
Choroid Plexus in Alzheimer’s Disease—The Current State of Knowledge



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Structure and Cell Organization of the Choroid Plexus
3. General Physiology of the CP
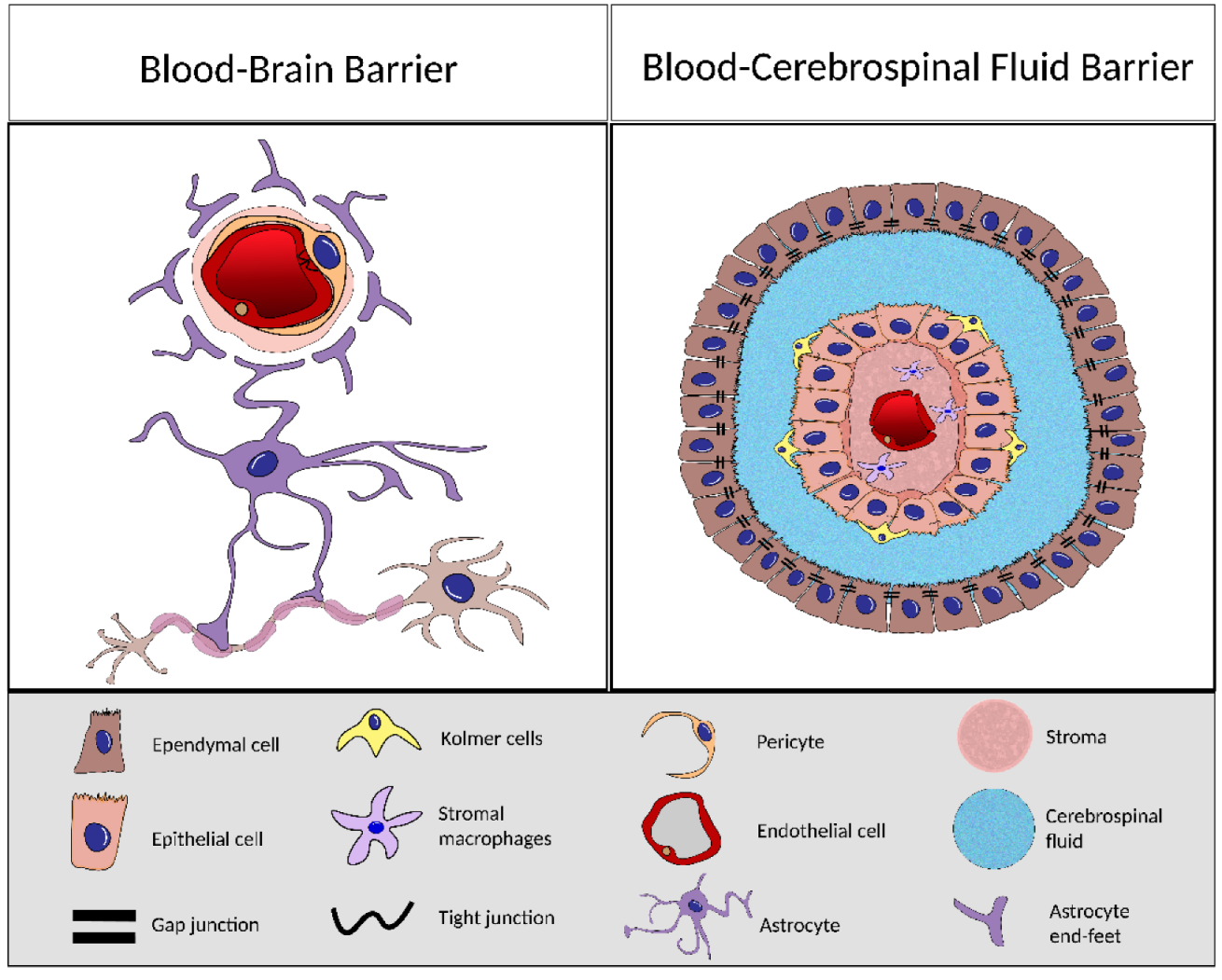
4. BCSFB and BBB Comparison
5. Aging of the BCSFB and Disease-Induced Alterations
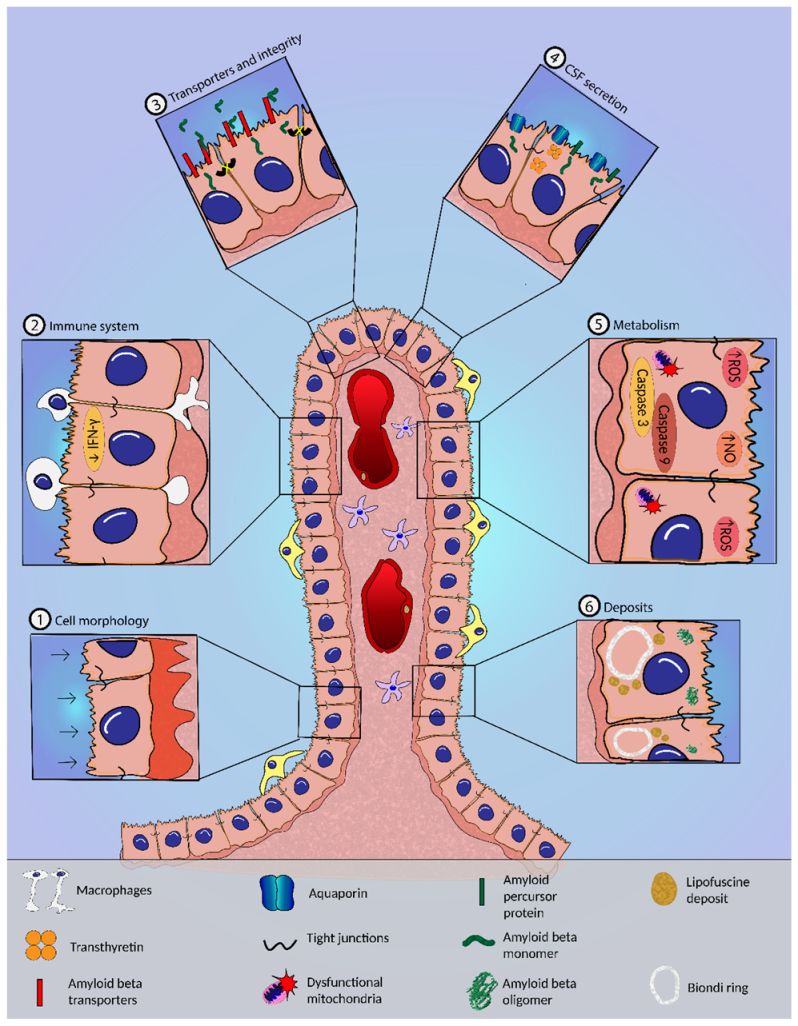
6. Alzheimer’s Disease and the CP
6.1. Morphological Alterations in the CP in AD
6.2. CSF Dynamics and Secretion in AD
6.3. BCSFB Integrity in AD
6.4. Transport of Aβ and Other Compounds across the BCSFB in AD
6.5. Metabolic Alterations and Oxidative Stress in the CP in AD
6.6. Inflammation and CP in AD
6.7. Features of CP Stem Cell in AD
6.8. Circadian Cycle Disturbed at CP in AD
6.9. CP Epithelial Cell Implants as a Therapy in AD
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghersi-Egea, J.-F.; Strazielle, N.; Catala, M.; Silva-Vargas, V.; Doetsch, F.; Engelhardt, B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol. 2018, 135, 337–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratzer, I.; Ek, J.; Stolp, H. The molecular anatomy and functions of the choroid plexus in healthy and diseased brain. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 183430. [Google Scholar] [CrossRef] [PubMed]
- Dillen, Y.; Kemps, H.; Gervois, P.; Wolfs, E.; Bronckaers, A. Adult Neurogenesis in the Subventricular Zone and Its Regulation After Ischemic Stroke: Implications for Therapeutic Approaches. Transl. Stroke Res. 2020, 11, 60–79. [Google Scholar] [CrossRef]
- Gonzalez-Marrero, I.; Hernández-Abad, L.G.; Castañeyra-Ruiz, L.; Carmona-Calero, E.M.; Castañeyra-Perdomo, A. Changes in the choroid plexuses and brain barriers associated with high blood pressure and ageing. Neurologia 2018. [Google Scholar] [CrossRef]
- Bothwell, S.W.; Janigro, D.; Patabendige, A. Cerebrospinal fluid dynamics and intracranial pressure elevation in neurological diseases. Fluids Barriers CNS 2019, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Cornford, E.M.; Varesi, J.B.; Hyman, S.; Damian, R.T.; Raleigh, M.J. Mitochondrial content of choroid plexus epithelium. Exp. Brain Res. 1997, 116, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Obernier, K.; Alvarez-Buylla, A. Neural stem cells: Origin, heterogeneity and regulation in the adult mammalian brain. Development 2019, 146, dev156059. [Google Scholar] [CrossRef] [Green Version]
- Solár, P.; Zamani, A.; Kubíčková, L.; Dubový, P.; Joukal, M. Choroid plexus and the blood–cerebrospinal fluid barrier in disease. Fluids Barriers CNS 2020, 17, 35. [Google Scholar] [CrossRef]
- Munro, D.A.D.; Bradford, B.M.; Mariani, S.A.; Hampton, D.W.; Vink, C.S.; Chandran, S.; Hume, D.A.; Pridans, C.; Priller, J. CNS macrophages differentially rely on an intronic Csf1r enhancer for their development. Development 2020, 147, dev194449. [Google Scholar] [CrossRef]
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035. [Google Scholar] [CrossRef]
- Redzic, Z.B.; Segal, M.B. The structure of the choroid plexus and the physiology of the choroid plexus epithelium. Adv. Drug Deliv. Rev. 2004, 56, 1695–1716. [Google Scholar] [CrossRef]
