
Orchestrated Cytokines Mediated by Biologics in Psoriasis and Its Mechanisms of Action


Abstract
:1. Introduction
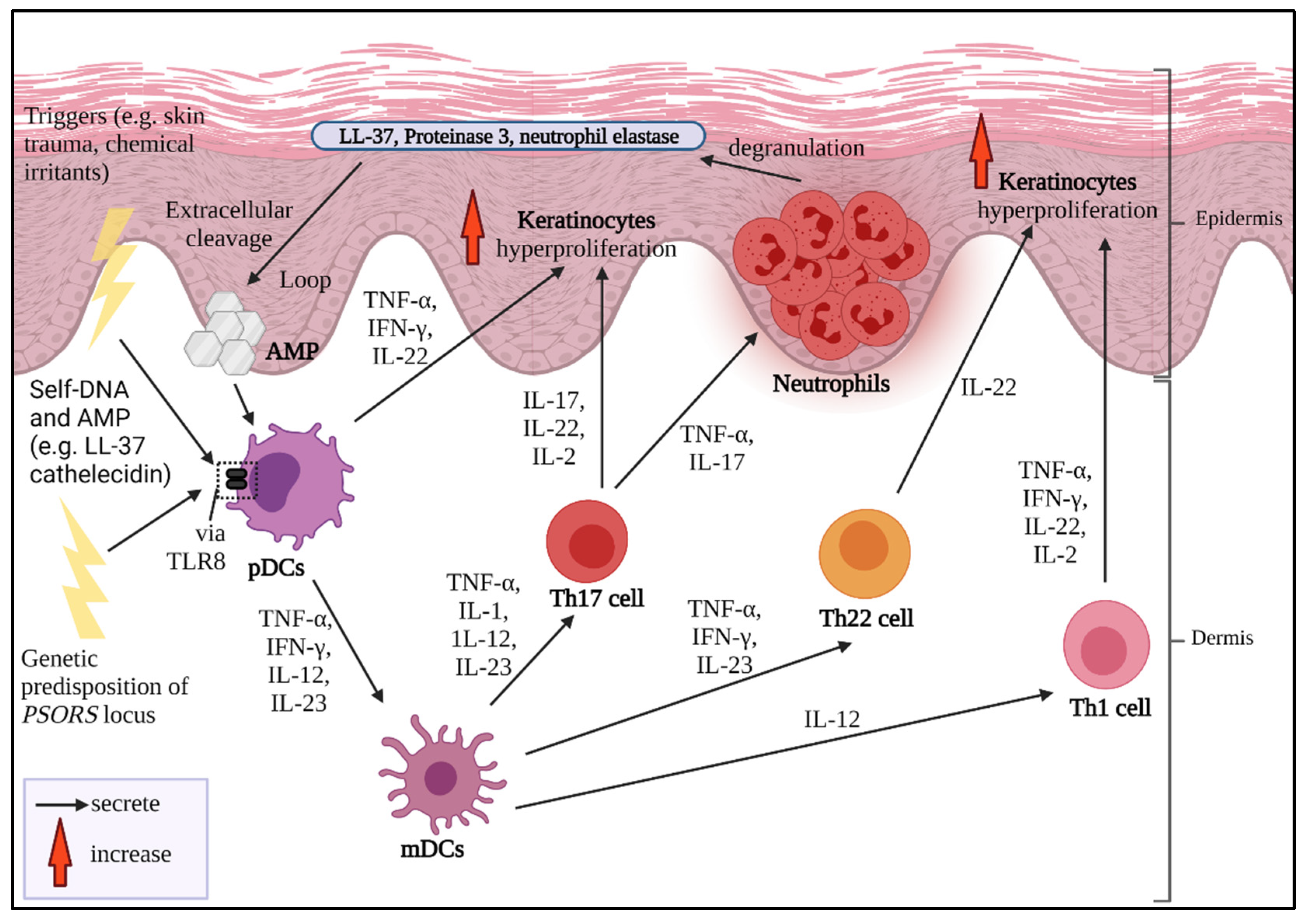
2. Pathogenesis of Psoriasis
3. Psoriasis and Cytokines as Biologics Target
4. Main Potential Cytokine Targets in Psoriasis
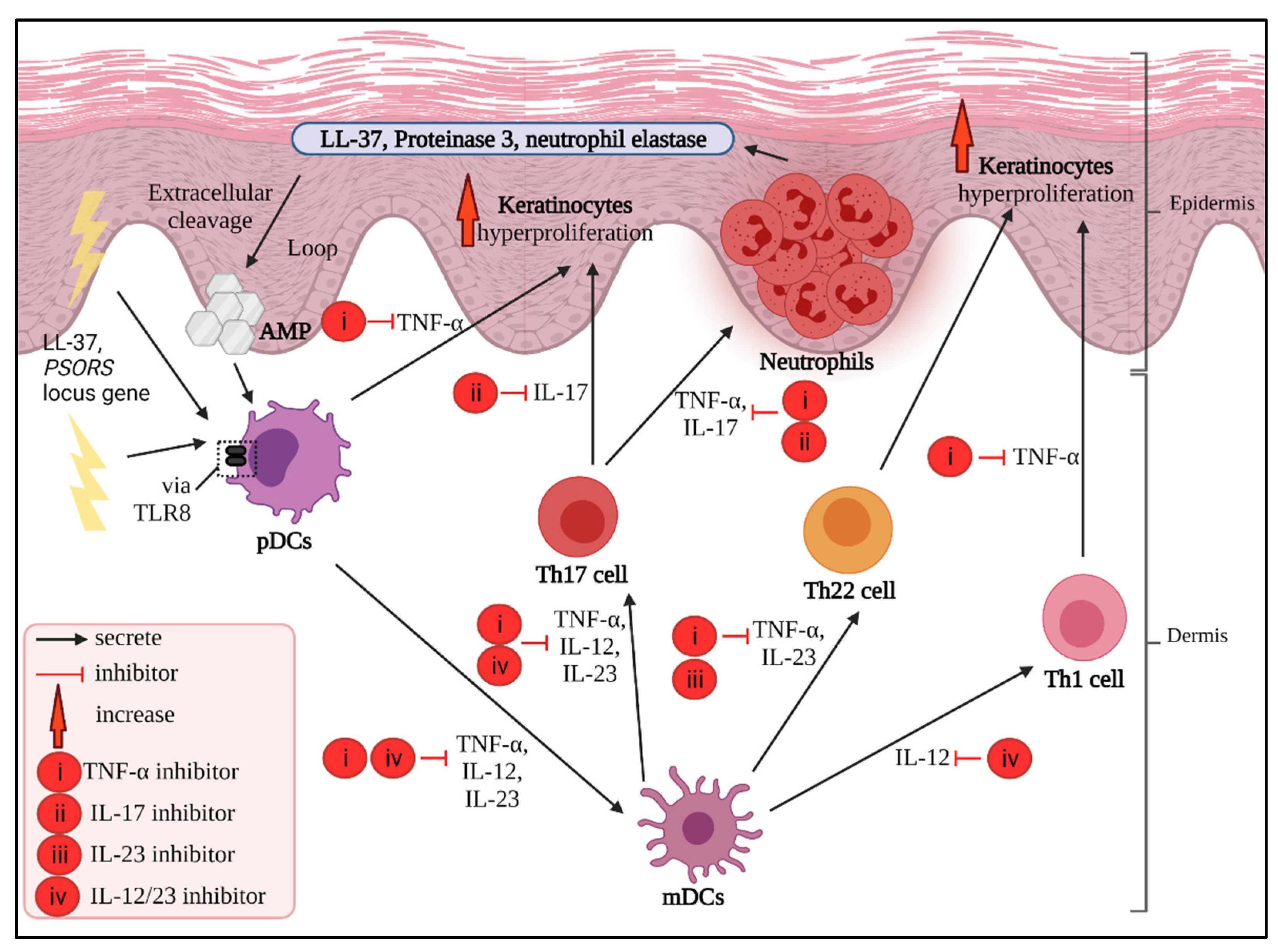
4.1. TNF-α Inhibitors
4.2. IL-17 Inhibitors
4.3. IL-23 Inhibitors
4.4. IL-12/23 Inhibitors
5. Other Pro-Inflammatory Cytokines Candidates
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamiya, K.; Kishimoto, M.; Sugai, J.; Komine, M.; Ohtsuki, M. Risk factors for the development of psoriasis. Int. J. Mol. Sci. 2019, 20, 4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Psoriasis Foundation. Get the Facts about Psoriasis and Psoriatic Arthritis. The National Psoriasis Foundation: National Psoriasis Foundation. 2017. Available online: https://www.psoriasis.org/psoriasis-statistics (accessed on 28 November 2021).
