
Uremic Toxin Lanthionine Induces Endothelial Cell Mineralization In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Experimental Design
2.3. Measurement of the Intracellular Ca2+
2.4. RNA Extraction
2.5. Reverse Transcription, PCR and qPCR
2.6. Protein Extraction
2.7. Western Blotting Analysis
2.8. Alizarin Red S Staining
2.9. Alkaline Phosphatase Activity Assay
2.10. Statistical Analysis
3. Results
3.1. Lanthionine Increases Intracellular Calcium Levels
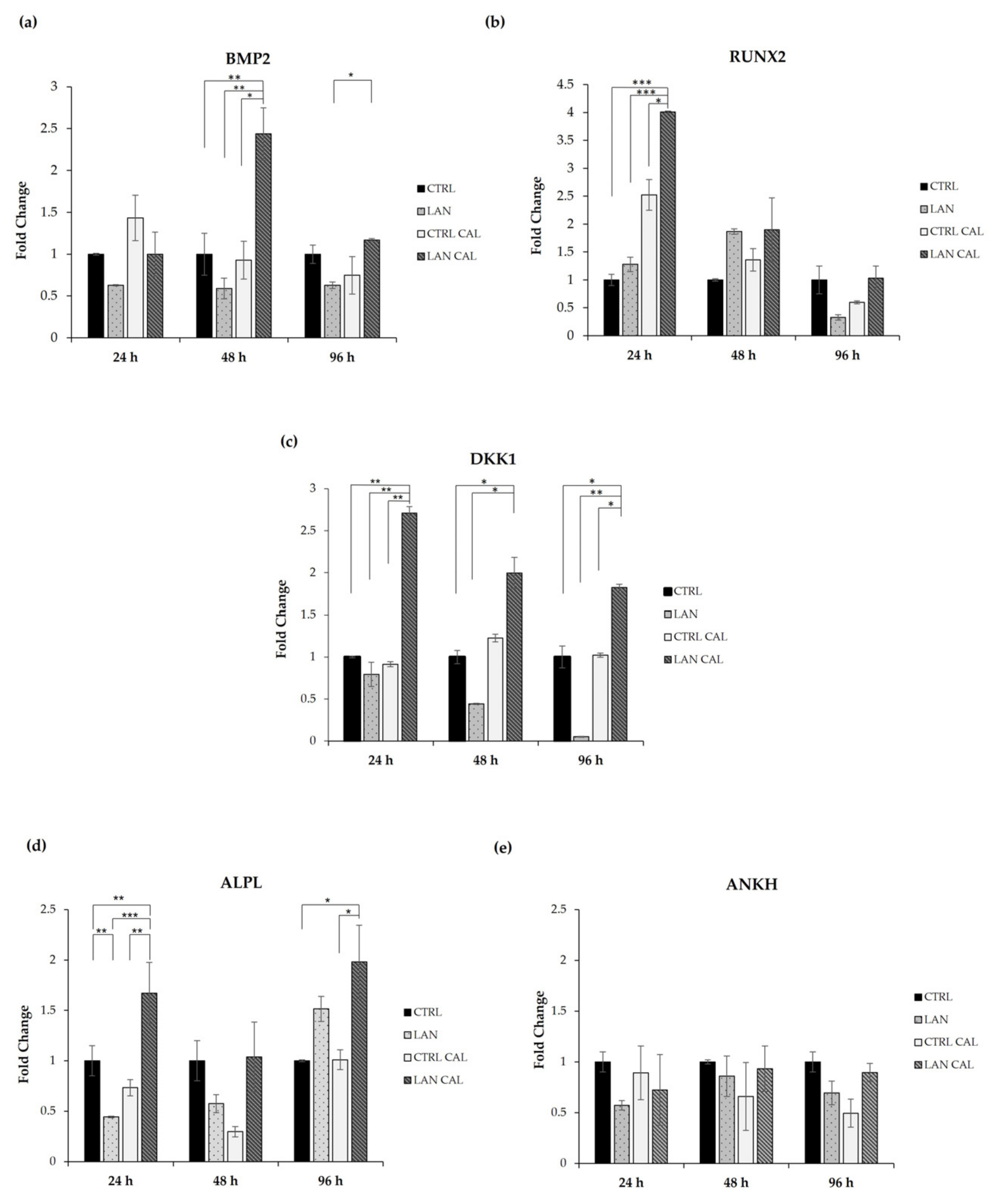
3.2. Changes in Gene Expression Involved in the Earliest Phase of Mineralizing Process
3.3. Effects of Lanthionine Alone and under Pro-Calcific Microenvironment on pERK/ERK and DKK1 Expression in EA.hy926 Cells
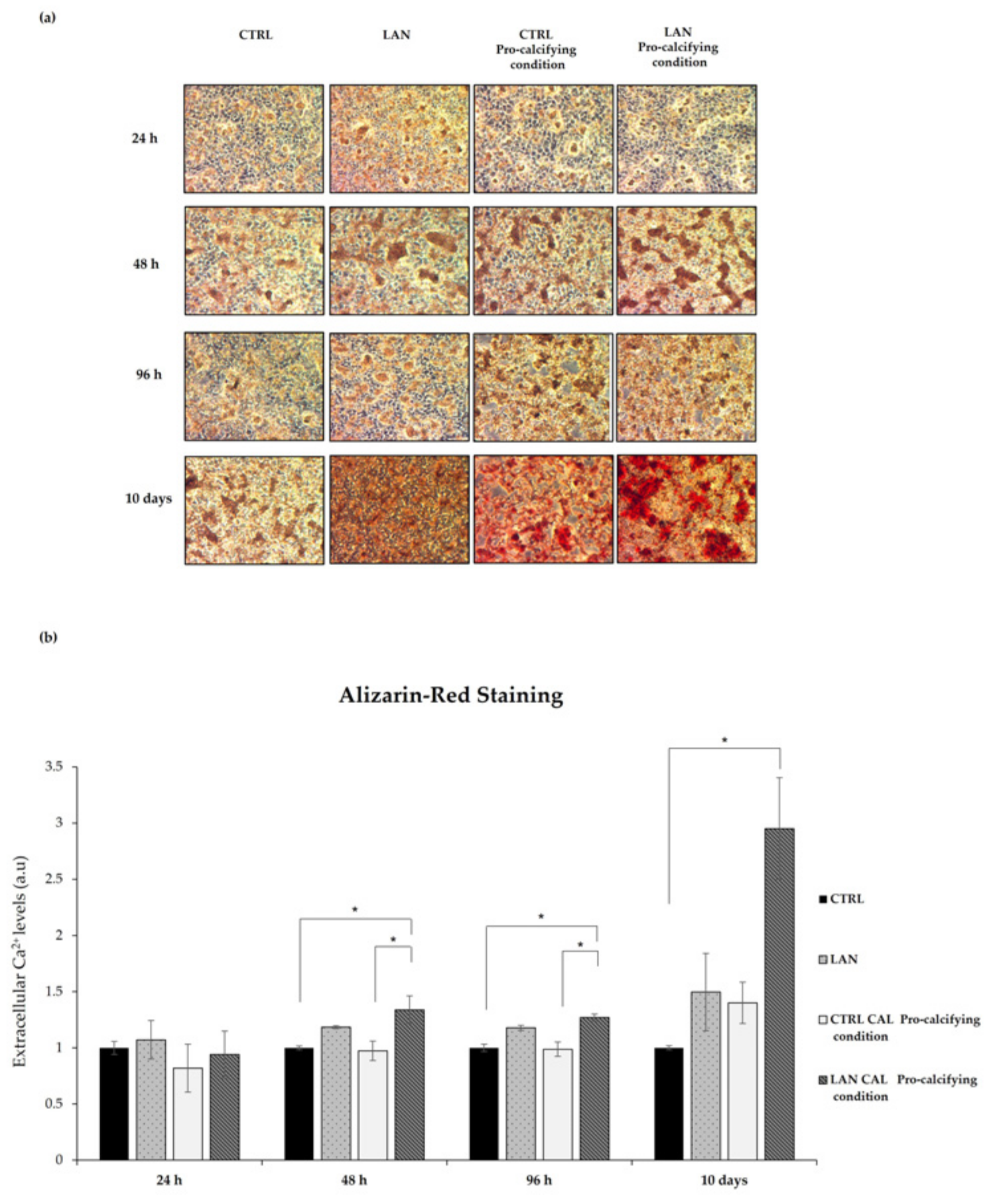
3.4. Lanthionine Effects on Calcium Deposition in EA.hy926 Cell
3.5. Alkaline Phosphatase Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jankowski, J.; Floege, J.; Böhm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, C.S.; Slatopolsky, E. Phosphate Toxicity in CKD: The Killer among Us. Clin. J. Am. Soc. Nephrol. 2016, 11, 1088–1100. [Google Scholar] [CrossRef] [Green Version]
- Kritmetapak, K.; Kumar, R. Phosphate as a Signaling Molecule. Calcif. Tissue Int. 2021, 108, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Bergwitz, C.; Harald, J. Phosphate sensing. Adv. Chronic Kidney Dis. 2011, 18, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Adeney, K.L.; Siscovick, D.S.; Ix, J.H.; Seliger, S.L.; Shlipak, M.G.; Jenny, N.S.; Kestenbaum, B.R. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J. Am. Soc. Nephrol. 2009, 20, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, N.; Fujimori, T.; Nishiguchi, S.; Tamori, A.; Shiomi, S.; Nakatani, T.; Sugimura, K.; Kishimoto, T.; Kinoshita, S.; Kuroki, T.; et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochem. Biophys. Res. Commun. 2001, 280, 1015–1020. [Google Scholar] [CrossRef]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutierrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [Green Version]
- Kovesdy, C.P.; Kuchmak, O.; Lu, J.L.; Kalantar-Zadeh, K. Outcomes associated with serum calcium level in men with non-dialysis-dependent chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 468–476. [Google Scholar] [CrossRef]
- Hannan, F.M.; Kallay, E.; Chang, W.; Brandi, M.L.; Thakker, R.V. The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat. Rev. Endocrinol. 2018, 15, 33–51. [Google Scholar] [CrossRef]