- Richardson, S.J.; Wijayagunaratne, R.C.; D’Souza, D.G.; Darras, V.M.; Van Herck, S.L.J. Transport of thyroid hormones via the choroid plexus into the brain: The roles of transthyretin and thyroid hormone transmembrane transporters. Front. Neurosci. 2015, 9, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, A.C.; Santos, J.; Costa, A.R.; Ferreira, C.L.; Tomás, J.; Quintela, T.; Ishikawa, H.; Schwerk, C.; Schroten, H.; Ferrer, I.; et al. Bitter taste receptors profiling in the human blood-cerebrospinal fluid-barrier. Biochem. Pharmacol. 2020, 177, 113954. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Baruch, K. The resolution of neuroinflammation in neurodegeneration: Leukocyte recruitment via the choroid plexus. EMBO J. 2014, 33, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, I.; Strazielle, N.; Saudrais, E.; Mönkkönen, K.; Malleval, C.; Blondel, S.; Ghersi-Egea, J.-F. Glutathione Conjugation at the Blood-CSF Barrier Efficiently Prevents Exposure of the Developing Brain Fluid Environment to Blood-Borne Reactive Electrophilic Substances. J. Neurosci. 2018, 38, 3466–3479. [Google Scholar] [CrossRef]
- Kumar, S.; Trivedi, P.K. Glutathione S-Transferases: Role in Combating Abiotic Stresses Including Arsenic Detoxification in Plants. Front. Plant. Sci. 2018, 9, 751. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A. Development of the choroid plexus and blood-CSF barrier. Front. Neurosci. 2015, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Blanchette, M.; Daneman, R. Formation and maintenance of the BBB. Mech. Dev. 2015, 138 Pt 1, 8–16. [Google Scholar] [CrossRef]
- Santos, C.R.A.; Duarte, A.C.; Quintela, T.; Tomás, J.; Albuquerque, T.; Marques, F.; Palha, J.A.; Gonçalves, I. The choroid plexus as a sex hormone target: Functional implications. Front. Neuroendocrinol. 2017, 44, 103–121. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Välitalo, P.A.; Huntjens, D.R.; Proost, J.H.; Vermeulen, A.; Krauwinkel, W.; Beukers, M.W.; van den Berg, D.-J.; Hartman, R.; Wong, Y.C.; et al. Predicting Drug Concentration-Time Profiles in Multiple CNS Compartments Using a Comprehensive Physiologically-Based Pharmacokinetic Model. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 765–777. [Google Scholar] [CrossRef] [Green Version]
- Strazielle, N.; Ghersi-Egea, J.-F. Factors affecting delivery of antiviral drugs to the brain. Rev. Med. Virol. 2005, 15, 105–133. [Google Scholar] [CrossRef]
- Vandenhaute, E.; Stump-Guthier, C.; Losada, M.L.; Tenenbaum, T.; Rudolph, H.; Ishikawa, H.; Schwerk, C.; Schroten, H.; Dürken, M.; März, M.; et al. The choroid plexus may be an underestimated site of tumor invasion to the brain: An in vitro study using neuroblastoma cell lines. Cancer Cell Int. 2015, 15, 102. [Google Scholar] [CrossRef] [Green Version]