- Hayes, J.; Koo, J. Psoriasis: Depression, anxiety, smoking, and drinking habits. Dermatol. Ther. 2010, 23, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Springate, D.A.; Parisi, R.; Kontopantelis, E.; Reeves, D.; Griffiths, C.E.M.; Ashcroft, D.M. Incidence, prevalence and mortality of patients with psoriasis: A UK population-based cohort study. Adv. Exp. Med. 2017, 176, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.-H.; Lin, C.-F.; Alalaiwe, A.; Yang, S.-C.; Fang, J.-Y. Apoptotic or antiproliferative activity of natural products against keratinocytes for the treatment of psoriasis. Int. J. Mol. Sci. 2019, 20, 2558. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.O.; Kang, S.Y.; Kim, J.C.; Park, C.W.; Chung, B.Y. Pediatric psoriasis: From new insights into pathogenesis to updates on treatment. Biomedicines 2021, 9, 940. [Google Scholar] [CrossRef]
- Pithadia, D.J.; Reynolds, K.A.; Lee, E.B.; Wu, J.J. Psoriasis-associated cutaneous pain: Etiology, assessment, impact, and management. J. Dermatol. Treat. 2019, 30, 435–440. [Google Scholar] [CrossRef]
- Rajguru, J.P.; Maya, D.; Kumar, D.; Suri, P.; Bhardwaj, S.; Patel, N.D. Update on psoriasis: A review. J. Fam. Med. Prim. Care Rev. 2020, 29, 20. [Google Scholar] [CrossRef]
- Kimmel, G.W.; Lebwohl, M. Psoriasis: Overview and diagnosis. evidence-based psoriasis: Diagnosis and treatment. In Updates in Clinical Dermatology; Springer: Cham, Swirzerland, 2021; pp. 1–16. [Google Scholar] [CrossRef]
- Heidenreich, R.; Röcken, M.; Ghoreschi, K. Angiogenesis drives psoriasis pathogenesis. Int. J. Clin. Exp. Pathol. 2009, 90, 232–248. [Google Scholar] [CrossRef]
- Talaee, R.; Hajheydari, Z.; Moghaddam, A.Y.; Seyyed Alireza Moraveji, S.A.; Ravandi, B.F. Prevalence of oral mucosal lesions and their association with severity of psoriasis among psoriatic patients referred to dermatology clinic: A cross-sectional study in Kashan/Iran. Open Access Maced. J. Med. Sci. 2017, 5, 978–982. [Google Scholar] [CrossRef] [Green Version]
- Leon, A.; Liao, W.J.; Gupta, R.; Koo, J.Y.; Wu, J.J. Tumor necrosis factor-α triad: Psoriasis, cardiovascular disease, and depression. Psoriasis Forum 2013, 19, 41–49. [Google Scholar] [CrossRef]
- Mattei, P.L.; Corey, K.C.; Kimball, A.B. Psoriasis Area Severity Index (PASI) and the Dermatology Life Quality Index (DLQI): The correlation between disease severity and psychological burden in patients treated with biological therapies. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.E.; Van Geel, M.J.; Hendriks, J.C.M.; van de Kerkhof, P.C.M.; Seyger, M.M.B.; de Jong, E.M.G.J. A pilot study on the Psoriasis Area and Severity Index (PASI) for small areas: Presentation and implications of the Low PASI score. J. Dermatol. Treat. 2015, 26, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Grjibovski, A.M.; Olsen, A.O.; Magnus, P.; Harris, J.R. Psoriasis in Norwegian twins: Contribution of genetic and environmental effects. J. Eur. Acad. Dermatol. 2007, 21, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Debbaneh, M.G.; Liao, W. Genetic epidemiology of psoriasis. Curr. Dermatol. Rep. 2014, 3, 61–78. [Google Scholar] [CrossRef]
- Coimbra, S.; Oliveira, H.; Reis, F.; Belo, L.; Rocha, S.; Quintanilha, A.; Figueiredo, A.; Teixeira, F.; Castro, E.; Rocha-Pereira, P.; et al. Interleukin (IL)-22, IL-17, IL-23, IL-8, vascular endothelial growth factor and tumour necrosis factor-α levels in patients with psoriasis before, during and after psoralen–ultraviolet A and narrowband ultraviolet B therapy. Br. J. Dermatol. Suppl. 2010, 163, 1282–1290. [Google Scholar] [CrossRef]
- Baliwag, J.; Barnes, D.H.; Johnston, A. Cytokines in psoriasis. Cytokine 2015, 73, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, M.D.F.S.P.D.; Rocha, B.D.O.; Duarte, G.V. Psoriasis: Classical and emerging comorbidities. An. Bras. Dermatol. 2015, 90, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Woo, Y.R.; Park, C.J.; Kang, H.; Kim, J.E. The risk of systemic diseases in those with psoriasis and psoriatic arthritis: From mechanisms to clinic. Int. J. Mol. Sci. 2020, 21, 7041. [Google Scholar] [CrossRef]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front. Immunol. 2018, 9, 1549. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Yamasaki, K. Psoriasis and antimicrobial peptides. Int. J. Mol. Sci. 2020, 21, 6791. [Google Scholar] [CrossRef]
- Dhar, S.; Banerjee, R.; Agrawal, N.; Chatterjee, S.; Malakar, R. Psoriasis in children: An insight. Indian J. Dermatol. 2011, 56, 262. [Google Scholar] [CrossRef] [PubMed]
- Rakhshan, A.; Zarrinpour, N.; Moradi, A.; Ahadi, M.; Omrani, M.D.; Ghafouri-Fard, S.; Taheri, M. Genetic variants within ANRIL (antisense non coding RNA in the INK4 locus) are associated with risk of psoriasis. Int. Immunopharmacol. 2020, 78, 106053. [Google Scholar] [CrossRef] [PubMed]
- Kara Polat, A.; Oguz Topal, I.; Karadag, A.S.; Aksoy, H.; Koku Aksu, A.E.; Ozkur, E.; Akbulut, T.O.; Demir, F.T.; Engin, B.; Uzuncakmak, T.K.; et al. The impact of COVID-19 in patients with psoriasis: A multicenter study in Istanbul. Dermatol. Ther. 2021, 34, e14691. [Google Scholar] [CrossRef] [PubMed]
- Campanati, A.; Marani, A.; Martina, E.; Diotallevi, F.; Radi, G.; Offidani, A. Psoriasis as an immune-mediated and inflammatory systemic disease: From pathophysiology to novel therapeutic approaches. Biomedicines 2021, 9, 1511. [Google Scholar] [CrossRef]
- Lew, W.; Bowcock, A.M.; Krueger, J.G. Psoriasis vulgaris: Cutaneous lymphoid tissue supports T-cell activation and ‘Type 1’inflammatory gene expression. Trends Immunol. 2004, 25, 295–305. [Google Scholar] [CrossRef]
- Schon, M.; Behmenburg, C.; Denzer, D.; Schon, M.P. Pathogenic function of IL-1beta in psoriasiform skin lesions of flaky skin (fsn/fsn) mice. Clin. Exp. Immunol. 2001, 123, 505–510. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. Heterogeneity and plasticity of T helper cells. Cell Res. 2010, 20, 4–12. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Farinas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J. Investig. Dermatol. 2011, 131, 677–687. [Google Scholar] [CrossRef]