- Brown, E. Clinical lessons from the calcium-sensing receptor. Nat. Rev. Endocrinol. 2007, 3, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D. Vitamin D: Production, metabolism, and mechanisms of action. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: Dartmouth, MA, USA, 2017. [Google Scholar]
- Franca, G.P.H.; Wolley, M.; Ranganathan, D.; Seguro, A.C. Vitamin D Deficiency in Chronic Kidney Disease: Recent Evidence and Controversies. Int. J. Environ. Res. Public Health 2018, 15, 1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, G.; Souberbielle, J.C.; Chazot, C. Vitamin D in Chronic Kidney Disease and Dialysis Patients. Nutrients 2017, 9, 328. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M. Fibroblast growth factor 23 and disordered vitamin D metabolism in chronic kidney disease: Updating the “trade-off” hypothesis. Clin. J. Am. Soc. Nephrol. 2010, 9, 710–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Jüppner, H.; Wolf, M. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2205–2215. [Google Scholar] [CrossRef]
- Perna, A.F.; Pizza, A.; Di Nunzio, A.; Bellantone, R.; Raffaelli, M.; Cicchella, T.; Conzo, G.; Santini, L.; Zacchia, M.; Trepiccione, F.; et al. ADAM17, a New Player in the Pathogenesis of Chronic Kidney Disease-Mineral and Bone Disorder. J. Ren. Nutr. 2017, 6, 453–457. [Google Scholar] [CrossRef]
- Munjal, A.; Bordoni, B. Histology, Vascular; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Yao, Y.; Jumabay, M.; Ly, A.; Radparvar, M.; Cubberly, M.R. Bostrom KI: A role for the endothelium in vascular calcification. Circ. Res. 2013, 11, 495–504. [Google Scholar] [CrossRef]
- Meng, F.; Zhao, Y.; Wang, B.; Li, B.; Sheng, Y.; Liu, M.; Li, H.; Xiu, R. Endothelial Cells Promote Calcification in Aortic Smooth Muscle Cells from Spontaneously Hypertensive Rats. Cell. Physiol. Biochem. 2018, 49, 2371–2381. [Google Scholar] [CrossRef]
- Alique, M.; Ruíz-Torres, M.P.; Bodega, G.; Noci, M.V.; Troyano, N.; Bohórquez, L.; Luna, C.; Luque, R.; Carmona, A.; Carracedo, J.; et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging 2017, 9, 778–789. [Google Scholar] [CrossRef] [Green Version]
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Choukroun, G.; Kamel, S.; Bennis, Y. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol. Dial. Transplant. 2019, 34, 1125–1134. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, J.; Yao, Y.; Boström, K.I. Contributions of the Endothelium to Vascular Calcification. Front. Cell Dev. Biol. 2021, 9, 620882. [Google Scholar] [CrossRef]