- Redzic, Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: Similarities and differences. Fluids Barriers CNS 2011, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohrmann, G.J.; Bucy, P.C. Human choroid plexus: A light and electron microscopic study. J. Neurosurg. 1970, 33, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Keep, R.F.; Jones, H.C. A morphometric study on the development of the lateral ventricle choroid plexus, choroid plexus capillaries and ventricular ependyma in the rat. Brain Res. Dev. Brain Res. 1990, 56, 47–53. [Google Scholar] [CrossRef]
- Gorlé, N.; Van Cauwenberghe, C.; Libert, C.; Vandenbroucke, R.E. The effect of aging on brain barriers and the consequences for Alzheimer’s disease development. Mamm. Genome 2016, 27, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Masseguin, C.; LePanse, S.; Corman, B.; Verbavatz, J.M.; Gabrion, J. Aging affects choroidal proteins involved in CSF production in Sprague-Dawley rats. Neurobiol. Aging 2005, 26, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Serot, J.M.; Christmann, D.; Dubost, T.; Couturier, M. Cerebrospinal fluid transthyretin: Aging and late onset Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 63, 506–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleine, T.O.; Hackler, R.; Lutcke, A.; Dauch, W.; Zofel, P. Transport and production of cerebrospinal fluid (CSF) change in aging humans under normal and diseased conditions. Z. Gerontol. 1993, 26, 251–255. [Google Scholar]
- Saul, J.; Hutchins, E.; Reiman, R.; Saul, M.; Ostrow, L.W.; Harris, B.T.; Van Keuren-Jensen, K.; Bowser, R.; Bakkar, N. Global alterations to the choroid plexus blood-CSF barrier in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2020, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Ayub, M.; Jin, H.K.; Bae, J.-S. The blood cerebrospinal fluid barrier orchestrates immunosurveillance, immunoprotection, and immunopathology in the central nervous system. BMB Rep. 2021, 54, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Batra, A.; Latour, L.L.; Ruetzler, C.A.; Hallenbeck, J.M.; Spatz, M.; Warach, S.; Henning, E.C. Increased plasma and tissue MMP levels are associated with BCSFB and BBB disruption evident on post-contrast FLAIR after experimental stroke. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2010, 30, 1188–1199. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, H.; Grant, S.C.; Petzold, L.R.; Harrington, M.G. Regulation of CSF and Brain Tissue Sodium Levels by the Blood-CSF and Blood-Brain Barriers During Migraine. Front. Comput. Neurosci. 2020, 14, 4. [Google Scholar] [CrossRef] [Green Version]
- Aldred, A.R.; Brack, C.M.; Schreiber, G. The cerebral expression of plasma protein genes in different species. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 1995, 111, 1–15. [Google Scholar] [CrossRef]
- Sharma, P.; Sharma, A.; Fayaz, F.; Wakode, S.; Pottoo, F.H. Biological Signatures of Alzheimer’s Disease. Curr. Top. Med. Chem. 2020, 20, 770–781. [Google Scholar] [CrossRef]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Serot, J.M.; Béné, M.C.; Foliguet, B.; Faure, G.C. Morphological alterations of the choroid plexus in late-onset Alzheimer’s disease. Acta Neuropathol. 2000, 99, 105–108. [Google Scholar] [CrossRef]