- Saeki, H.; Imafuku, S.; Abe, M.; Shintani, Y.; Onozuka, D.; Hagihara, A.; Katoh, N.; Murota, H.; Takeuchi, S.; Sugaya, M.; et al. Poor adherence to medication as assessed by the Morisky Medication Adherence Scale-8 and low satisfaction with treatment in 237 psoriasis patients. J. Dermatol. 2015, 42, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Nast, A.; Jacobs, A.; Rosumeck, S.; Werner, R.N. Efficacy and safety of systemic long-term treatments for moderate-to-severe psoriasis: A systematic review and meta-analysis. J. Investig. Dermatol. 2015, 135, 2641–2648. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.B.; Lebwohl, M.G. Review of safety and efficacy of approved systemic psoriasis therapies. Int. J. Dermatol. 2019, 58, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Torres, T.; Filipe, P. Small molecules in the treatment of psoriasis. Drug Dev. Res. 2015, 76, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Gisondi, P.; Del Giglio, M.; Girolomoni, G. Treatment approaches to moderate to severe psoriasis. Int. J. Mol. Sci. 2017, 18, 2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rønholt, K.; Iversen, L. Old and new biological therapies for psoriasis. Int. J. Mol. Sci. 2017, 18, 2297. [Google Scholar] [CrossRef] [Green Version]
- Ortonne, J.P.; Prinz, J.C. Alefacept: A novel and selective biologic agent for the treatment of chronic plaque psoriasis. Eur. J. Dermatol. 2004, 14, 41–45. [Google Scholar]
- Liu, C.M.; McKenna, J.K.; Krueger, G.G. Alefacept: A novel biologic in the treatment of psoriasis. Drugs Today 2004, 40, 961–974. [Google Scholar] [CrossRef]
- Langley, R.G.; Cherman, A.M.; Gupta, A.K. Alefacept: An expert review concerning the treatment of psoriasis. Expert Opin. Pharmacother. 2005, 6, 2327–2333. [Google Scholar] [CrossRef]
- Jenneck, C.; Novak, N. The safety and efficacy of alefacept in the treatment of chronic plaque psoriasis. Ther. Clin. Risk Manag. 2007, 3, 411–420. [Google Scholar]
- Lebwohl, M. Psoriasis. Lancet 2003, 361, 1197–1204. [Google Scholar] [CrossRef]
- Sivamani, R.K.; Correa, G.; Ono, Y.; Bowen, M.P.; Raychaudhuri, S.P.; Maverakis, E. Biological therapy of psoriasis. Indian J. Dermatol. 2010, 55, 161. [Google Scholar] [CrossRef]
- Bak, R.O.; Mikkelsen, J.G. Regulation of cytokines by small RNAs during skin inflammation. J. Biomed. Sci. 2010, 17, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zheng, H.; Zhou, H.; Huang, N.; Wei, X.; Liu, X.; Teng, X.; Hu, Z.; Zhang, J.; Zhou, X.; et al. Systematic screening and identification of novel psoriasis-specific genes from the transcriptome of psoriasis-like keratinocytes. Mol. Med. Rep. 2019, 19, 1529–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, A.B.; Chamian, F.; Masud, S.; Cardinale, I.; Abello, M.V.; Lowes, M.A.; Chen, F.; Magliocco, M.; Krueger, J.G. TNF inhibition rapidly down-regulates multiple proinflammatory pathways in psoriasis plaques. J. Immunol. 2005, 175, 2721–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015, 26, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, A.; Conrad, C. Psoriasis: Classical vs. paradoxical. the yin-yang of TNF and Type I interferon. Front. Immunol. 2018, 9, 2746. [Google Scholar] [CrossRef] [PubMed]
- Calzascia, T.; Pellegrini, M.; Hall, H.; Sabbagh, L.; Ono, N.; Elford, A.R.; Ohashi, P.S. TNF-α is critical for antitumor but not antiviral T cell immunity in mice. J. Clin. Investig. 2007, 117, 3833–3845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Hamano, R.; Subleski, J.J.; Hurwitz, A.A.; Howard, O.Z.; Oppenheim, J.J. Expression of costimulatory TNFR2 induces resistance of CD4+ FoxP3− conventional T cells to suppression by CD4+ FoxP3+ regulatory T cells. J. Immunol. 2010, 185, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Perez, R.; Cabaleiro, T.; Dauden, E.; Abad-Santos, F. Gene polymorphisms that can predict response to anti-TNF therapy in patients with psoriasis and related autoimmune diseases. Pharm. J. 2013, 13, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, L.; Ma, W.; Cai, D.; Zhong, H.; Sun, Q. Associations between tumor necrosis factor-a polymorphisms and risk of psoriasis: A meta-analysis. PLoS ONE 2013, 8, e68827. [Google Scholar] [CrossRef] [Green Version]
- Murdaca, G.; Gulli, R.; Spano, F.; Lantieri, F.; Burlando, M.; Parodi, A.; Mandich, P.; Puppo, F. TNF-α gene polymorphisms: Association with disease susceptibility and response to anti-TNF-α treatment in psoriatic arthritis. J. Investig. Dermatol. 2014, 134, 2503–2509. [Google Scholar] [CrossRef] [Green Version]
- Mazloom, S.E.; Yan, D.; Hu, J.Z.; Ya, J.; Husni, M.E.; Warren, C.B.; Fernandez, A.P. TNF-α inhibitor–induced psoriasis: A decade of experience at the Cleveland Clinic. JAAD 2020, 83, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Ruano, J.; Suárez-Fariñas, M.; Shemer, A.; Oliva, M.; Guttman-Yassky, E.; Krueger, J.G. Molecular and cellular profiling of scalp psoriasis reveals differences and similarities compared to skin psoriasis. PLoS ONE 2016, 11, e0148450. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Spain, S.L.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Knight, J.; Tejasvi, T.; Kang, H.M.; Allen, M.H.; Lambert, S.; et al. Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci. Nat. Commun. 2015, 6, 7001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.; Fritz, Y.; Dawes, S.M.; Diaconu, D.; Al-Attar, P.M.; Guzman, A.M.; Chen, C.S.; Fu, W.; Gudjonsson, J.E.; McCormick, T.S.; et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J. Immunol. 2013, 190, 2252–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banno, T.; Gazel, A.; Blumenberg, M. Effects of tumor necrosis factor-α (TNFα) in epidermal keratinocytes revealed using global transcriptional profiling. J. Biol. Chem. 2004, 279, 32633–32642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yost, J.; Gudjonsson, J.E. The role of TNF inhibitors in psoriasis therapy: New implications for associated comorbidities. Med. Rep. 2009, 1, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownstone, N.D.; Hong, J.; Mosca, M.; Hadeler, E.; Liao, W.; Bhutani, T.; Koo, J. Biologic treatments of psoriasis: An update for the clinician. Biol. Targets Ther. 2019, 15, 39. [Google Scholar] [CrossRef]
- Nguyen, T.U.; Koo, J. Etanercept in the treatment of plaque psoriasis. Clinical, cosmetic and investigational dermatology. Clin. Cosmet. Investig. 2009, 19, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants–past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [Green Version]