- Alique, M.; Bodega, G.; Corchete, E.; García-Menéndez, E.; de Sequera, P.; Luque, R.; Rodríguez-Padrón, D.; Marqués, M.; Portolés, J.; Carracedo, J.; et al. Microvesicles from indoxyl sulfate-treated endothelial cells induce vascular calcification in vitro. Comput. Struct. Biotechnol. J. 2020, 9, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.C.; Aikawa, E. Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front. Cardiovasc. 2018, 5, 187. [Google Scholar] [CrossRef] [Green Version]
- Cola, C.; Almeida, M.; Li, D.; Romeo, F.; Mehta, J.L. Regulatory role of endothelium in the expression of genes affecting arterial calcification. Biochem. Biophys. Res. Commun. 2004, 320, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Osako, M.K.; Nakagami, H.; Koibuchi, N.; Shimizu, H.; Nakagami, F.; Koriyama, H.; Shimamura, M.; Miyake, T.; Rakugi, H.; Morishita, R. Estrogen inhibits vascular calcification via vascular RANKL system: Common mechanism of osteoporosis and vascular calcification. Circ. Res. 2010, 107, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Lomashvili, K.A.; Garg, P.; Narisawa, S.; Millan, J.L.; O’Neill, W.C. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: Potential mechanism for uremic vascular calcification. Kidney Int. 2008, 73, 1024–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Hortells, L.; Sosa, C.; Guillén, N.; Lucea, S.; Millán, Á.; Sorribas, V. Identifying early pathogenic events during vascular calcification in uremic rats. Kidney Int. 2017, 92, 1384–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teplyuk, N.M.; Haupt, L.M.; Ling, L.; Dombrowski, C.; Mun, F.K.; Nathan, S.S.; Lian, J.B.; Stein, J.L.; Stein, G.S.; Cool, S.M.; et al. The osteogenic transcription factor Runx2 regulates components of the fibroblast growth factor/proteoglycan signaling axis in osteoblasts. J. Cell. Biochem. 2009, 107, 144–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Chen, C.; Xue, R.; Wang, Y.; Dong, B.; Li, J.; Chen, C.; Jiang, J.; Fan, W.; Liang, Z. Maf1 ameliorates cardiac hypertrophy by inhibiting RNA polymerase III through ERK1/2. Theranostics 2019, 9, 7268–7281. [Google Scholar] [CrossRef]
- Ge, C.; Xiao, G.; Jiang, D.; Yang, Q.; Hatch, N.E.; Roca, H.; Franceschi, R.T. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J. Biol. Chem. 2009, 284, 32533–32543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, A.F.; Russo, L.; D’Esposito, V.; Formisano, P.; Bruzzese, D.; Vigorito, C.; Coppola, A.; Lombari, P.; Russo, D.; Ingrosso, D. Lanthionine, a Novel Uremic Toxin, in the Vascular Calcification of Chronic Kidney Disease: The Role of Proinflammatory Cytokines. Int. J. Mol. Sci. 2021, 22, 6875. [Google Scholar] [CrossRef] [PubMed]
