 2009, 104, 131–139. [Google Scholar] [CrossRef]
- Carpi, A.; Menabò, R.; Kaludercic, N.; Pelicci, P.; Di Lisa, F.; Giorgio, M. The cardioprotective effects elicited by p66Shc ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 774–780. [Google Scholar] [CrossRef]
- Villeneuve, C.; Guilbeau-Frugier, C.; Sicard, P.; Lairez, O.; Ordener, C.; Duparc, T.; De Paulis, D.; Couderc, B.; Spreux-Varoquaux, O.; Tortosa, F.; et al. p53-PGC-1α Pathway Mediates Oxidative Mitochondrial Damage and Cardiomyocyte Necrosis Induced by Monoamine Oxidase-A Upregulation: Role in Chronic Left Ventricular Dysfunction in Mice. Antioxid. Redox Signal. 2013, 18, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Umbarkar, P.; Singh, S.; Arkat, S.; Bodhankar, S.; Lohidasan, S.; Sitasawad, S.L. Monoamine oxidase-A is an important source of oxidative stress and promotes cardiac dysfunction, apoptosis, and fibrosis in diabetic cardiomyopathy. Free Radic. Biol. Med. 2015, 87, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaludercic, N.; Carpi, A.; Nagayama, T.; Sivakumaran, V.; Zhu, G.; Lai, E.W.; Bedja, D.; De Mario, A.; Chen, K.; Gabrielson, K.L.; et al. Monoamine Oxidase B Prompts Mitochondrial and Cardiac Dysfunction in Pressure Overloaded Hearts. Antioxid. Redox Signal. 2014, 20, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manni, M.E.; Rigacci, S.; Borchi, E.; Bargelli, V.; Miceli, C.; Giordano, C.; Raimondi, L.; Nediani, C. Monoamine Oxidase Is Overactivated in Left and Right Ventricles from Ischemic Hearts: An Intriguing Therapeutic Target. Oxidative Med. Cell. Longev. 2016, 2016, 4375418. [Google Scholar] [CrossRef]
- Sharma, A.; Fonarow, G.C.; Butler, J.; Ezekowitz, J.A.; Felker, G.M. Coenzyme Q10 and Heart Failure: A State-of-the-Art Review. Circ. Heart Fail. 2016, 9, e002639. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.D.; Mahoney, D.E.; Hiebert, J.B.; Thimmesch, A.R.; Diaz, F.J.; Smith, C.; Shen, Q.; Mudaranthakam, D.P.; Clancy, R.L. Study protocol, randomized controlled trial: Reducing symptom burden in patients with heart failure with preserved ejection fraction using ubiquinol and/or D-ribose. BMC Cardiovasc. Disord. 2018, 18, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—Biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef]
- McCarthy, S.; Somayajulu, M.; Sikorska, M.; Borowy-Borowski, H.; Pandey, S. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water-soluble Coenzyme Q10. Toxicol. Appl. Pharmacol. 2004, 201, 21–31. [Google Scholar] [CrossRef]
- Hathcock, J.N.; Shao, A. Risk assessment for coenzyme Q10 (Ubiquinone). Regul. Toxicol. Pharmacol. 2006, 45, 282–288. [Google Scholar] [CrossRef]
- Von Hardenberg, A.; Maack, C. Mitochondrial Therapies in Heart Failure. Handb. Exp. Pharmacol. 2017, 243, 491–514. [Google Scholar] [PubMed]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: Results from Q-SYMBIO: A randomized double-blind trial. JACC Heart Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. J. Cereb. Blood Flow Metab. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [Green Version]