- Kivelevitch, D.; Mansouri, B.; Menter, A. Long term efficacy and safety of etanercept in the treatment of psoriasis and psoriatic arthritis. Biol. Targets Ther. 2014, 8, 169. [Google Scholar] [CrossRef] [Green Version]
- Knight, D.M.; Trinh, H.; Le, J.; Siegel, S.; Shealy, D.; McDonough, M.; Ghrayeb, J. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol. Immunol. 1993, 30, 1443–1453. [Google Scholar] [CrossRef]
- Kaymakcalan, Z.; Sakorafas, P.; Bose, S.; Scesney, S.; Xiong, L.; Hanzatian, D.K.; Salfeld, J.; Sasso, E.H. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin. Immunol. 2009, 131, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Talbot, C.; Sagar, P.M.; Johnston, M.J.; Finan, P.J.; Burke, D. Infliximab in the surgical management of complex fistulating anal Crohn’s disease. Int. J. Colorectal Dis. 2005, 7, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef] [PubMed]
- Antoni, C.; Krueger, G.G.; de Vlam, K.; Birbara, C.; Beutler, A.; Guzzo, C.; Zhou, B.; Dooley, L.T.; Kavanaugh, A. Infliximab improves signs and symptoms of psoriatic arthritis: Results of the IMPACT 2 trial. Ann. Rheum. Dis. 2005, 64, 1150–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, S.; Gong, Y.; Chen, Y.; Shi, Y. Infliximab and biosimilar infliximab in psoriasis: Efficacy, loss of efficacy, and adverse events. Drug Des. Devel. Ther. 2019, 13, 2491–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazza, J.; Rossi, A.; Weinberg, J.M. Innovative uses of tumor necrosis factor α inhibitors. Dermatol. Clin. 2010, 28, 559–575. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Zangrilli, A.; Bavetta, M.; Bianchi, L.; Chimenti, S.; Saraceno, R. Real-life 9-year experience with adalimumab in psoriasis and psoriatic arthritis: Results of a single-centre, retrospective study. J. Eur. Acad. Dermatol. 2017, 31, 304–311. [Google Scholar] [CrossRef]
- Kamata, M.; Tada, Y. Efficacy and safety of biologics for psoriasis and psoriatic arthritis and their impact on comorbidities: A literature review. Int. J. Mol. Sci. 2020, 21, 1690. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Vu, T.; Lee, H.; Hu, C.; Ling, J.; Yan, H.; Baker, D.; Beutler, A.; Pendley, C.; Wagner, C.; et al. Population pharmacokinetics of golimumab, an anti-tumor necrosis factor-α human monoclonal antibody, in patients with psoriatic arthritis. J. Clin. Pharmacol. 2009, 49, 1056–1070. [Google Scholar] [CrossRef]
- Shealy, D.J.; Cai, A.; Staquet, K.; Baker, A.; Lacy, E.R.; Johns, L.; Vafa, O.; Gunn, G.; Tam, S.; Sague, S.; et al. Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor α. MAbs Taylor Fr. 2010, 2, 428–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, K.; Burden, A.D.; Eaton, J.N.; Hawkins, N.S. Efficacy of biologics in the treatment of moderate to severe psoriasis: A network meta-analysis of randomized controlled trials. Br. J. Dermatol. 2012, 166, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Förger, F.; Abraham, B.; Flynn, A.D.; Moltó, A.; Flipo, R.M.; van Tubergen, A.; Shaughnessy, L.; Simpson, J.; Teil, M.; et al. Lack of placental transfer of certolizumab pegol during pregnancy: Results from CRIB, a prospective, postmarketing, pharmacokinetic study. ARD 2018, 77, 228–233. [Google Scholar] [CrossRef]
- Nesbitt, A.; Fossati, G.; Bergin, M.; Stephens, P.; Stephens, S.; Foulkes, R.; Brown, D.; Robinson, M.; Bourne, T. Mechanism of action of certolizumab pegol (CDP870): In vitro comparison with other anti-tumor necrosis factor α agents. Inflamm. Bowel Dis. 2007, 13, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.R.; Burls, A.; Cabello, J.B.; Casasempere, P.V.; Bort-Marti, S.; Bernal, J.A. Certolizumab pegol (CDP870) for rheumatoid arthritis in adults. Cochrane Database Syst. Rev. 2017, 9, CD007649. [Google Scholar] [CrossRef]
- Esposito, M.; Carubbi, F.; Giunta, A.; Alunno, A.; Giacomelli, R.; Fargnoli, M.C. Certolizumab pegol for the treatment of psoriatic arthritis and plaque psoriasis. Expert Rev. Clin. Immunol. 2020, 16, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.M.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis—Results of two phase 3 trials. NEJM 2014, 371, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Malakouti, M.; Brown, G.E.; Wang, E.; Koo, J.; Levin, E.C. The role of IL-17 in psoriasis. J. Dermatol. Treat. 2015, 26, 41–44. [Google Scholar] [CrossRef]
- Li, H.; Chen, J.; Huang, A.; Stinson, J.; Heldens, S.; Foster, J.; Dowd, P.; Gurney, A.L.; Wood, W.I. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc. Natl. Acad. Sci. USA 2000, 97, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 2001, 15, 985–995. [Google Scholar] [CrossRef] [Green Version]
- Hymowitz, S.G.; Filvaroff, E.H.; Yin, J.; Lee, J.; Cai, L.; Risser, P.; Maruoka, M.; Mao, W.; Foster, J.; Kelley, R.F.; et al. IL-17s adopt a cystine knot fold: Structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001, 20, 5332–5341. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. Recent advances in the IL-17 cytokine family. Curr. Opin. 2011, 23, 613–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieder, J.; Kivelevitch, D.; Menter, A. Secukinumab: A review of the anti-IL-17A biologic for the treatment of psoriasis. TACD 2018, 9, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, S.; Zheng, G.; Huang, J.; Songyang, Z.; Zhao, X.; Lin, X. Gain-of-function mutation of CARD14 leads to spontaneous psoriasis-like skin inflammation through enhanced keratinocyte response to IL-17A. Immunity 2018, 49, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Erbel, C.; Akhavanpoor, M.; Okuyucu, D.; Wangler, S.; Dietz, A.; Zhao, L.; Stellos, K.; Little, K.M.; Lasitschka, F.; Doesch, A.; et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J. Immunol. 2014, 193, 4344–4355. [Google Scholar] [CrossRef] [Green Version]
- Von Stebut, E.; Boehncke, W.H.; Ghoreschi, K.; Gori, T.; Kaya, Z.; Thaci, D.; Schäffler, A. IL-17A in psoriasis and beyond: Cardiovascular and metabolic implications. Front. Immunol. 2020, 10, 3096. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.A.; Towne, J.E.; Kricorian, G.; Klekotka, P.; Gudjonsson, J.E.; Krueger, J.G.; Russell, C.B. The emerging role of IL-17 in the pathogenesis of psoriasis: Preclinical and clinical findings. J. Investig. Dermatol. 2013, 133, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Gordon, K.B.; Blauvelt, A.; Papp, K.A.; Langley, R.G.; Luger, T.; Ohtsuki, M.; Reich, K.; Amato, D.; Ball, S.G.; Braun, D.K.; et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. NEJM 2016, 375, 345–356. [Google Scholar] [CrossRef]