- Vigorito, C.; Anishchenko, E.; Mele, L.; Capolongo, G.; Trepiccione, F.; Zacchia, M.; Lombari, P.; Capasso, R.; Ingrosso, D.; Perna, A.F. Uremic Toxin Lanthionine Interferes with the Transsulfuration Pathway, Angiogenetic Signaling and Increases Intracellular Calcium. Int. J. Mol. Sci. 2019, 20, 2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winslow, R.L.; Walker, M.A.; Greenstein, J.L. Modeling calcium regulation of contraction, energetics, signaling, and transcription in the cardiac myocyte. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 37–67. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.K. Calcium Signaling and Gene Expression. Adv. Exp. Med. Biol. 2020, 1131, 537–545. [Google Scholar] [PubMed]
- Ewence, A.E.; Bootman, M.; Roderick, H.L.; Skepper, J.N.; McCarthy, G.; Epple, M.; Neumann, M.; Shanahan, C.M.; Proudfoot, D. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: A potential mechanism in atherosclerotic plaque destabilization. Circ. Res. 2008, 103, e28–e34. [Google Scholar] [CrossRef] [Green Version]
- Zeng, P.; Yang, J.; Liu, L.; Yang, X.; Yao, Z.; Ma, C.; Zhu, H.; Su, J.; Zhao, Q.; Feng, K.; et al. ERK1/2 inhibition reduces vascular calcification by activating miR-126-3p-DKK1/LRP6 pathway. Theranostics 2021, 11, 1129–1146. [Google Scholar] [CrossRef]
- Pinzone, J.J.; Hall, B.M.; Thudi, N.K.; Vonau, M.; Qiang, Y.W.; Rosol, T.J.; Shaughnessy, J.D., Jr. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 2009, 113, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Mao, B.; Wu, W.; Li, Y.; Hoppe, D.; Stannek, P.; Glinka, A.; Niehrs, C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 2001, 411, 321–325. [Google Scholar] [CrossRef]
- Srikanth, M.P.; Feldman, R.A. Elevated Dkk1 Mediates Downregulation of the Canonical Wnt Pathway and Lysosomal Loss in an iPSC Model of Neuronopathic Gaucher Disease. Biomolecules 2020, 10, 1630. [Google Scholar] [CrossRef]
- Ueland, T.; Otterdal, K.; Lekva, T.; Halvorsen, B.; Gabrielsen, A.; Sandberg, W.J.; Paulsson-Berne, G.; Pedersen, T.M.; Folkersen, L.; Gullestad, L.; et al. Dickkopf-1 enhances inflammatory interaction between platelets and endothelial cells and shows increased expression in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1228–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, D.; Perna, A.F.; Oliva, A.; Vanholder, R.; Pletinck, A.; Guastafierro, S.; Di Nunzio, A.; Vigorito, C.; Capasso, G.; Jankowski, V.; et al. Impact of the uremic milieu on the osteogenic potential of mesenchymal stem cells. PLoS ONE 2015, 10, e0116468. [Google Scholar]