- Bertero, E.; Kutschka, I.; Maack, C.; Dudek, J. Cardiolipin remodeling in Barth syndrome and other hereditary cardiomyopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165803. [Google Scholar] [CrossRef] [PubMed]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Zhu, M. Free radical oxidation of cardiolipin: Chemical mechanisms, detection and implication in apoptosis, mitochondrial dysfunction and human diseases. Free Radic. Res. 2012, 46, 959–974. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Hachem, S.; Zhang, K. Chronic Therapy with Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs with Advanced Heart Failure. Circ. Heart Fail. 2016, 9, e002206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatfield, K.C.; Sparagna, G.C.; Chau, S.; Phillips, E.K.; Ambardekar, A.V.; Aftab, M.; Mitchell, M.B.; Sucharov, C.C.; Miyamoto, S.D.; Stauffer, B.L. Elamipretide Improves Mitochondrial Function in the Failing Human Heart. JACC Basic Transl. Sci. 2019, 4, 147–157. [Google Scholar] [CrossRef]
- Brown, D.A.; Hale, S.L.; Baines, C.; del Rio, C.; Hamlin, R.L.; Yueyama, Y.; Kijtawornrat, A.; Yeh, S.T.; Frasier, C.R.; Stewart, L.M.; et al. Reduction of Early Reperfusion Injury with the Mitochondria-Targeting Peptide Bendavia. J. Cardiovasc. Pharmacol. Ther. 2013, 19, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ. Heart Fail. 2017, 10, e004389. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Khan, M.S.; Anker, S.D.; Fonarow, G.C.; Kim, R.J.; Nodari, S.; O’Connor, C.M.; Pieske, B.; Pieske-Kraigher, E.; Sabbah, H.N.; et al. Effects of Elamipretide on Left Ventricular Function in Patients with Heart Failure with Reduced Ejection Fraction: The PROGRESS-HF Phase 2 Trial. J. Card. Fail. 2020, 26, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, A.B.; Boen, J.R.; Segers, V.F.; Van Craenenbroeck, E.M. Heart Failure with Preserved Ejection Fraction: A Review of Cardiac and Noncardiac Pathophysiology. Front. Physiol. 2019, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Q.; Irvine, J.C.; Cao, A.H.; Alexander, A.E.; Love, J.E.; Patel, R.; McMullen, J.R.; Kaye, D.M.; Kemp-Harper, B.K.; Ritchie, R.H. Nitroxyl (HNO) Stimulates Soluble Guanylyl Cyclase to Suppress Cardiomyocyte Hypertrophy and Superoxide Generation. PLoS ONE 2012, 7, e34892. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, H.N.; Tocchetti, C.G.; Wang, M.; Daya, S.; Gupta, R.C.; Tunin, R.S.; Mazhari, R.; Takimoto, E.; Paolocci, N.; Cowart, D.; et al. Nitroxyl (HNO). Circ. Heart Fail. 2013, 6, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheorghiade, M.; Greene, S.J.; Filippatos, G.; Erdmann, E.; Ferrari, R.; Levy, P.D.; Maggioni, A.; Nowack, C.; Mebazaa, A.; COMPOSE Investigators and Coordinators. Cinaciguat, a soluble guanylate cyclase activator: Results from the randomized, controlled, phase IIb COMPOSE programme in acute heart failure syndromes. Eur. J. Heart Fail. 2012, 14, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.