- González-Marrero, I.; Giménez-Llort, L.; Johanson, C.E.; Carmona-Calero, E.M.; Castañeyra-Ruiz, L.; Brito-Armas, J.M.; Castañeyra-Perdomo, A.; Castro-Fuentes, R. Choroid plexus dysfunction impairs beta-amyloid clearance in a triple transgenic mouse model of alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Brkic, M.; Balusu, S.; Van Wonterghem, E.; Gorlé, N.; Benilova, I.; Kremer, A.; Van Hove, I.; Moons, L.; De Strooper, B.; Kanazir, S.; et al. Amyloid β Oligomers Disrupt Blood-CSF Barrier Integrity by Activating Matrix Metalloproteinases. J. Neurosci. 2015, 35, 12766–12778. [Google Scholar] [CrossRef] [Green Version]
- Miklossy, J.; Kraftsik, R.; Pillevuit, O.; Lepori, D.; Genton, C.; Bosman, F.T. Curly Fiber and Tangle-like Inclusions in the Ependyma and Choroid Plexus—A Pathogenetic Relationship with the Cortical Alzheimer-type Changes? J. Neuropathol. Exp. Neurol. 1998, 57, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Wen, G.Y.; Wisniewski, H.M.; Kascsak, R.J. Biondi ring tangles in the choroid plexus of Alzheimer’s disease and normal aging brains: A quantitative study. Brain Res. 1999, 832, 40–46. [Google Scholar] [CrossRef]
- Oksche, A.; Liesner, R.; Tigges, J.; Tigges, M. Intraepithelial inclusions resembling human Biondi bodies in the choroid plexus of an aged chimpanzee. Cell Tissue Res. 1984, 235, 467–469. [Google Scholar] [CrossRef]
- Kiktenko, A.I. Biondi bodies in the choroid plexus epithelium of the human brain-A scanning electron-microscopic study. Cell Tissue Res. 1986, 244, 239–240. [Google Scholar] [CrossRef]
- Reeg, S.; Grune, T. Protein Oxidation in Aging: Does It Play a Role in Aging Progression? Antioxid. Redox Signal. 2015, 23, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, M.O.; Spuch, C.; Antequera, D.; Rodal, I.; de Yébenes, J.G.; Molina, J.A.; Bermejo, F.; Carro, E. Megalin mediates the transport of leptin across the blood-CSF barrier. Neurobiol. Aging 2008, 29, 902–912. [Google Scholar] [CrossRef]
- Vargas, T.; Ugalde, C.; Spuch, C.; Antequera, D.; Morán, M.J.; Martín, M.A.; Ferrer, I.; Bermejo-Pareja, F.; Carro, E. Aβ accumulation in choroid plexus is associated with mitochondrial-induced apoptosis. Neurobiol. Aging 2010, 31, 1569–1581. [Google Scholar] [CrossRef]
- Silverberg, G.D.; Messier, A.A.; Miller, M.C.; Machan, J.T.; Majmudar, S.S.; Stopa, E.G.; Donahue, J.E.; Johanson, C.E. Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J. Neuropathol. Exp. Neurol. 2010, 69, 1034–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Marrero, I.; Castañeyra-Ruiz, L.; González-Toledo, J.M.; Castañeyra-Ruiz, A.; de Paz-Carmona, H.; Castro, R.; Hernandez-Fernaud, J.R.; Castañeyra-Perdomo, A.; Carmona-Calero, E.M. High Blood Pressure Effects on the Blood to Cerebrospinal Fluid Barrier and Cerebrospinal Fluid Protein Composition: A Two-Dimensional Electrophoresis Study in Spontaneously Hypertensive Rats. Int. J. Hypertens. 2013, 2013, 164653. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Zhong, Z.; Jiang, W.; Du, E.; Dodel, R.; Farlow, M.R.; Zheng, W.; Du, Y. The role of choroid plexus in IVIG-induced beta-amyloid clearance. Neuroscience 2014, 270, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Silverberg, G.D.; Heit, G.; Huhn, S.; Jaffe, R.A.; Chang, S.D.; Bronte-Stewart, H.; Rubenstein, E.; Possin, K.; Saul, T.A. The cerebrospinal fluid production rate is reduced in dementia of the Alzheimer’s type. Neurology 2001, 57, 1763–1766. [Google Scholar] [CrossRef]