- Huang, X.D.; Zhang, H.; He, M.X. Comparative and evolutionary analysis of the interleukin 17 gene family in invertebrates. PLoS ONE 2015, 10, e0132802. [Google Scholar] [CrossRef]
- Angkasekwinai, P.; Park, H.; Wang, Y.H.; Wang, Y.H.; Chang, S.H.; Corry, D.B.; Liu, Y.J.; Zhu, Z.; Dong, C. Interleukin 25 promotes the initiation of proallergic type 2 responses. Exp. Med. 2007, 204, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Wasilewska, A.; Winiarska, M.; Olszewska, M.; Rudnicka, L. Interleukin-17 inhibitors. A new era in treatment of psoriasis and other skin diseases. Postepy Dermatol Alergol. 2016, 33, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, J.G.; Fretzin, S.; Suárez-Fariñas, M.; Haslett, P.A.; Phipps, K.M.; Cameron, G.S.; McColm, J.; Katcherian, A.; Cueto, I.; White, T.; et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J. Allergy Clin. Immunol. 2012, 130, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkham, B.W.; Kavanaugh, A.; Reich, K. Interleukin-17A: A unique pathway in immune-mediated diseases: Psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology 2014, 141, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijnen, D.; Knol, E.F.; Gent, Y.Y.; Giovannone, B.; Beijn, S.J.; Kupper, T.S.; Bruijnzeel-Koomen, C.A.F.M.; Clark, R.A. CD8+ T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-γ, IL-13, IL-17, and IL-22. J. Investig. Dermatol. 2013, 133, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.T.; Hardeland, R.; Zmijewski, M.A.; Slominski, R.M.; Reiter, R.J.; Paus, R. Melatonin: A cutaneous perspective on its production, metabolism, and functions. J. Investig. Dermatol. 2018, 138, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Starnes, T.; Broxmeyer, H.E.; Robertson, M.J.; Hromas, R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J. Immunol. 2002, 2169, 642–646. [Google Scholar] [CrossRef] [Green Version]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and Th 17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef]
- Ruddy, M.J.; Wong, G.C.; Liu, X.K.; Yamamoto, H.; Kasayama, S.; Kirkwood, K.L.; Gaffen, S.L. Functional cooperation between interleukin-17 and tumor necrosis factor-α is mediated by CCAAT/enhancer-binding protein family members. J. Biol. Chem. 2004, 279, 2559–2567. [Google Scholar] [CrossRef] [Green Version]
- Laan, M.; Cui, Z.H.; Hoshino, H.; Lötvall, J.; Sjöstrand, M.; Gruenert, D.C.; Skoogh, B.E.; Lindén, A. Neutrophil recruitment by human IL-17 via CXC chemokine release in the airways. J. Immunol. 1999, 162, 2347–2352. [Google Scholar]
- Moseley, T.A.; Haudenschild, D.R.; Rose, L.; Reddi, A.H. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003, 14, 155–174. [Google Scholar] [CrossRef]
- Harper, E.G.; Guo, C.; Rizzo, H.; Lillis, J.V.; Kurtz, S.E.; Skorcheva, I.; Purdy, D.; Fitch, E.; Iordanov, M.; Blauvelt, A. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: Implications for psoriasis pathogenesis. J. Investig. Dermatol. 2009, 129, 2175–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, C.; Usher, P.A.; Kjellerup, R.B.; Lundsgaard, D.; Iversen, L.; Kragballe, K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br. J. Dermatol. Suppl. 2009, 160, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, S.B.; Cicek, N.; Coskun, M.; Yegin, O.; Alpsoy, E. Serum and tissue levels of IL-17 in different clinical subtypes of psoriasis. Arch. Dermatol. 2012, 304, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Fujishima, S.; Watanabe, H.; Kawaguchi, M.; Suzuki, T.; Matsukura, S.; Homma, T.; Howell, B.G.; Hizawa, N.; Mitsuya, T.; Huang, S.K.; et al. Involvement of IL-17F via the induction of IL-6 in psoriasis. Arch. Dermatol. 2010, 302, 499–505. [Google Scholar] [CrossRef]
- Pantelyushin, S.; Haak, S.; Ingold, B.; Kulig, P.; Heppner, F.L.; Navarini, A.A.; Becher, B. Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. J. Clin. Investig. 2012, 122, 2252–2256. [Google Scholar] [CrossRef] [Green Version]
- Soderstrom, C.; Berstein, G.; Zhang, W.; Valdez, H.; Fitz, L.; Kuhn, M.; Fraser, S. Ultra-sensitive measurement of IL-17A and IL-17F in psoriasis patient serum and skin. AAPS J. 2017, 19, 1218–1222. [Google Scholar] [CrossRef]
- Ramirez-Carrozzi, V.; Sambandam, A.; Luis, E.; Lin, Z.; Jeet, S.; Lesch, J.; Hackney, J.; Kim, J.; Zhou, M.; Lai, J.; et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 2011, 12, 1159–1166. [Google Scholar] [CrossRef]
- Chang, S.H.; Reynolds, J.M.; Pappu, B.P.; Chen, G.; Martinez, G.J.; Dong, C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity 2011, 35, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Zhu, S.; Shi, P.; Liu, Y.; Shi, Y.; Levin, S.D.; Qian, Y. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat. Immunol. 2011, 12, 1151–1158. [Google Scholar] [CrossRef]
- Song, X.; Gao, H.; Qian, Y. Th17 differentiation and their pro-inflammation function. Adv. Exp. Med. 2014, 841, 99–151. [Google Scholar] [CrossRef]
- Reszke, R.; Szepietowski, J.C. Secukinumab in the treatment of psoriasis: An update. Immunotherapy 2017, 9, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, A.; Mease, P.J.; McInnes, I.B.; Baraliakos, X.; Reich, K.; Blauvelt, A.; Leonardi, C.; Porter, B.; Gupta, A.D.; Widmer, A.; et al. Long-term safety of secukinumab in patients with moderate-to-severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis: Integrated pooled clinical trial and post-marketing surveillance data. Arthritis Res. Ther. 2019, 21, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.J.; Beck, K.M.; Liao, W. Secukinumab in the treatment of psoriasis: Patient selection and perspectives. Psoriasis (Auckl.) 2018, 8, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolbinger, F.; Loesche, C.; Valentin, M.A.; Jiang, X.; Cheng, Y.; Jarvis, P.; Peters, T.; Calonder, C.; Bruin, G.; Polus, F.; et al. β-Defensin 2 is a responsive biomarker of IL-17A–driven skin pathology in patients with psoriasis. J. Allergy Clin. Immunol. 2017, 139, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Toussirot, E. Ixekizumab: An anti-IL-17A monoclonal antibody for the treatment of psoriatic arthritis. Expert Opin. Biol. Ther. 2018, 18, 101–107. [Google Scholar] [CrossRef]