-A.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieske, B.; Butler, J.; Filippatos, G.; Lam, C.; Maggioni, A.P.; Ponikowski, P.; Shah, S.; Solomon, S.; Kraigher-Krainer, E.; Samano, E.T.; et al. Rationale and design of the SOluble guanylate Cyclase stimulatoR in heArT failurE Studies (SOCRATES). Eur. J. Heart Fail. 2014, 16, 1026–1038. [Google Scholar] [CrossRef]
- Zhazykbayeva, S.; Pabel, S.; Mügge, A.; Sossalla, S.; Hamdani, N. The molecular mechanisms associated with the physiological responses to inflammation and oxidative stress in cardiovascular diseases. Biophys. Rev. 2020, 12, 947–968. [Google Scholar] [CrossRef] [PubMed]
- Redfield, M.M.; Borlaug, B.A.; Lewis, G.D.; Mohammed, S.F.; Semigran, M.J.; Lewinter, M.M.; Deswal, A.; Hernandez, A.F.; Lee, K.L.; Braunwald, E.; et al. PhosphdiesteRasE-5 Inhibition to Improve CLinical Status and EXercise Capacity in Diastolic Heart Failure (RELAX) trial: Rationale and design. Circ. Heart Fail. 2012, 5, 653–659. [Google Scholar] [CrossRef] [Green Version]
- Corbin, J.D. Mechanisms of action of PDE5 inhibition in erectile dysfunction. Int. J. Impot. Res. 2004, 16, S4–S7. [Google Scholar] [CrossRef] [Green Version]
- Lewis, G.D.; Lachmann, J.; Camuso, J.; Lepore, J.J.; Shin, J.; Martinovic, M.E.; Systrom, D.M.; Bloch, K.D.; Semigran, M.J. Sildenafil Improves Exercise Hemodynamics and Oxygen Uptake in Patients with Systolic Heart Failure. Circulation 2007, 115, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; Vicenzi, M.; Arena, R.; Guazzi, M.D. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: Results of a 1-year, prospective, randomized, placebo-controlled study. Circ. Heart Fail. 2011, 4, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guazzi, M.; Vicenzi, M.; Arena, R. Phosphodiesterase 5 inhibition with sildenafil reverses exercise oscillatory breathing in chronic heart failure: A long-term cardiopulmonary exercise testing placebo-controlled study. Eur. J. Heart Fail. 2012, 14, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; Vicenzi, M.; Arena, R.; Guazzi, M.D. Pulmonary hypertension in heart failure with preserved ejection fraction: A target of phosphodiesterase-5 inhibition in a 1-year study. Circulation 2011, 124, 164–174. [Google Scholar] [CrossRef]
- Pinilla-Vera, M.; Hahn, V.S.; Kass, D.A. Leveraging Signaling Pathways to Treat Heart Failure with Reduced Ejection Fraction. Circ. Res. 2019, 124, 1618–1632. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Stanley, B.A.; Murray, C.I.; Sivakumaran, V.; Donzelli, S.; Mancardi, D.; Pagliaro, P.; Gao, W.D.; Van Eyk, J.; Kass, D.A.; et al. Playing with cardiac “redox switches”: The “HNO way” to modulate cardiac function. Antioxid. Redox Signal 2011, 114, 1687–1698. [Google Scholar] [CrossRef] [Green Version]