- Johanson, C.E.; Duncan, J.A.; Klinge, P.M.; Brinker, T.; Stopa, E.G.; Silverberg, G.D. Multiplicity of cerebrospinal fluid functions: New challenges in health and disease. Cereb. Fluid Res. 2008, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Silverberg, G.; Mayo, M.; Saul, T.; Fellmann, J.; McGuire, D. Elevated cerebrospinal fluid pressure in patients with Alzheimer’s disease. Cereb. Fluid Res. 2006, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.; Miller, M.C.; Caralopoulos, I.N.; Worden, M.S.; Brinker, T.; Gordon, Z.N.; Johanson, C.E.; Silverberg, G.D. Temporal course of cerebrospinal fluid dynamics and amyloid accumulation in the aging rat brain from three to thirty months. Fluids Barriers CNS 2012, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Speake, T.; Freeman, L.J.; Brown, P.D. Expression of aquaporin 1 and aquaporin 4 water channels in rat choroid plexus. Biochim. Biophys. Acta 2003, 1609, 80–86. [Google Scholar] [CrossRef]
- Bergen, A.A.; Kaing, S.; ten Brink, J.B.; Gorgels, T.G.; Janssen, S.F.; Bank, T.N.B. Gene expression and functional annotation of human choroid plexus epithelium failure in Alzheimer’s disease. BMC Genom. 2015, 16, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kant, S.; Stopa, E.G.; Johanson, C.E.; Baird, A.; Silverberg, G.D. Choroid plexus genes for CSF production and brain homeostasis are altered in Alzheimer’s disease. Fluids Barriers CNS 2018, 15, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.P.C.; Chen, R.L.; Preston, J.E. The influence of cerebrospinal fluid turnover on age-related changes in cerebrospinal fluid protein concentrations. Neurosci. Lett. 2010, 476, 138–141. [Google Scholar] [CrossRef]
- Sousa, J.C.; Cardoso, I.; Marques, F.; Saraiva, M.J.; Palha, J.A. Transthyretin and Alzheimer’s disease: Where in the brain? Neurobiol. Aging 2007, 28, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Antequera, D.; Vargas, T.; Ugalde, C.; Spuch, C.; Molina, J.A.; Ferrer, I.; Bermejo-Pareja, F.; Carro, E. Cytoplasmic gelsolin increases mitochondrial activity and reduces Abeta burden in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 36, 42–50. [Google Scholar] [CrossRef]
- Stopa, E.G.; Tanis, K.Q.; Miller, M.C.; Nikonova, E.V.; Podtelezhnikov, A.A.; Finney, E.M.; Stone, D.J.; Camargo, L.M.; Parker, L.; Verma, A.; et al. Comparative transcriptomics of choroid plexus in Alzheimer’s disease, frontotemporal dementia and Huntington’s disease: Implications for CSF homeostasis. Fluids Barriers CNS 2018, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Premkumar, D.R.D.; Pax, A.B.; Cohen, D.L.; Lieberburg, I. Production and increased detection of amyloid β protein and amyloidogenic fragments in brain microvessels, meningeal vessels and choroid plexus in Alzheimer’s disease. Mol. Brain Res. 1996, 35, 58–68. [Google Scholar] [CrossRef]
