
Study Designs for Evaluation of Combination Treatment: Focus on Individual Patient Benefit



Abstract
:1. Introduction
2. General Considerations, Current Practice, and Regulatory Recommendations
2.1. General Considerations
2.2. Paralell Group vs. Add-On Study Designs
2.3. Additional Considerations for Study Design
2.4. Recommendations from Regulatory Authorities
3. Treatment Benefit at the Group vs. Individual Patient Level
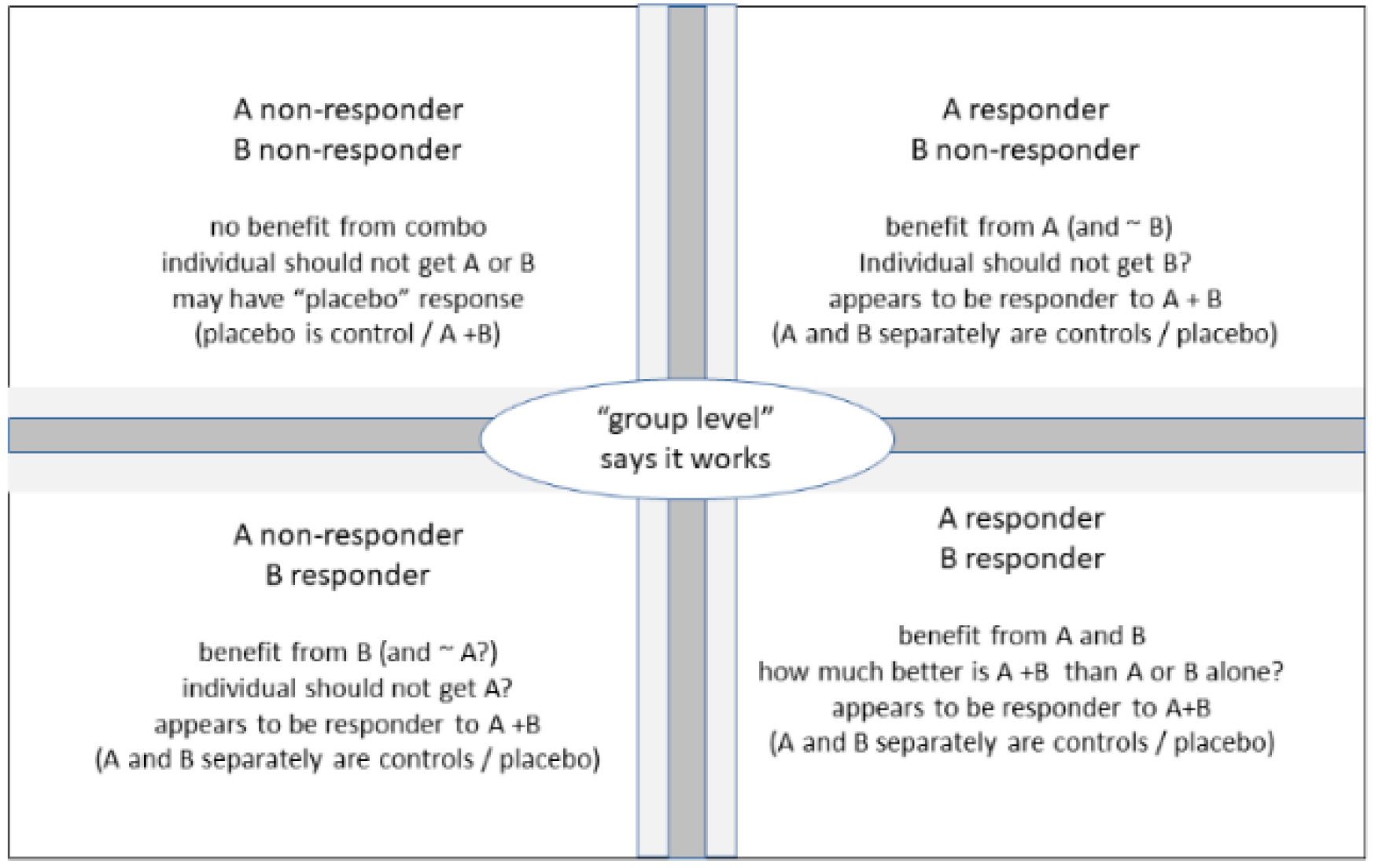
3.1. Individual Benefit/Risk Considerations
3.2. Expressing Outcomes as Responder Rates and Infering True Beneficiaries of Combination Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weber, M.A.; Schiffrin, E.L.; White, W.B.; Mann, S.; Lindholm, L.H.; Kenerson, J.G.; Flack, J.M.; Carter, B.L.; Materson, B.J.; Ram, C.V.; et al. Clinical practice guidelines for the management of hypertension in the community: A statement by the Amcerican Society of Hypertension and the International Society of Hypertension. J. Clin. Hypertens. 2014, 16, 14–26. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, M.M.; Baptist, A.P.; Blake, K.V.; Brooks, E.G.; Bryant-Stephens, T.; DiMango, E.; Dixon, A.E.; Elward, K.S.; Hartert, T.; Krishnan, J.A.; et al. 2020 Focused updates to the asthma management guidelines: A report from the National Asthma Education and Prevention Program Coordinating Committee Expert Panel working group. J. Allergy Clin. Immunol. 2020, 146, 1217–1270. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aggarwal, C.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; et al. NCCN guidelines insights: Non-small cell lung cancer, Version 1.2020. J. Natl. Compr. Cancer Netw. 2019, 17, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lightner, D.J.; Gomelsky, A.; Souter, L.; Vasavada, S.P. Diagnosis and treatment of overactive bladder (non-neurogenic) in adults: AUA/SUFU Guideline Amendment 2019. J. Urol. 2019, 202, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Gravas, S.; Cornu, J.N.; Gacci, M.; Gratzke, C.; Herrmann, T.R.W.; Mamoulakis, C.; Rieken, M.; Speakman, M.; Tikkinen, K.A. Management of Non-Neurogenic Male LUTS. Available online: https://uroweb.org/guideline/treatment-of-non-neurogenic-male-luts/ (accessed on 22 December 2021).
- European Medicines Agency. ICH Guideline M3(R2) on Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authoritzation for Pharmaceuticals. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m3r2-non-clinical-safety-studies-conduct-human-clinical-trials-marketing-authorisation_en.pdf (accessed on 25 December 2021).
- Lepor, H.; Williford, W.O.; Barry, M.J.; Brawer, M.K.; Dixon, C.M.; Gormley, G.; Haakenson, C.; Machi, M.; Narayan, P.; Padley, R.J. The efficacy of terazosin, finasteride, or both in benign prostatic hyperplasia. N. Engl. J. Med. 1996, 335, 533–539. [Google Scholar] [CrossRef]