- Tita, C.; Gilbert, E.M.; Van Bakel, A.B.; Grzybowski, J.; Haas, G.J.; Jarrah, M.; Dunlap, S.H.; Gottlieb, S.S.; Klapholz, M.; Patel, P.C.; et al. A Phase 2a dose-escalation study of the safety, tolerability, pharmacokinetics and haemodynamic effects of BMS-986231 in hospitalized patients with heart failure with reduced ejection fraction. Eur. J. Heart Fail. 2017, 19, 1321–1332. [Google Scholar] [CrossRef] [Green Version]
- Hartman, J.C.; del Rio, C.; Reardon, J.E.; Zhang, K.; Sabbah, H.N. Intravenous Infusion of the Novel HNO Donor BMS-986231 Is Associated with Beneficial Inotropic, Lusitropic, and Vasodilatory Properties in 2 Canine Models of Heart Failure. JACC Basic Transl. Sci. 2018, 3, 625–638. [Google Scholar] [CrossRef]
- Felker, G.M.; Borentain, M.; Cleland, J.G.; DeSouza, M.M.; Kessler, P.D.; O’Connor, C.M.; Seiffert, D.; Teerlink, J.R.; Voors, A.A.; McMurray, J.J. Rationale and design for the development of a novel nitroxyl donor in patients with acute heart failure. Eur. J. Heart Fail. 2019, 21, 1022–1031. [Google Scholar] [CrossRef]
- Kemp-Harper, B.; Schmidt, H.H.H.W. cGMP in the vasculature. Handb. Exp. Pharmacol. 2009, 191, 447–467. [Google Scholar]
- Ivanova, J.; Salama, G.; Clancy, R.M.; Schor, N.F.; Nylander, K.D.; Stoyanovsky, D.A. Formation of nitroxyl and hydroxyl radical in solutions of sodium trioxodinitrate: Effects of pH and cytotoxicity. J. Biol. Chem. 2003, 278, 42761–42768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiva, S.; Crawford, J.H.; Ramachandran, A.; Ceaser, E.K.; Hillson, T.; Brookes, P.; Patel, R.P.; Darley-Usmar, V.M. Mechanisms of the interaction of nitroxyl with mitochondria. Biochem. J. 2004, 379, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Tocchetti, C.G.; Wang, W.; Froehlich, J.P.; Huke, S.; Aon, M.A.; Wilson, G.M.; Di Benedetto, G.; O’Rourke, B.; Gao, W.D.; Wink, D.A.; et al. Nitroxyl Improves Cellular Heart Function by Directly Enhancing Cardiac Sarcoplasmic Reticulum Ca2+ Cycling. Circ. Res. 2007, 100, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolocci, N.; Saavedra, W.F.; Miranda, K.M.; Martignani, C.; Isoda, T.; Hare, J.M.; Espey, M.G.; Fukuto, J.M.; Feelisch, M.; Wink, D.A.; et al. Nitroxyl anion exerts redox-sensitive positive cardiac inotropy in vivo by calcitonin gene-related peptide signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 10463–10468. [Google Scholar] [CrossRef] [Green Version]