- Liu, F.; Xue, Z.-Q.; Deng, S.-H.; Kun, X.; Luo, X.-G.; Patrylo, P.R.; Rose, G.M.; Cai, H.; Struble, R.G.; Cai, Y.; et al. γ-Secretase binding sites in aged and Alzheimer’s disease human cerebrum: The choroid plexus as a putative origin of CSF Aβ. Eur. J. Neurosci. 2013, 37, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, D.R.D.; Kalaria, R.N. Altered expression of amyloid β precursor mRNAs in cerebral vessels, meninges, and choroid plexus in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1996, 777, 288–292. [Google Scholar] [CrossRef]
- Nakamura, T.; Shoji, M.; Harigaya, Y.; Watanabe, M.; Hosoda, K.; Cheung, T.T.; Shaffer, L.M.; Golde, T.E.; Younkin, L.H.; Younkin, S.G.; et al. Amyloid β protein levels in cerebrospinal fluid are elevated in early-onset Alzheimer’s disease. Ann. Neurol. 1994, 36, 903–911. [Google Scholar] [CrossRef]
- Crossgrove, J.S.; Li, G.J.; Zheng, W. The Choroid Plexus Removes β-Amyloid from Brain Cerebrospinal Fluid. Exp. Biol. Med. 2005, 230, 771–776. [Google Scholar] [CrossRef]
- Pascale, C.L.; Miller, M.C.; Chiu, C.; Boylan, M.; Caralopoulos, I.N.; Gonzalez, L.; Johanson, C.E.; Silverberg, G.D. Amyloid-beta transporter expression at the blood-CSF barrier is age-dependent. Fluids Barriers CNS 2011, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Shibata, M.; Yamada, S.; Ram Kumar, S.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-β1-40 peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: Preliminary observations. Neurobiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef]
- Carro, E.; Trejo, J.L.; Spuch, C.; Bohl, D.; Heard, J.M.; Torres-Aleman, I. Blockade of the insulin-like growth factor I receptor in the choroid plexus originates Alzheimer’s-like neuropathology in rodents: New cues into the human disease? Neurobiol. Aging 2006, 27, 1618–1631. [Google Scholar] [CrossRef] [Green Version]
- Carro, E.; Spuch, C.; Trejo, J.L.; Antequera, D.; Torres-Aleman, I. Choroid Plexus Megalin Is Involved in Neuroprotection by Serum Insulin-Like Growth Factor I. J. Neurosci. 2005, 25, 10884–10893. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative Damage Is the Earliest Event in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Perez-Gracia, E.; Blanco, R.; Carmona, M.; Carro, E.; Ferrer, I. Oxidative stress damage and oxidative stress responses in the choroid plexus in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 497–504. [Google Scholar] [CrossRef]
- Anthony, S.G.; Schipper, H.M.; Tavares, R.; Hovanesian, V.; Cortez, S.C.; Stopa, E.G.; Johanson, C.E. Stress protein expression in the Alzheimer-diseased choroid plexus. J. Alzheimers. Dis. 2003, 5, 171–177. [Google Scholar] [CrossRef]
- Cottrell, D.A.; Blakely, E.L.; Johnson, M.A.; Ince, P.G.; Turnbull, D.M. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology 2001, 57, 260–264. [Google Scholar] [CrossRef]