- Blegvad, C.; Skov, L.; Zachariae, C. Ixekizumab for the treatment of psoriasis: An update on new data since first approval. Expert Rev. Clin. Immunol. 2019, 15, 111–121. [Google Scholar] [CrossRef]
- Zaba, L.C.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Nograles, K.E.; Guttman-Yassky, E.; Cardinale, I.; Lowes, M.A.; Krueger, J.G. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J. Allergy Clin. Immunol. 2009, 124, 1022–1030. [Google Scholar] [CrossRef] [Green Version]
- Blauvelt, A.; Reich, K.; Tsai, T.F.; Tyring, S.; Vanaclocha, F.; Kingo, K.; Ziv, M.; Pinter, A.; Vender, R.; Hugot, S.; et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate-to-severe plaque psoriasis up to 1 year: Results from the CLEAR study. JAAD 2017, 76, 60–69. [Google Scholar] [CrossRef]
- Puig, L. Brodalumab: The first anti-IL-17 receptor agent for psoriasis. Drugs Today 2017, 53, 283–297. [Google Scholar] [CrossRef]
- Foulkes, A.C.; Warren, R.B. Brodalumab in psoriasis: Evidence to date and clinical potential. Drugs Context 2019, 8, 212570. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.B.; Rand, H.; Bigler, J.; Kerkof, K.; Timour, M.; Bautista, E.; Krueger, J.G.; Salinger, D.H.; Welcher, A.A.; Martin, D.A. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti–IL-17 receptor monoclonal antibody. J. Immunol. 2014, 192, 3828–3836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirula, A.; Nilsen, J.; Klekotka, P.; Kricorian, G.; Erondu, N.; Towne, J.E.; Russell, C.B.; Martin, D.A.; Budelsky, A.L. Effect of IL-17 receptor A blockade with brodalumab in inflammatory diseases. Rheumatology 2016, 55, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. NEJM 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Saraceno, R.; Chimenti, M.S.; Guttman-Yassky, E.; Krueger, J.G. Role of IL-23 in the pathogenesis of psoriasis: A novel potential therapeutic target? Expert Opin. Ther. Targets 2014, 18, 513–525. [Google Scholar] [CrossRef]
- Chan, T.C.; Hawkes, J.E.; Krueger, J.G. Interleukin 23 in the skin: Role in psoriasis pathogenesis and selective interleukin 23 blockade as treatment. Ther. Adv. Chronic Dis. 2018, 9, 111–119. [Google Scholar] [CrossRef]
- Chan, J.R.; Blumenschein, W.; Murphy, E.; Diveu, C.; Wiekowski, M.; Abbondanzo, S.; Bowman, E.P. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2–dependent mechanisms with implications for psoriasis pathogenesis. Exp. Med. 2006, 203, 2577–2587. [Google Scholar] [CrossRef]
- Chen, L.; Deshpande, M.; Grisotto, M.; Smaldini, P.; Garcia, R.; He, Z.; Gulko, P.S.; Lira, S.A.; Furtado, G.C. Skin expression of IL-23 drives the development of psoriasis and psoriatic arthritis in mice. Sci. Rep. 2020, 10, 8259. [Google Scholar] [CrossRef]
- Di Meglio, P.; Nestle, F.O. The role of IL-23 in the immunopathogenesis of psoriasis. F1000 Biol. 2010, 2, 40. [Google Scholar] [CrossRef] [Green Version]
- Fotiadou, C.; Lazaridou, E.; Sotiriou, E.; Ioannides, D. Targeting IL-23 in psoriasis: Current perspectives. Psoriasis: Targets Ther. 2018, 8, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Papp, K.; Thaçi, D.; Reich, K.; Riedl, E.; Langley, R.G.; Krueger, J.G.; Gottlieb, A.B.; Nakagawa, H.; Bowman, E.P.; Mehta, A.; et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br. J. Dermatol. 2015, 173, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Banaszczyk, K. Tildrakizumab in the treatment of psoriasis–literature review. Reumatologia 2019, 57, 234. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Papp, K.A.; Blauvelt, A.; Tyring, S.K.; Sinclair, R.; Thaçi, D.; Nograles, K.; Mehta, A.; Cichanowitz, N.; Li, Q.; et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): Results from two randomised controlled, phase 3 trials. Lancet 2017, 390, 276–288. [Google Scholar] [CrossRef]
- Gordon, K.B.; Duffin, K.C.; Bissonnette, R.; Prinz, J.C.; Wasfi, Y.; Li, S.; Shen, Y.K.; Szapary, P.; Randazzo, B.; Reich, K. A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. NEJM 2015, 373, 136–144. [Google Scholar] [CrossRef]
- Sweet, K.; Song, Q.; Loza, M.J.; McInnes, I.B.; Ma, K.; Leander, K.; Franks, C.; Cooper, P.; Siebert, S. Guselkumab induces robust reduction in acute phase proteins and type 17 effector cytokines in active psoriatic arthritis: Results from phase 3 trials. RMD Open 2021, 7, e001679. [Google Scholar] [CrossRef]
- Jones, L.L.; Vignali, D.A. Molecular interactions within the IL-6/IL-12 cytokine/receptor superfamily. Immunol. Res. 2011, 51, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef]
- Jeon, C.; Sekhon, S.; Yan, D.; Afifi, L.; Nakamura, M.; Bhutani, T. Monoclonal antibodies inhibiting IL-12, -23, and-17 for the treatment of psoriasis. Hum. Vaccines Immunother. 2017, 13, 2247–2259. [Google Scholar] [CrossRef] [Green Version]
- Smeltz, R.B.; Chen, J.; Ehrhardt, R.; Shevach, E.M. Role of IFN-γ in Th1 differentiation: IFN-γ regulates IL-18Rα expression by preventing the negative effects of IL-4 and by inducing/maintaining IL-12 receptor β2 expression. J. Immunol. 2002, 168, 6165–6172. [Google Scholar] [CrossRef] [Green Version]
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17–producing helper T cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef]
- El-behi, M.; Ciric, B.; Yu, S.; Zhang, G.X.; Fitzgerald, D.C.; Rostami, A. Differential effect of IL-27 on developing versus committed Th17 cells. J. Immunol. 2009, 183, 4957–4967. [Google Scholar] [CrossRef] [PubMed]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaghi, D.; Krueger, G.G. Ustekinumab: A review in the treatment of plaque psoriasis and psoriatic arthritis. J. Drugs Dermatol. 2012, 11, 160–167. [Google Scholar] [PubMed]
- Brodmerkel, C.; Li, K.; Garcet, S.; Hayden, K.; Chiricozzi, A.; Novitskaya, I.; Krueger, J.G. Modulation of inflammatory gene transcripts in psoriasis vulgaris: Differences between ustekinumab and etanercept. J. Allergy Clin. Immunol. 2019, 143, 1965–1969. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, M.; Yang, L.; Hanakawa, Y.; Dai, X.; Shirakata, Y. IFN-α enhances IL-22 receptor expression in keratinocytes: A possible role in the development of psoriasis. J. Investig. Dermatol. 2012, 2132, 1933–1937. [Google Scholar] [CrossRef] [Green Version]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Wang, Y.H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef]