- Paolocci, N.; Katori, T.; Champion, H.C.; St John, M.E.; Miranda, K.M.; Fukuto, J.M. Positive inotropic and lusitropic effects of HNO/NO- in failing hearts: Independence from beta-adrenergic signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 5537–5542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziolo, M.T.; Kohr, M.J.; Kaludercic, N.; Tocchetti, C.G.; Gao, W.D.; A Kass, D.; Janssen, P.M.L.; Paolocci, N. Nitroxyl enhances myocyte Ca2 transients by exclusively targeting SR Ca2+-cycling. Front. Biosci. 2010, E2, 614–626. [Google Scholar] [CrossRef] [Green Version]
- Dai, T.; Tian, Y.; Tocchetti, C.G.; Katori, T.; Murphy, A.M.; Kass, D.A.; Paolocci, N.; Gao, W.D. Nitroxyl increases force development in rat cardiac muscle. J. Physiol. 2007, 580, 951–960. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Mercurio, V.; Maack, C. The multifaceted mechanisms of nitroxyl in heart failure: Inodilator or “only” vasodilator? Eur. J. Heart Fail. 2021, 123, 1156–1159. [Google Scholar] [CrossRef]
- Hare, J.M. Nitroso–Redox Balance in the Cardiovascular System. N. Engl. J. Med. 2004, 351, 2112–2114. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Daiber, A.; Gori, T. More answers to the still unresolved question of nitrate tolerance. Eur. Heart J. 2013, 34, 2666–2673. [Google Scholar] [CrossRef]
- Daiber, A.; Oelze, M.; Coldewey, M.; Kaiser, K.; Huth, C.; Schildknecht, S.; Bachschmid, M.M.; Nazirisadeh, Y.; Ullrich, V.; Mülsch, A.; et al. Hydralazine is a powerful inhibitor of peroxynitrite formation as a possible explanation for its beneficial effects on prognosis in patients with congestive heart failure. Biochem. Biophys. Res. Commun. 2005, 338, 1865–1874. [Google Scholar] [CrossRef]
- Münzel, T.; Kurz, S.; Rajagopalan, S.; Thoenes, M.; Berrington, W.R.; A Thompson, J.; A Freeman, B.; Harrison, D.G. Hydralazine prevents nitroglycerin tolerance by inhibiting activation of a membrane-bound NADH oxidase. A new action for an old drug. J. Clin. Investig. 1996, 98, 1465–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münzel, T.; Steven, S.; Daiber, A. Organic nitrates: Update on mechanisms underlying vasodilation, tolerance and endothelial dysfunction. Vasc. Pharmacol. 2014, 63, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P. From molecules to patients: Exploring the therapeutic role of soluble guanylate cyclase stimulators. Biol. Chem. 2018, 399, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.L.; Ziesche, S.; Yancy, C.; Carson, P.; D’Agostino, R.; Ferdinand, K.; Taylor, M.; Adams, K.; Sabolinski, M.; Worcel, M.; et al. Combination of Isosorbide Dinitrate and Hydralazine in Blacks with Heart Failure. N. Engl. J. Med. 2004, 351, 2049–2057. [Google Scholar] [CrossRef] [Green Version]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Haskó, G.; Schmidt, H.H.H.W.; Stasch, J.P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Montfort, W.R.; Wales, J.A.; Weichsel, A. Structure and Activation of Soluble Guanylyl Cyclase, the Nitric Oxide Sensor. Antioxid. Redox Signal. 2017, 26, 107–121. [Google Scholar] [CrossRef]