- Baruch, K.; Rosenzweig, N.; Kertser, A.; Deczkowska, A.; Sharif, A.M.; Spinrad, A.; Tsitsou-Kampeli, A.; Sarel, A.; Cahalon, L.; Schwartz, M. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 2015, 6, 7967. [Google Scholar] [CrossRef]
- Kunis, G.; Baruch, K.; Rosenzweig, N.; Kertser, A.; Miller, O.; Berkutzki, T.; Schwartz, M. IFN-γ-dependent activation of the brain’s choroid plexus for CNS immune surveillance and repair. Brain 2013, 136, 3427–3440. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, S.D.; Ferreira, A.C.; Gao, F.; Coppola, G.; Geschwind, D.H.; Sousa, J.C.; Correia-Neves, M.; Sousa, N.; Palha, J.A.; Marques, F. The choroid plexus transcriptome reveals changes in type I and II interferon responses in a mouse model of Alzheimer’s disease. Brain. Behav. Immun. 2015, 49, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Baruch, K.; Deczkowska, A.; Rosenzweig, N.; Tsitsou-Kampeli, A.; Sharif, A.M.; Matcovitch-Natan, O.; Kertser, A.; David, E.; Amit, I.; Schwartz, M. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat. Med. 2016, 22, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Kertser, A.; Porat, Z.; Schwartz, M. Cerebral nitric oxide represses choroid plexus NF κB-dependent gateway activity for leukocyte trafficking. EMBO J. 2015, 34, 1816–1828. [Google Scholar] [CrossRef] [Green Version]
- Steeland, S.; Gorlé, N.; Vandendriessche, C.; Balusu, S.; Brkic, M.; Van Cauwenberghe, C.; Van Imschoot, G.; Van Wonterghem, E.; De Rycke, R.; Kremer, A.; et al. Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer’s disease. EMBO Mol. Med. 2018, 10, e8300. [Google Scholar] [CrossRef] [PubMed]
- Jurkowski, M.P.; Bettio, L.; Woo, E.K.; Patten, A.; Yau, S.-Y.; Gil-Mohapel, J. Beyond the Hippocampus and the SVZ: Adult Neurogenesis Throughout the Brain. Front. Cell. Neurosci. 2020, 14, 576444. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, J.; Chopp, M. Cell proliferation and differentiation from ependymal, subependymal and choroid plexus cells in response to stroke in rats. J. Neurol. Sci. 2002, 193, 137–146. [Google Scholar] [CrossRef]
- Itokazu, Y.; Kitada, M.; Dezawa, M.; Mizoguchi, A.; Matsumoto, N.; Shimizu, A.; Ide, C. Choroid plexus ependymal cells host neural progenitor cells in the rat. Glia 2006, 53, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Bolos, M.; Spuch, C.; Ordoñez-Gutierrez, L.; Wandosell, F.; Ferrer, I.; Carro, E. Neurogenic effects of β-amyloid in the choroid plexus epithelial cells in Alzheimer’s disease. Cell. Mol. Life Sci. 2013, 70, 2787–2797. [Google Scholar] [CrossRef]
- Kim, H.J.; Chae, S.C.; Lee, D.K.; Chromy, B.; Lee, S.C.; Park, Y.C.; Klein, W.L.; Krafft, G.A.; Hong, S.T. Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J. 2003, 17, 118–120. [Google Scholar] [CrossRef]
- Dibner, C.; Schibler, U.; Albrecht, U. The mammalian circadian timing system: Organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 2009, 72, 517–549. [Google Scholar] [CrossRef] [Green Version]
- Myung, J.; Schmal, C.; Hong, S.; Tsukizawa, Y.; Rose, P.; Zhang, Y.; Holtzman, M.J.; De Schutter, E.; Herzel, H.; Bordyugov, G.; et al. The choroid plexus is an important circadian clock component. Nat. Commun. 2018, 9, 1062. [Google Scholar] [CrossRef]