- Debruyne, F.M.; Jardin, A.; Colloi, D.; Resel, L.; Witjes, W.P.; Delauche-Cavallier, M.C.; McCarthy, C.; Geffriaud-Ricouard, C. Sustained-release alfuzosin, finasteride and the combination of both in the treatment of benign prostatic hyperplasia. Eur. Urol. 1998, 34, 169–175. [Google Scholar] [CrossRef]
- Kirby, R.; Roehrborn, C.G.; Boyle, P.; Bartsch, G.; Jardin, A.; Cary, M.M.; Sweeney, M.; Grossman, E.B. Efficacy and tolerability of doxazosin and finasteride, alone or in combination, in treatment of symptomatic benign prostatic hyperplasia: The Prospective European Doxazosin and Combination Therapy (PREDICT) trial. Urology 2003, 61, 119–126. [Google Scholar] [CrossRef]
- McConnell, J.D.; Roehrborn, C.G.; Bautista, O.; Andriole, G.L.; Dixon, C.M.; Kusek, J.W.; Lepor, H.; McVary, K.T.; Nyberg, L.M.; Clarke, H.S.; et al. The long-term effect of doxazosin, finasteride, and combination therapy on the clinical progression of benign prostatic hyperplasia. N. Engl. J. Med. 2003, 349, 2387–2398. [Google Scholar] [CrossRef] [Green Version]
- Roehrborn, C.G.; Siami, P.; Barkin, J.; Damiao, R.; Major-Walker, K.; Nandy, I.; Morrill, B.B.; Gagnier, R.P.; Montorsi, F. The effects of combination therapy with dutasteride and tamsulosin on clinical outcomes in men with symptomatic benign prostatic hyperplasia: 4-year results from the CombAT study. Eur. Urol. 2010, 57, 123–131. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Maeda, T.; Kikuchi, E.; Hasegawa, M.; Ishioka, K.; Hagiwara, M.; Miyazaki, Y.; Shinojima, T.; Miyajima, A.; Oya, M. Solifenacin or mirabegron could improve persistent overactive bladder symptoms after dutasteride treatment in patients with benign prostatic hyperplasia. Urology 2015, 85, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Chapple, C.R.; Cardozo, L.; Nitti, V.W.; Siddiqui, E.; Michel, M.C. Mirabegron in overactive bladder: A review of efficacy, safety, and tolerability. Neurourol. Urodyn. 2014, 33, 17–30. [Google Scholar] [CrossRef]
- Staskin, D.R.; Michel, M.C.; Sun, F.; Guan, Z.; Morrow, J.D. The effect of elective sham dose escalation on the palcebo response during an antimuscarinic trial for overactive bladder symptoms. J. Urol. 2012, 187, 1721–1726. [Google Scholar] [CrossRef]
- Yamaguchi, O.; Kikizaki, H.; Homma, Y.; Igawa, Y.; Takeda, M.; Nishizawa, O.; Gotoh, M.; Yoshida, M.; Yokoyama, O.; Seki, N.; et al. Safety and efficacy of mirabegron as ‘add-on’ therapy in patients with overactive bladder treated with solifenacin: A post-marketing, open-label study in Japan (MILAI study). BJU Int. 2015, 1146, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Athanasopoulos, A.; Gyftopoulos, K.; Giannitsas, K.; Fisfis, J.; Perimenis, P.; Barbalias, G. Combination treatment with an α-blocker plus an anticholinergic for bladder outlet obstruction: A prospective, randomized, controlled study. J. Urol. 2003, 169, 2253–2256. [Google Scholar] [CrossRef]