- Breitenstein, S.; Roessig, L.; Sandner, P.; Lewis, K.S. Novel sGC Stimulators and sGC Activators for the Treatment of Heart Failure. Handb. Exp. Pharmacol. 2017, 243, 225–247. [Google Scholar] [CrossRef]
- Follmann, M.; Ackerstaff, J.; Redlich, G.; Wunder, F.; Lang, D.; Kern, A.; Fey, P.; Griebenow, N.; Kroh, W.; Becker-Pelster, E.-M.; et al. Discovery of the Soluble Guanylate Cyclase Stimulator Vericiguat (BAY 1021189) for the Treatment of Chronic Heart Failure. J. Med. Chem. 2017, 60, 5146–5161. [Google Scholar] [CrossRef] [Green Version]
- Premer, C.; Kanelidis, A.J.; Hare, J.M.; Schulman, I.H. Rethinking Endothelial Dysfunction as a Crucial Target in Fighting Heart Failure. Mayo Clinic Proc. Innov. Qual. Outcomes 2019, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kolijn, D.; Kovács, Á.; Herwig, M.; Lódi, M.; Sieme, M.; Alhaj, A.; Sandner, P.; Papp, Z.; Reusch, P.H.; Haldenwang, P.; et al. Enhanced Cardiomyocyte Function in Hypertensive Rats with Diastolic Dysfunction and Human Heart Failure Patients After Acute Treatment with Soluble Guanylyl Cyclase (sGC) Activator. Front. Physiol. 2020, 11, 345. [Google Scholar] [CrossRef]
- Archer, S.L.; Huang, J.M.; Hampl, V.; Nelson, D.P.; Shultz, P.J.; Weir, E.K. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1994, 91, 7583–7587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buys, E.; Zimmer, D.; Chickering, J.; Graul, R.; Chien, Y.; Profy, A.; Hadcock, J.; Masferrer, J.; Milne, G. Discovery and development of next generation sGC stimulators with diverse multidimensional pharmacology and broad therapeutic potential. Nitric Oxide 2018, 78, 72–80. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Rocca, H.-P.B.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Lang, N.N.; Ahmad, F.A.; Cleland, J.G.; O’Connor, C.M.; Teerlink, J.R.; Voors, A.A.; Taubel, J.; Hodes, A.R.; Anwar, M.; Karra, R.; et al. Haemodynamic effects of the nitroxyl donor cimlanod (BMS -986231) in chronic heart failure: A randomized trial. Eur. J. Heart Fail. 2021, 23, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; Greene, S.J.; Butler, J.; Filippatos, G.; Lam, C.S.P.; Maggioni, A.P.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; Kraigher-Krainer, E.; et al. Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: The SOCRATES-REDUCED randomized trial. JAMA 2015, 314, 2251–2262. [Google Scholar] [CrossRef] [PubMed]
- Pieske, B.; Maggioni, A.P.; Lam, C.S.; Pieske-Kraigher, E.; Filippatos, G.; Butler, J.; Ponikowski, P.; Shah, S.; Solomon, S.D.; Scalise, A.-V.; et al. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: Results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur. Heart J. 2017, 38, 1119–1127. [Google Scholar] [CrossRef]
- Armstrong, P.W.; Lam, C.S.P.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; O’Connor, C.M. Effect of Vericiguat vs Placebo on Quality of Life in Patients with Heart Failure and Preserved Ejection Fraction: The VITALITY-HFpEF Randomized Clinical Trial. JAMA 2020, 324, 1512–1521. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