- Zhang, L.J.; Sen, G.L.; Ward, N.L.; Johnston, A.; Chun, K.; Chen, Y.; Adase, C.; Sanford, J.A.; Gao, N.; Chensee, M.; et al. Antimicrobial peptide LL37 and MAVS signaling drive interferon-β production by epidermal keratinocytes during skin injury. Immunity 2016, 45, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Conrad, C.; Di Domizio, J.; Mylonas, A.; Belkhodja, C.; Demaria, O.; Navarini, A.A.; Lapointe, A.K.; French, L.E.; Vernez, M.V.; Gilliet, M. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat. Commun. 2018, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Gui, J.; Gober, M.; Yang, X.; Katlinski, K.V.; Marshall, C.M.; Sharma, M.; Werth, V.P.; Baker, D.P.; Rui, H.; Seykora, J.T.; et al. Therapeutic elimination of the type 1 interferon receptor for treating psoriatic skin inflammation. J. Investig. Dermatol. 2016, 136, 1990–2002. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Krueger, J.G.; Bowcock, A.M. Psoriasis: Genetic associations and immune system changes. Genes Immun. 2007, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shoeib, M.A.; El-Shafey, E.N.; Sonbol, A.A.; Lashin, S.E.R. Assessment of serum interferon-γ in psoriasis. Menoufia Med. J. 2015, 28, 488. [Google Scholar] [CrossRef]
- Kurtovic, N.O.; Halilovic, E.K. Serum concentrations of interferon gamma (IFN-γ) in patients with psoriasis: Correlation with clinical type and severity of the disease. Med. Arch. 2018, 72, 410. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Imai, Y.; Sakaguchi, Y.; Haneda, T.; Yamanishi, K. Serum cytokines correlated with the disease severity of generalized pustular psoriasis. Dis. Markers 2013, 34, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; Teague, H.L.; Swindell, W.R.; Baumer, Y.; Ward, N.L.; Xing, X.; Baugous, B.; Stuart, P.E.; Playford, M.; Voorhees, J.J.; et al. IFN-γ and TNF-α synergism may provide a link between psoriasis and inflammatory atherogenesis. Sci. Rep. 2017, 7, 13831. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Banno, T.; Walsh, R.; Blumenberg, M. Transcriptional responses of human epidermal keratinocytes to cytokine interleukin-1. J. Cell. Physiol. 2008, 214, 1–13. [Google Scholar] [CrossRef]
- Perera, G.K.; Di Meglio, P.; Nestle, F.O. Psoriasis. Annu. Rev. Pathol. 2012, 7, 385–422. [Google Scholar] [CrossRef]
- Cai, Y.; Xue, F.; Quan, C.; Qu, M.; Liu, N.; Zhang, Y.; Fleming, C.; Hu, X.; Zhang, H.; Weichselbaum, R.; et al. A critical role of the IL-1β–IL-1R signaling pathway in skin inflammation and psoriasis pathogenesis. J. Investig. Dermatol. 2019, 139, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Johnston, A.; Xing, X.; Guzman, A.M.; Riblett, M.; Loyd, C.M.; Ward, N.L.; Gudjonsson, J.E. IL-1F5, -F6, -F8, and-F9: A novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J. Immunol. Res. 2011, 186, 2613–2622. [Google Scholar] [CrossRef] [Green Version]
- Foster, A.M.; Baliwag, J.; Chen, C.S.; Guzman, A.M.; Stoll, S.W.; Gudjonsson, J.E.; Ward, N.L.; Johnston, A. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J. Immunol. 2014, 192, 6053–6061. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, L.; Morelli, M.; Scarponi, C.; Eisenmesser, E.Z.; Doti, N.; Pagnanelli, G.; Gubinelli, E.; Mazzanti, C.; Cavani, A.; Ruvo, M.; et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis. 2018, 9, 1104. [Google Scholar] [CrossRef]
- Saggini, A.; Chimenti, S.; Chiricozzi, A. IL-6 as a druggable target in psoriasis: Focus on pustular variants. J. Immunol. Res. 2014, 2014, 964069. [Google Scholar] [CrossRef] [PubMed]
- Lise, M.L.Z.; Baptista, T.S.A.; Petersen, L.E.; Bauer, M.E.; Ungaretti, C.A.L.; Torres, E.; Harter, K.; Staub, H.L. Subclinical atherogenesis in patients with mild psoriasis: A role for IL-6? Rev. Assoc. Med. Bras. 2017, 63, 747–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, H.; Koga, T.; Kohda, F.; Hara, H.; Urabe, K.; Furue, M. Interleukin-8-positive neutrophils in psoriasis. J. Dermatol. Sci. 2001, 26, 119–124. [Google Scholar] [CrossRef]
- Zalewska, A.; Głowacka, E.; Wyczółkowska, J.; Tchórzewski, H.; Narbutt, J.; Sysa-Jȩdrzejowska, A. Interleukin 6 and 8 levels in plasma and fibroblast cultures in psoriasis. Mediat. Inflamm. 2006, 2006, 081767. [Google Scholar] [CrossRef] [Green Version]
- Villanova, F.; Di Meglio, P.; Nestle, F.O. Biomarkers in psoriasis and psoriatic arthritis. ARD 2013, 72, ii104–ii110. [Google Scholar] [CrossRef] [PubMed]
- Lüthje, K.; Kallies, A.; Shimohakamada, Y.; Belz, G.T.; Light, A.; Tarlinton, D.M.; Nutt, S.L. The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat. Immunol. 2012, 13, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.L.; Yang, H.Y.; Wang, F.F.; Zhang, X.X.; Bai, Y.P. Interleukin-21 is associated with the severity of psoriasis vulgaris through promoting CD4+ T cells to differentiate into Th17 cells. Am. J. Transl. Res. 2016, 8, 3188. [Google Scholar]
- Shi, Y.; Chen, Z.; Zhao, Z.; Yu, Y.; Fan, H.; Xu, X.; Gu, J. IL-21 induces an imbalance of Th17/Treg cells in moderate-to-severe plaque psoriasis patients. Front. Immunol. 2019, 10, 1865. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, V.; Rus, H.; Chen, C.; Rus, V. CTL-promoting effects of IL-21 counteract murine lupus in the parent→ F1 graft-versus-host disease model. J. Immunol. 2016, 196, 1529–1540. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.J.; Xu, Z.; Parthasarathy, U.; Drashansky, T.T.; Helm, E.Y.; Zuniga, A.N.; Lorentsen, K.J.; Mansouri, S.; Cho, J.Y.; Edelmann, M.J.; et al. Hectd3 promotes pathogenic Th17 lineage through Stat3 activation and Malt1 signaling in neuroinflammation. Nat. Commun. 2019, 10, 701. [Google Scholar] [CrossRef] [Green Version]