- Drake, M.J.; Chapple, C.; Esen, A.A.; Athanasiou, S.; Cambronero, J.; Mitcheson, D.; Herschorn, S.; Saleem, T.; Huang, M.; Siddiqui, E.; et al. Efficacy and safety of mirabegon add-on therapy to solifenacin in incontinent overactive bladder patients with an iInadequate response to initial 4-week solifenacin monotherapy: A randomised double-blind multicentre phase 3B study (BESIDE). Eur. Urol. 2016, 70, 136–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, P.; Kelleher, C.; Staskin, D.; Rechberger, T.; Kay, R.; Martina, R.; Newgreen, D.; Paireddy, A.; van Maanen, R.; Ridder, A. Combination treatment with mirabegron and solifenacin in patients with overactive bladder: Efficacy and safety results from a randomised, double-blind, dose-ranging, phase 2 study (Symphony). Eur. Urol. 2015, 67, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Casabe, A.; Roehrborn, C.G.; Da Pozzo, L.F.; Zepeda, S.; Henderson, R.J.; Sorsaburu, S.; Hennerges, C.; Wong, D.G.; Viktrup, L. Efficacy and safety of the coadministration of tadalafil once daily with finasteride for 6 months in men with lower urinary tract symptoms and prostatic enlargement secondary to benign prostatic hyperplasia. J. Urol. 2014, 191, 727–733. [Google Scholar] [CrossRef]
- Barry, M.J.; Williford, W.O.; Chang, Y.; Machi, M.; Jones, K.M.; Walker-Corkery, E.; Lepor, H. Benign prostatic hyperplasia specific healthy status measures in clinical research: How much change in the American Urological Association Symptom Index and the Benign Prostatic Hyerplasia Impact Index is perceptible to patients? J. Urol. 1995, 154, 1770–1774. [Google Scholar] [CrossRef]
- van Kerrebroeck, P.; Chapple, C.; Drogendijk, T.; Klaver, M.; Sokol, R.; Speakman, M.; Traudtner, K.; Drake, M.J. Combination therapy with solifenacin and tamsulosin oral controlled absorption system in a single table for lower urinary tract symptoms in men: Efficacy and safety results from the randomised controlled NEPTUNE trial. Eur. Urol. 2013, 64, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Peng, B.; Lei, G.-L.; Wei, Q.; Yang, L. Study of phosphodiesterase 5 inhibitors and a-adrenoceptor antagonists used alone or in combination for the treatment of lower urinary tract symptoms due to benign prostatic hyperplasia. Minerva Urol. Nefrol. 2020, 72, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Kallidonis, P.; Adamou, C.; Kotsiris, D.; Ntasiotis, P.; Verze, P.; Athanasopoulos, A. Combination therapy with alpha-blocker and phosphodiesterase-5 inhibitor for improving lower urinary tract symptoms and erectile dysfunction in comparison with monotherapy: A systematic review and meta-analysis. Eur. Urol. Focus 2020, 6, 537–558. [Google Scholar] [CrossRef]