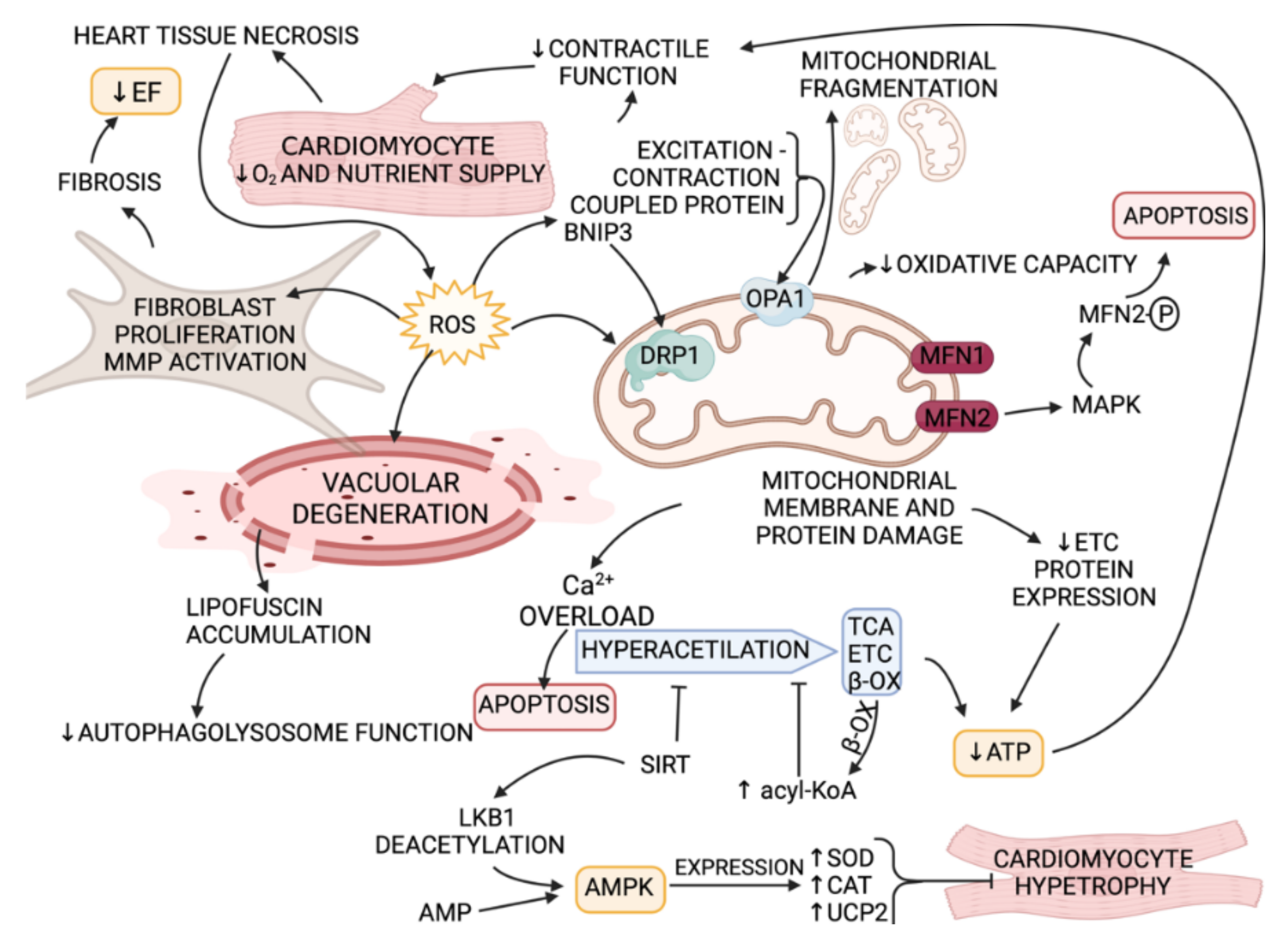{kind=link}
{kind=link}
| Medicine | Patients | Appl., Dose and Duration | Results | Pathophysiological Mechanism | Reference |
|---|---|---|---|---|---|
| Valsartan | CHF, NYHA functional class II–IV, n = 83 | 6-week study, 80 or 160 mg bid | Produced hemodynamic and hormonal effects. | Blocks angiotensin AT1 receptor leading to NOX2 activity reduction. | [191] |
| Sacubitril/valsartan (LCZ696) | HFrEF (NYHA II–IV) and LVEF ≤ 40% n = 4822 | 5 years | Decreased levels of NT-proBNP or improved left atrial volumes. | Inhibits neprilysin/ ATR | [192] |
| Sacubitril/valsartan | HFrEF, n = 54 | Twice a day 24/26, 49/51, 97/103 mg | Improved NYHA class, decreased NT-pro BNP concentration, reduced mortality. | Inhibits neprilysin/ ATR1 | [193] |
| Candesartan | HFpEF n = 1958, HFrEF, n = 1959 | 2.9 years | Improved outcomes in both groups. | ATR1 inhibitor | [194] |
| Medicine | Patients | Appl., Dose and Duration | Results | Pathophysiological Mechanism | Reference |
|---|---|---|---|---|---|
| OTC OCs | Patients with heart failure with preserved ejection fraction (HFpEF), n = 16. | 600 mg of α-lipoic acid, 1000 mg of vitamin C, and 600 IU of vitamin E | Improved peripheral vascular function regardless of changes in global markers of oxidative stress in HFpEF. | Alterations in redox balance as a result of attenuated endogenous AOx capacity and/or elevated oxidative stress might be an underlying mechanism. | [216] |
| AOx | Patients with HFrEF, n = 14. | 1 g of vitamin C, 600 IU of vitamin E, and 0 mg/day). 6 g α). -lipoic acid | Improved macrovascular function, reduced oxidative stress, and increased AOx capacity in patients with HFrEF | Changes redox balance; Increases oxidative stress; Decreases endogenous AOx protection. | [221] |
| Medicine | Patients | Appl., Dose and Duration | Results | Pathophysiological Mechanism | Reference |
|---|---|---|---|---|---|
| Elamipretide (SS-31) | HFrEF(EF ≤ 35%), n = 24 and placebo n = 12. | i.v., 4-h infusion 0.25 mg × kg−1 × h−1 | ↓LVESV, ↓LVEDV | By binding to cardiolipin, decreases ROS production. | [251] |
| Elamipretide (SS-31) | HFrEF(EF≤40%), n = 48 and placebo n = 23. | p.o., 4 mg or 40 mg once daily for 28 days. | Did not improve LVESV. | By binding to cardiolipin, decreases ROS production. | [252] |
| Coenzyme Q10 | Moderate to severe HFrEF, n = 420. | p.o., 100 mg 3 times daily, 2 years. | Significantly improved NYHA class, CV events ↓by 50%. | Q10 is involved in eNOS regulation | [243] |
| Medicine | Patients | Appl., Dose and Duration | Results | Pathophysiological Mechanism | Reference |
|---|---|---|---|---|---|
| Empagliflozin | HFpEF II–IV class (EF > 40%), n = 2997, placebo n = 2991. | 10 mg once daily or placebo 36 months | Reduced cardiovascular death and hospitalization | SLGT2 inhibitor and sGC activator | [295] |
