
Amyloid Disassembly: What Can We Learn from Chaperones?

Abstract
:1. Introduction
2. Chaperones in Amyloid Disassembly
2.1. Molecular Chaperones in Amyloid Disassembly
2.1.1. Hsp70 Chaperone Network
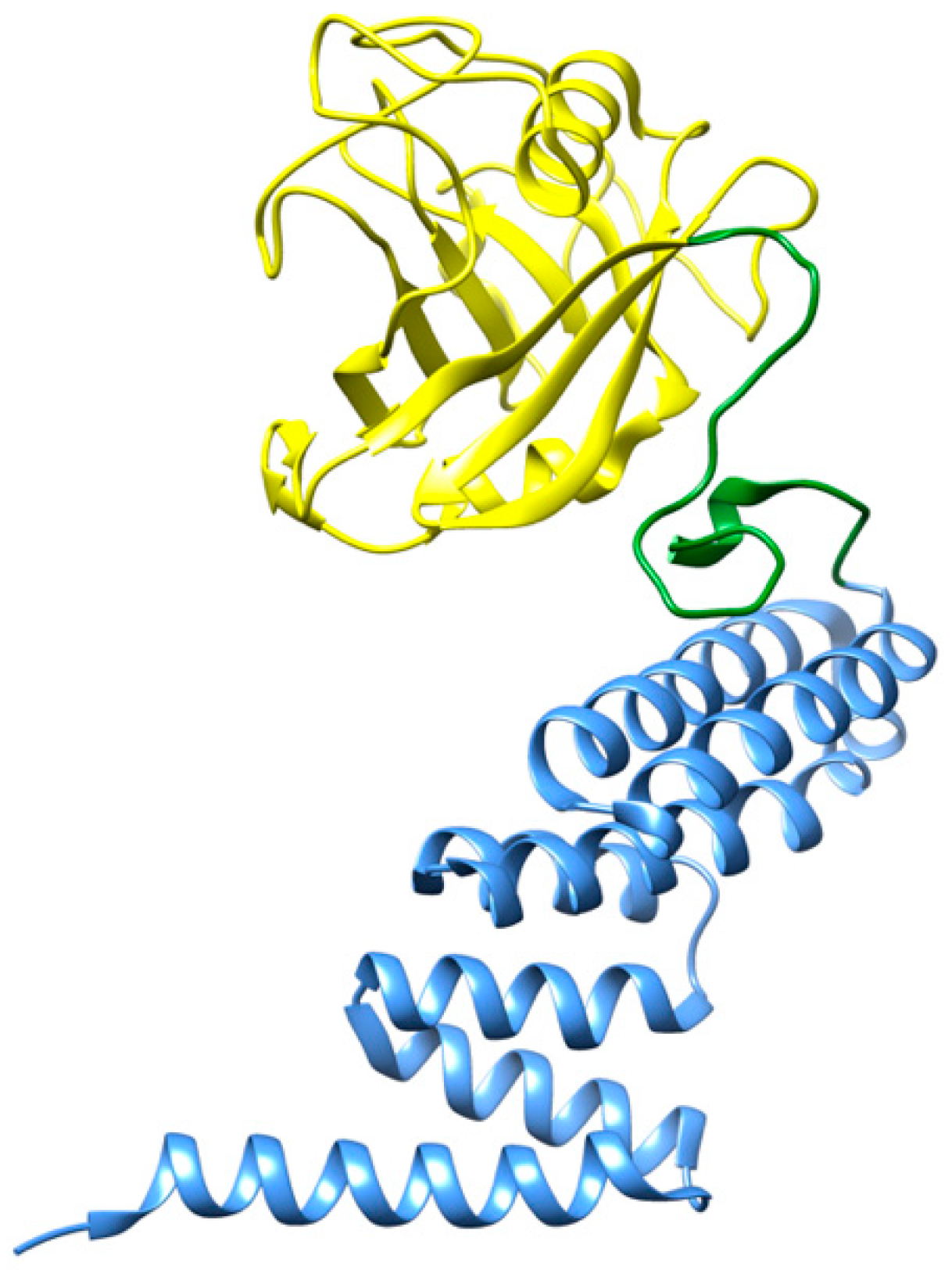
2.1.2. Hsp90
2.1.3. Tripartite Motif Proteins (TRIMs)