- Quintela, T.; Sousa, C.; Patriarca, F.M.; Gonçalves, I.; Santos, C.R.A. Gender associated circadian oscillations of the clock genes in rat choroid plexus. Brain Struct. Funct. 2015, 220, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Quintela, T.; Albuquerque, T.; Lundkvist, G.; Carmine Belin, A.; Talhada, D.; Gonçalves, I.; Carro, E.; Santos, C.R.A. The choroid plexus harbors a circadian oscillator modulated by estrogens. Chronobiol. Int. 2018, 35, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Furtado, A.; Astaburuaga, R.; Costa, A.; Duarte, A.C.; Goncalves, I.; Cipolla-Neto, J.; Lemos, M.C.; Carro, E.; Relogio, A.; Santos, C.R.A.; et al. The Rhythmicity of Clock Genes is Disrupted in the Choroid Plexus of the APP/PS1 Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 77, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.C.; Furtado, A.; Hrynchak, M.V.; Costa, A.R.; Talhada, D.; Gonçalves, I.; Lemos, M.C.; Quintela, T.; Santos, C.R.A. Age, Sex Hormones, and Circadian Rhythm Regulate the Expression of Amyloid-Beta Scavengers at the Choroid Plexus. Int. J. Mol. Sci. 2020, 21, 6813. [Google Scholar] [CrossRef]
- Bolos, M.; Antequera, D.; Aldudo, J.; Kristen, H.; Bullido, M.J.; Carro, E. Choroid plexus implants rescue Alzheimer’s disease-like pathologies by modulating amyloid-β degradation. Cell. Mol. Life Sci. 2014, 71, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- Aliaghaei, A.; Digaleh, H.; Khodagholi, F.; Ahmadiani, A. Encapsulated Choroid Plexus Epithelial Cells Actively Protect Against Intrahippocampal Aβ-induced Long-Term Memory Dysfunction; Upregulation of Effective Neurogenesis with the Abrogated Apoptosis and Neuroinflammation. J. Mol. Neurosci. 2015, 56, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.B.; Wang, R.; Yi, Y.F.; Gao, Z.; Chen, Y.Z. Lycopene mitigates β-amyloid induced inflammatory response and inhibits NF-κB signaling at the choroid plexus in early stages of Alzheimer’s disease rats. J. Nutr. Biochem. 2018, 53, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Liu, C.; Wang, R.; Gao, X.; Hao, C.; Liu, C. A combination of lycopene and human amniotic epithelial cells can ameliorate cognitive deficits and suppress neuroinflammatory signaling by choroid plexus in Alzheimer’s disease rat. J. Nutr. Biochem. 2021, 88, 108558. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gião, T.; Teixeira, T.; Almeida, M.R.; Cardoso, I. Choroid Plexus in Alzheimer’s Disease—The Current State of Knowledge. Biomedicines 2022, 10, 224. https://doi.org/10.3390/biomedicines10020224
Gião T, Teixeira T, Almeida MR, Cardoso I. Choroid Plexus in Alzheimer’s Disease—The Current State of Knowledge. Biomedicines. 2022; 10(2):224. https://doi.org/10.3390/biomedicines10020224
Chicago/Turabian StyleGião, Tiago, Tiago Teixeira, Maria Rosário Almeida, and Isabel Cardoso. 2022. "Choroid Plexus in Alzheimer’s Disease—The Current State of Knowledge" Biomedicines 10, no. 2: 224. https://doi.org/10.3390/biomedicines10020224