| Sildenafil | Stable outpatient individuals with HFpEF, n = 160. | 24 weeks | Did not improve exercise capacity and clinical status. | Inhibits cGMP breakdown | [265] |
| Nitroxyl | HFpEF, n = 65. | 23 months | HNO increased cGMP concentrations and had NOX inhibitory and antihypertrophic effects in rat cardiomyocytes. | NO donor. | [255] |
| Cymlanod | HFrEF, n = 45 | 5 h i.v infusion or placebo | Slightly reduced LV and LA volumes | NO donor | [296] |
| Hydralazine | HFrEF, NYHA class III–IV (EF ≤ 35% or < 45% with LVIDd > 2.9 cm/m), n = 1050. | 18 months | Improved survival. | ROS scavenger; Inhibitor of O2−generation; normalizes endogenous rates of vascular O2−production [122]. | [286] |
| Cinaciguat | HFrEF. n = 62 | 1 year | Did not significantly improve dyspnea or cardiac index. | Increases cGMP production by targeting guanylate cyclase at the NO receptor (sGC). | [256] |
| Vericiguat | HFrEF, (LVEF < 45%, history of decompensation within the last four weeks), n = 456 | 1.25 mg, 2.5 mg, 5 mg, or 10 mg for 12 weeks | Was well-tolerated and higher doses were associated with a greater reduction in NT-pro BNP level. | Triggers cGMP production by binding the heme-containing non-oxidized form of sGC regulatory domain. | [290] |
| HFpEF ( LVEF > 45% and a history of decompensation within the last four weeks), n = 477 | from 1.25 mg to 10 mg once daily for 12 weeks | Appeared to be well-tolerated and improved patients with HFpEF quality of life; however, had no significant impact on NT-proBNP level. | [297] | ||
| HFpEF (LVEF > 45%), n = 789 | 15 mg or 10 mg daily | No significant changes were observed. | [298] | ||
| HFpEF (NYHA II to IV) with an LVEF < 45%, history of decompensation over the last six months, elevated NT-proBNP or BNP), n = 5050 | 10 mg once daily for 10.8 months | Hospitalization for heart failure and death from cardiovascular causes were reduced compared to placebo. | [299] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mongirdienė, A.; Skrodenis, L.; Varoneckaitė, L.; Mierkytė, G.; Gerulis, J. Reactive Oxygen Species Induced Pathways in Heart Failure Pathogenesis and Potential Therapeutic Strategies. Biomedicines 2022, 10, 602. https://doi.org/10.3390/biomedicines10030602
Mongirdienė A, Skrodenis L, Varoneckaitė L, Mierkytė G, Gerulis J. Reactive Oxygen Species Induced Pathways in Heart Failure Pathogenesis and Potential Therapeutic Strategies. Biomedicines. 2022; 10(3):602. https://doi.org/10.3390/biomedicines10030602
Chicago/Turabian StyleMongirdienė, Aušra, Laurynas Skrodenis, Leila Varoneckaitė, Gerda Mierkytė, and Justinas Gerulis. 2022. "Reactive Oxygen Species Induced Pathways in Heart Failure Pathogenesis and Potential Therapeutic Strategies" Biomedicines 10, no. 3: 602. https://doi.org/10.3390/biomedicines10030602