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol. Renal. Physiol. 2014, 307, F891–F900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zununi Vahed, S.; Mostafavi, S.; Hosseiniyan Khatibi, S.M.; Shoja, M.M.; Ardalan, M. Vascular Calcification: An Important Understanding in Nephrology. Vasc. Health Risk Manag. 2020, 16, 167–180. [Google Scholar] [CrossRef]
- Lumps, B.; Hamartomas, J.S. Prendiville, in neonatal dermatology. In Metastatic Calcification, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Perna, A.F.; D’Aniello, A.; Lowenson, J.D.; Clarke, S.; De Santo, N.G.; Ingrosso, D. D-Aspartate Content of Erytrocyte Membrane Proteins id Decreased in Uremia: Implication of the Repair of Damages Proteins. J. Am. Soc. Nephrol. 1997, 8, 95–104. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; Satta, E.; Lombardi, C.; Galletti, P.; D’Aniello, A.; De Santo, N.G. Plasma protein aspartyl damage is increased in hemodialysis patients: Studies on causes and consequences. J. Am. Soc. Nephrol. 2004, 15, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Perna, A.F.; Ingrosso, D. Low hydrogen sulphide and chronic kidney disease: A dangerous liaison. Nephrol. Dial. Transplant. 2012, 27, 486–493. [Google Scholar] [CrossRef]
- Perna, A.F.; Di Nunzio, A.; Amoresano, A.; Pane, F.; Fontanarosa, C.; Pucci, P.; Vigorito, C.; Cirillo, G.; Zacchia, M.; Trepiccione, F.; et al. Divergent behavior of hydrogen sulfide pools and of the sulfur metabolite lanthionine, a novel uremic toxin, in dialysis patients. Biochimie 2016, 126, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Perna, A.F.; Anishchenko, E.; Vigorito, C.; Zacchia, M.; Trepiccione, F.; D’Aniello, S.; Ingrosso, D. Zebrafish, a Novel Model System to Study Uremic Toxins: The Case for the Sulfur Amino Acid Lanthionine. Int. J. Mol. Sci. 2018, 19, 1323. [Google Scholar] [CrossRef] [Green Version]
- Freise, C.; Querfeld, U.; Ludwig, A.; Hamm, B.; Schnorr, J.; Taupitz, M. Uraemic extracellular vesicles augment osteogenic transdifferentiation of vascular smooth muscle cells via enhanced AKT signalling and PiT-1 expression. J. Cell. Mol. Med. 2021, 25, 5602–5614. [Google Scholar] [CrossRef]
- Dongdong, W.; Zhu, C.; Andy, W.K.Y.; Atanas, G. Differences between common endothelial cell models primary human aortic endothelial cells and EA. hy926 cells revealed through transcriptomics, bioinformatics, and functional analysis. Curr. Res. Biotechnol. 2021, 3, 2590–2628. [Google Scholar]
- Mackenzie, N.C.; Zhu, D.; Longley, L.; Patterson, C.S.; Kommareddy, S.; MacRae, V.E. MOVAS-1 cell line: A new in vitro model of vascular calcification. Int. J. Mol. Med. 2011, 27, 663–668. [Google Scholar] [PubMed] [Green Version]