- Venken, K.; Jacques, P.; Mortier, C.; Labadia, M.E.; Decruy, T.; Coudenys, J.; Hoyt, K.; Wayne, A.; Hughes, R.; Turner, M.; et al. RORγt inhibition selectively targets IL-17 producing iNKT and γδ-T cells enriched in Spondyloarthritis patients. Nat. Commun. 2019, 10, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Li, S.; Ying, S.; Tang, S.; Ding, Y.; Li, Y.; Qiao, J.; Fang, H. The IL-23/IL-17 pathway in inflammatory skin diseases: From bench to bedside. Front. Immunol. 2020, 11, 2971. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Eidenschenk, C.; Ouyang, W. IL-22, not simply a Th17 cytokine. Immunol. Rev. 2013, 252, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Sa, S.M.; Valdez, P.A.; Wu, J.; Jung, K.; Zhong, F.; Hall, L.; Kasman, I.; Winer, J.; Modrusan, Z.; Danilenko, D.M.; et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J. Immunol. 2007, 178, 2229–2240. [Google Scholar] [CrossRef]
- Sabat, R.; Ouyang, W.; Wolk, K. Therapeutic opportunities of the IL-22–IL-22R1 system. Nat. Rev. Drug Discov. 2014, 13, 21–38. [Google Scholar] [CrossRef]
- Cordoro, K.M.; Hitraya-Low, M.; Taravati, K.; Sandoval, P.M.; Kim, E.; Sugarman, J.; Pauli, M.L.; Liao, W.; Rosenblum, M.D. Skin-infiltrating, interleukin-22–producing T cells differentiate pediatric psoriasis from adult psoriasis. JAAD 2017, 77, 417–424. [Google Scholar] [CrossRef]
- Zhuang, L.; Ma, W.; Yan, J.; Zhong, H. Evaluation of the effects of IL-22 on the proliferation and differentiation of keratinocytes in vitro. Mol. Med. Rep. 2020, 22, 2715–2722. [Google Scholar] [CrossRef]
- Nikamo, P.; Cheuk, S.; Lysell, J.; Enerbäck, C.; Bergh, K.; Landén, N.X.; Eidsmo, L.; Ståhle, M. Genetic variants of the IL22 promoter associate to onset of psoriasis before puberty and increased IL-22 production in T cells. J. Investig. Dermatol. 2014, 134, 1535–1541. [Google Scholar] [CrossRef] [Green Version]
- Voglis, S.; Moos, S.; Kloos, L.; Wanke, F.; Zayoud, M.; Pelczar, P.; Giannou, A.D.; Pezer, S.; Albers, M.; Luessi, F.; et al. Regulation of IL-22BP in psoriasis. Sci. Rep. 2018, 8, 5085. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.C.; Tsai, T.F. Anti-interleukin and interleukin therapies for psoriasis: Current evidence and clinical usefulness. TAMD 2017, 9, 277–294. [Google Scholar] [CrossRef]
- Nickoloff, B.J.; Nestle, F.O. Recent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunities. J. Clin. Investig. 2004, 113, 166. [Google Scholar] [CrossRef]
- Olson, J.L.; Courtney, R.J.; Rouhani, B.; Mandava, N.; Dinarello, C.A. Intravitreal anakinra inhibits choroidal neovascular membrane growth in a rat model. Ocul. Immunol. Inflamm. 2009, 17, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-α production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cytokine Targets | Biologic Drug Name (Brand) | Year of FDA Approval for Psoriasis Treatment | Molecular Structure | Mode of Action | Possible Side Effects | References |
|---|---|---|---|---|---|---|
| TNF-α | Infliximab (Remicade®) | 2006 | Human-mouse chimeric combination of mAb IgG1 | Inhibit circulating and transmembrane-bound TNF-α | Upper respiratory tract infection, hepatotoxicity, tuberculosis risk, worsening psoriasis | [63,65,66] |
| Etanercept (Enbrel®) | 2004 | Extracellular region of TNFR2 fusion with humanized mAb IgG1 | Inhibit soluble and non-membrane-bound circulatory TNF-α from binding to TNFR2 receptor | Upper and lower respiratory tract infections, rhinitis, pharyngitis, tuberculosis risk | [60,61] | |
| Adalimumab (Humira®) | 2008 | Humanized mAb IgG1 | Inhibit circulating and transmembrane-bound TNF-α | Upper respiratory tract infection, sinusitis, urinary tract infection | [64,69] | |
| Golimumab (Simponi®) | Not applicable * | Humanized mAb IgG1κ | Inhibit circulating and transmembrane-bound TNF-α | Recurring psoriasis flare | [72] | |
| Certolizumab pegol (Cimzia®) | Not applicable * | Humanized Fab subunit to mAb fusion, with Fc-free PEGylation and no Fc region | Inhibit circulating and transmembrane-bound TNF-α | Urinary tract infections, gastroenteritis, nasopharyngitis, headache, pruritus, tuberculosis risk | [75] | |
| IL-17 | Secukinumab (Cosentyx®) | 2015 | Humanized mAb IgG1 | Inhibit IL-17A and IL-17F | Nasopharyngitis, diarrhea, mucocutaneous candidiasis, upper respiratory tract infection, neutropenia | [114,117] |
| Ixekizumab (Taltz®) | 2016 | Humanized mAb IgG4 | Inhibit IL-17A | Candidiasis, irritable bowel syndrome, neutropenia | [118] | |
| Brodalumab (Siliq®) | 2017 | Humanized mAb IgG2 | Block IL-17A and IL-17C receptors | Arthralgia, headaches, fatigue | [122,124,125] | |
| IL-23 | Tildrakizumab (Ilumya®) | 2018 | Humanized mAb IgG1κ | Inhibit IL-23 alpha subunit; p19 subunit | Inflammatory bowel syndrome, acute myocardial infarction | [122,136] |
| Guselkumab (Tremfya®) | 2017 | Humanized mAb IgG1λ | Inhibit IL-23 alpha subunit; p19 subunit | Upper respiratory tract, nasopharyngitis, headaches, infection | [122,136] | |
| Risankizumab (Skyrizi®) | 2019 | Humanized mAb IgG1 | Inhibit IL-23A | Nasopharyngitis, upper respiratory tract infection, headache, arthralgia, back pain, diarrhea | [122,136] | |
| IL-12/23 | Ustekinumab (Stelara®) | 2009 | Humanized mAb IgG1 | Simultaneously inhibit p40 subunit of IL-12 and IL-23 | Tuberculosis risk | [140,145] |
| Cytokines Target | Mode of Action in Psoriasis | Expected Biological Inhibitory Activities in Psoriasis | Expected Side Effects | References |
|---|---|---|---|---|
| Type I IFN (-α, -β) |
|
| Not yet documented | [47,147,182,183,184,185] |
| Type II IFN (-γ) |
|
| Not yet documented | [154,182,183] |
| IL-1β |
|
| Recurring psoriasis flare | [159,182,183] |
| IL-36 |
|
| Not yet documented | [161,182] |
| IL-6 |
|
| Inducing psoriasis onset | [30,161,182] |
| IL-8 |
|
| Irritation, pain, itch, edema | [10,161,167,182] |
| IL-21 |
|
| Not yet documented | [169,170,171,182] |
| IL-17/IL-23 combination |
|
| Not yet documented | [86,174,182] |
| IL-22 |
|
| Not yet documented | [176,177,182] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohd Noor, A.A.; Azlan, M.; Mohd Redzwan, N. Orchestrated Cytokines Mediated by Biologics in Psoriasis and Its Mechanisms of Action. Biomedicines 2022, 10, 498. https://doi.org/10.3390/biomedicines10020498
Mohd Noor AA, Azlan M, Mohd Redzwan N. Orchestrated Cytokines Mediated by Biologics in Psoriasis and Its Mechanisms of Action. Biomedicines. 2022; 10(2):498. https://doi.org/10.3390/biomedicines10020498
Chicago/Turabian StyleMohd Noor, Aina Akmal, Maryam Azlan, and Norhanani Mohd Redzwan. 2022. "Orchestrated Cytokines Mediated by Biologics in Psoriasis and Its Mechanisms of Action" Biomedicines 10, no. 2: 498. https://doi.org/10.3390/biomedicines10020498