- U. S. Food and Drug Administration. Guidance for Industry. Nonclinical Safetry Evaluation of Drug or Biologic Combinations. Available online: https://www.fda.gov/media/119657/download (accessed on 25 December 2021).
- European Medicines Agency. Guideline on Clinical Development of Fixed Combination Medical Procuts. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-development-fixed-combination-medicinal-products-revision-2_en.pdf (accessed on 25 December 2021).
- U. S. Food and Drug Administration. Guide for Industry. Codevelopment of Two or More New Investigational Drugs for Use in Combination. Available online: https://www.fda.gov/media/80100/download (accessed on 25 December 2021).
- Madsen, F.A.; Bruskewitz, R.C. Benign prostatic hyperplasia: Pathophysiology and pharmacological treatment. Curr. Opin. Nephrol. Hypertens. 1995, 4, 455–459. [Google Scholar] [CrossRef]
- Amiri, M.; Murgas, S.; Stang, A.; Michel, M.C. Do overactive bladder symptoms and their treatment-associated changes exhibit a normal distribution? Implications for analysis and reporting. Neurourol. Urodyn. 2020, 39, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Michel, M.C.; Bressel, H.U.; Goepel, M.; Rübben, H. A 6-months large-scale study into the safety of tamsulosin. Br. J. Clin. Pharmacol. 2001, 51, 609–614. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Resp A | Resp B | Resp C | Resp T | P Harm |
|---|---|---|---|---|
| Assumed values of A, B and C | ||||
| 50 | 50 | 0 | 100 | 100 |
| 50 | 50 | 50 | 50 | 50 |
| 70 | 70 | 50 | 90 | 50 |
| 70 | 40 | 40 | 70 | 60 |
| 70 | 40 | 30 | 80 | 70 |
| 50 | 50 | 30 | 70 | 70 |
| 50 | 50 | 10 | 90 | 90 |
| Measured values of A, B and T | ||||
| 43 | 33 | 34 | 42 | 66 |
| 52 | 61 | 46 | 67 | 54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michel, M.C.; Staskin, D. Study Designs for Evaluation of Combination Treatment: Focus on Individual Patient Benefit. Biomedicines 2022, 10, 270. https://doi.org/10.3390/biomedicines10020270
Michel MC, Staskin D. Study Designs for Evaluation of Combination Treatment: Focus on Individual Patient Benefit. Biomedicines. 2022; 10(2):270. https://doi.org/10.3390/biomedicines10020270
Chicago/Turabian StyleMichel, Martin C., and David Staskin. 2022. "Study Designs for Evaluation of Combination Treatment: Focus on Individual Patient Benefit" Biomedicines 10, no. 2: 270. https://doi.org/10.3390/biomedicines10020270