- Zhang, P.; Wang, A.P.; Yang, H.P.; Ai, L.; Zhang, H.J.; Wang, Y.M.; Bi, Y.L.; Fan, H.H.; Gao, J.; Zhang, H.Y.; et al. Apelin-13 attenuates high glucose-induced calcification of MOVAS cells by regulating MAPKs and PI3K/AKT pathways and ROS-mediated signals. Biomed. Pharmacother. 2020, 128, 110271. [Google Scholar] [CrossRef] [PubMed]
- Awuah, B.E.; Shin, S.; Bandyopadhyay, B.C. Tannic acid attenuates vascular calcification-induced proximal tubular cells damage through paracrine signaling. Biomed. Pharmacother. 2021, 140, 111762. [Google Scholar] [CrossRef]
- Tsang, H.; Cui, L.; Farquharson, C.; Corcoran, B.M.; Summers, K.M.; Macrae, V.E. Exploiting novel valve interstitial cell lines to study calcific aortic valve disease. Mol. Med. Rep. 2018, 17, 2. [Google Scholar] [CrossRef] [Green Version]
- Lomashvili, K.A.; Cobbs, S.; Hennigar, R.A.; Hardcastle, K.I.; O’Neill, W.C. Phosphate-induced vascular calcification: Role of pyrophosphate and osteopontin. J. Am. Soc. Nephrol. 2004, 15, 1392–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Lihua, N.; Changjiang, Z.; Xiaorong, H.; Xiaoyan, W. Vascular calcification: New insights into endothelial cells. Microvas. Res. 2021, 134, 104105. [Google Scholar]
- Sun, L.; I Vitolo, M.I.; Qiao, M.; Anglin, I.E.; Passaniti, A. Regulation of TGFβ1-mediated growth inhibition and apoptosis by RUNX2 isoforms in endothelial cells. Oncogene 2004, 23, 4722–4734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helbing, T.; Rothweiler, R.; Ketterer, E.; Goetz, L.; Heinke, J.; Grundmann, S.; Duerschmied, D.; Patterson, C.; Bode, C.; Moser, M. BMP activity controlled by BMPER regulates the proinflammatory phenotype of endothelium. Blood 2011, 118, 5040–5049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, L.A.; Pi, X.; Patterson, C. The role of BMPs in endothelial cell function and dysfunction. Trends Endocrinol. Metab. 2014, 25, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; De Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 10, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Che, X.; Jeong, J.H.; Choi, J.Y.; Lee, Y.J.; Lee, Y.H.; Bae, S.C.; Lee, Y.M. Runx2 protein stabilizes hypoxia-inducible factor-1α through competition with von Hippel-Lindau protein (pVHL) and stimulates angiogenesis in growth plate hypertrophic chondrocytes. J. Biol. Chem. 2012, 287, 14760–14771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millán, J.L. Alkaline Phosphatases: Structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic. Signal 2006, 2, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.; Millán, J.L.; Savinova, O.V. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J. Am. Heart Assoc. 2015, 4, e002499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Xu, M.-J.; Zhao, M.-M.; Dai, X.-Y.; Kong, W.; Wilson, G.; Guan, Y.; Wang, C.-Y.; Wang, X. Activation of nuclear factor-kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int. 2012, 82, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, M.; Zhang, Y.; Zeng, R.; Liu, X.; Chen, W.; Zhang, M.; Zhang, C.; Li, M.; Zhang, M. The pro-angiogenesis effect of miR33a-5p/Ets-1/DKK1 signaling in ox-LDL induced HUVECs. Int. J. Biol. Sci. 2021, 17, 4122–4139. [Google Scholar] [CrossRef]
- Tang, X.; Lin, J.; Wang, G.; Lu, J. MicroRNA-433-3p promotes osteoblast differentiation through targeting DKK1 expression. PLoS ONE 2017, 12, e0179860. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.U.; Kirton, J.P.; Wilkinson, F.L.; Towers, E.; Sinha, S.; Rouhi, M.; Vizard, T.N.; Sage, A.P.; Martin, D.; Ward, D.T.; et al. Calcification is associated with loss of functional calcium-sensing receptor in vascular smooth muscle cells. Cardiovasc. Res. 2009, 81, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Abbasian, N.; Burton, J.O.; Herbert, K.E.; Tregunna, B.E.; Brown, J.R.; Ghaderi-Najafabadi, M.; Brunskill, N.J.; Goodall, A.H.; Bevington, A. Hyperphosphatemia, Phosphoprotein Phosphatases, and Microparticle Release in Vascular Endothelial Cells. J. Am. Soc. Nephrol. 2015, 26, 2152–2162. [Google Scholar] [CrossRef] [Green Version]
- Marco, G.S.; Hausberg, M.; Hillebrand, U.; Rustemeyer, P.; Wittkowski, W.; Lang, D.; Pavenstädt, H. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am. J. Physiol. Renal. Physiol. 2008, 294, F1381–F1387. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csiszar, A.; Ahmad, M.; Smith, K.E.; Labinskyy, N.; Gao, Q.; Kaley, G.; Edwards, J.G.; Wolin, M.S.; Ungvari, Z. Bone morphogenetic protein-2 induces proinflammatory endothelial phenotype. Am. J. Pathol. 2006, 168, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goettsch, C.; Strzelecka-Kiliszek, A.; Bessueille, L.; Quillard, T.; Mechtouff, L.; Pikula, S.; Canet-Soulas, E.; Millán, J.L.; Fonta, C.; Magne, D. TNAP as a therapeutic target for cardiovascular calcification—A discussion of its pleiotropic functions in the body. Cardiovasc. Res. 2020, 117, 1605. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Hwang, S.Y.; Nam, Y.J.; Kim, D.; Shin, S.J.; Yoon, H.E. The combined prognostic significance of alkaline phosphatase and vascular calcification in patients with end-stage kidney disease. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Opdebeeck, B.; Neven, E.; Millán, J.L.; Pinkerton, A.B.; D’Haese, P.C.; Verhulst, A. Pharmacological TNAP inhibition efficiently inhibits arterial media calcification in a warfarin rat model but deserves careful consideration of potential physiological bone formation/mineralization impairment. Bone 2020, 137, 115392. [Google Scholar] [CrossRef]
- Ahn, K.; Pan, S.; Beningo, K.; Hupe, D. A permanent human cell line (EA. hy926) preserves the characteristics of endothelin converting enzyme from primary human umbilical vein endothelial cells. Life Sci. 1995, 56, 2331–2341. [Google Scholar] [CrossRef]
- Stinghen, A.E.; Gonçalves, S.M.; Martines, E.G.; Nakao, L.S.; Riella, M.C.; Aita, C.A.; Pecoits-Filho, R. Increased plasma and endothelial cell expression of chemokines and adhesion molecules in chronic kidney disease. Nephron. Clin. Pract. 2009, 111, c117–c126. [Google Scholar] [CrossRef] [PubMed]
- Papayianni, A.; Alexopoulos, E.; Giamalis, P.; Gionanlis, L.; Belechri, A.M.; Koukoudis, P.; Memmos, D. Circulating levels of ICAM-1, VCAM-1, and MCP-1 are increased in haemodialysis patients: Association with inflammation, dyslipidaemia, and vascular events. Nephrol. Dialysis Transplant. 2002, 17, 435–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marc, V.; Cozzolino, M. Vascular calcification in chronic kidney disease: Different bricks in the wall? Kidney Int. 2017, 3, 808–817. [Google Scholar]
- Gregg, L.P.; Tio, M.C.; Li, X.; Adams-Huet, B.; De Lemos, J.A.; Hedayati, S.S. Association of Monocyte Chemoattractant Protein-1 with Death and Atherosclerotic Events in Chronic Kidney Disease. Am. J. Nephrol. 2018, 47, 395–405. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coppola, A.; Vigorito, C.; Lombari, P.; Martínez, Y.G.; Borriello, M.; Trepiccione, F.; Ingrosso, D.; Perna, A.F. Uremic Toxin Lanthionine Induces Endothelial Cell Mineralization In Vitro. Biomedicines 2022, 10, 444. https://doi.org/10.3390/biomedicines10020444
Coppola A, Vigorito C, Lombari P, Martínez YG, Borriello M, Trepiccione F, Ingrosso D, Perna AF. Uremic Toxin Lanthionine Induces Endothelial Cell Mineralization In Vitro. Biomedicines. 2022; 10(2):444. https://doi.org/10.3390/biomedicines10020444
Chicago/Turabian StyleCoppola, Annapaola, Carmela Vigorito, Patrizia Lombari, Yuselys García Martínez, Margherita Borriello, Francesco Trepiccione, Diego Ingrosso, and Alessandra F. Perna. 2022. "Uremic Toxin Lanthionine Induces Endothelial Cell Mineralization In Vitro" Biomedicines 10, no. 2: 444. https://doi.org/10.3390/biomedicines10020444