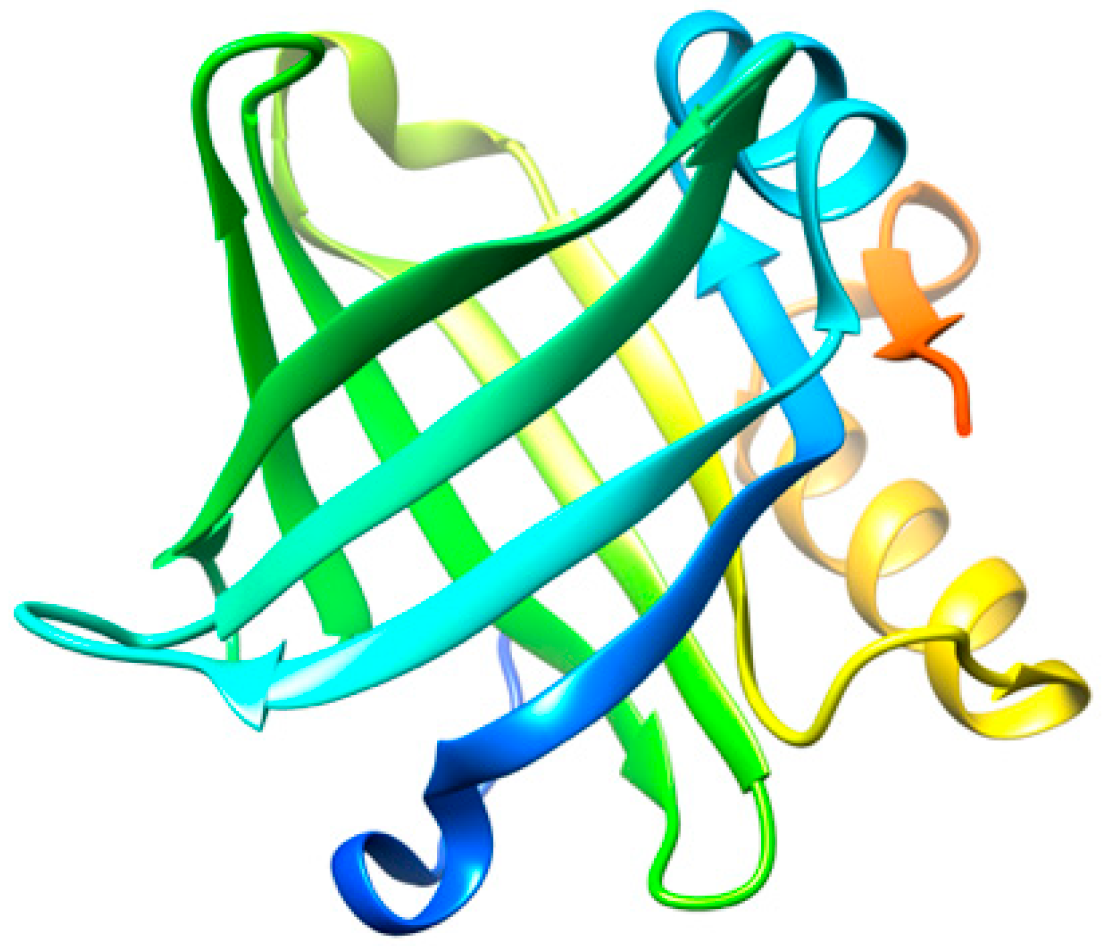
2.1.4. Lipocalin-Prostaglandin D Synthase (L-PGDS)
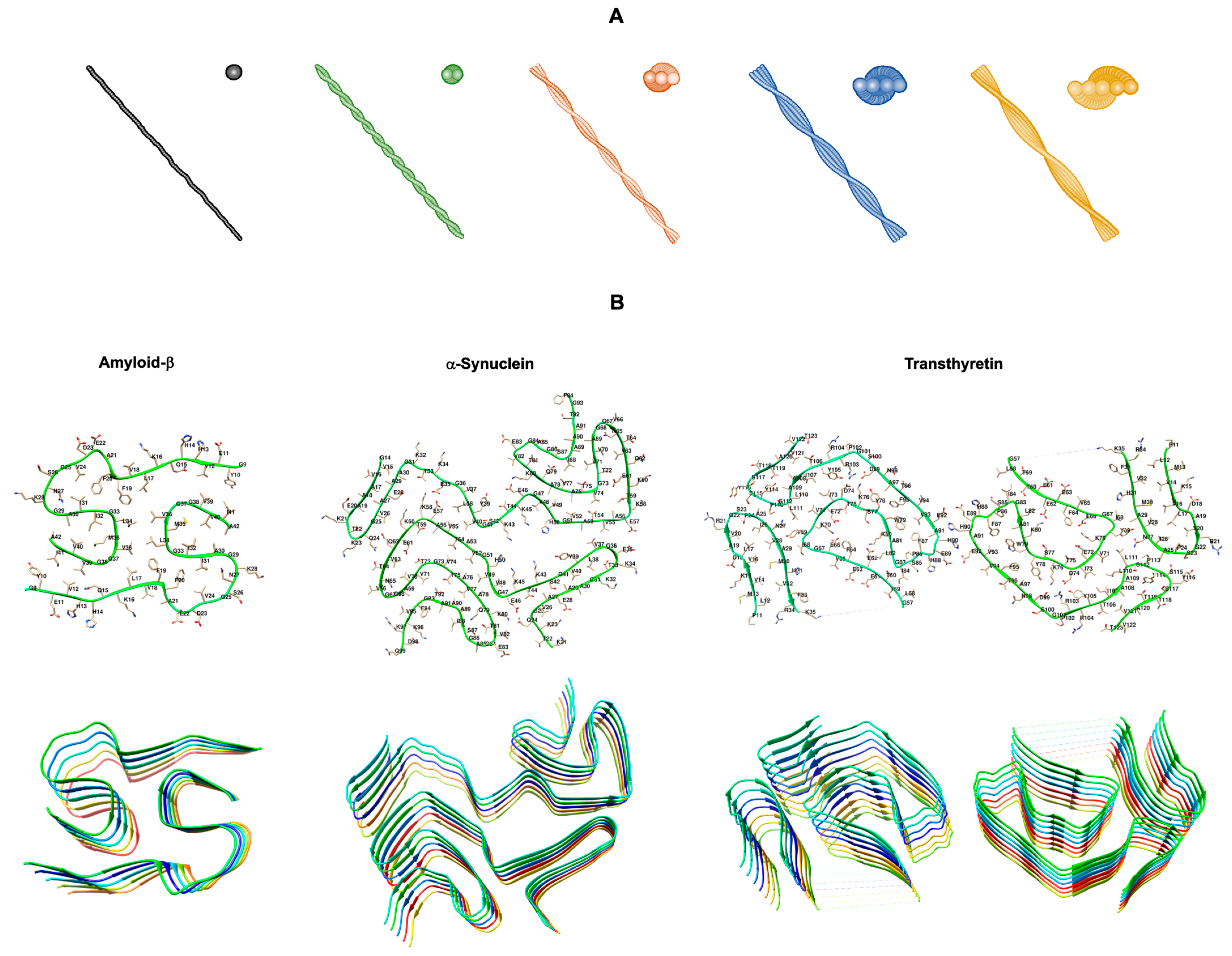
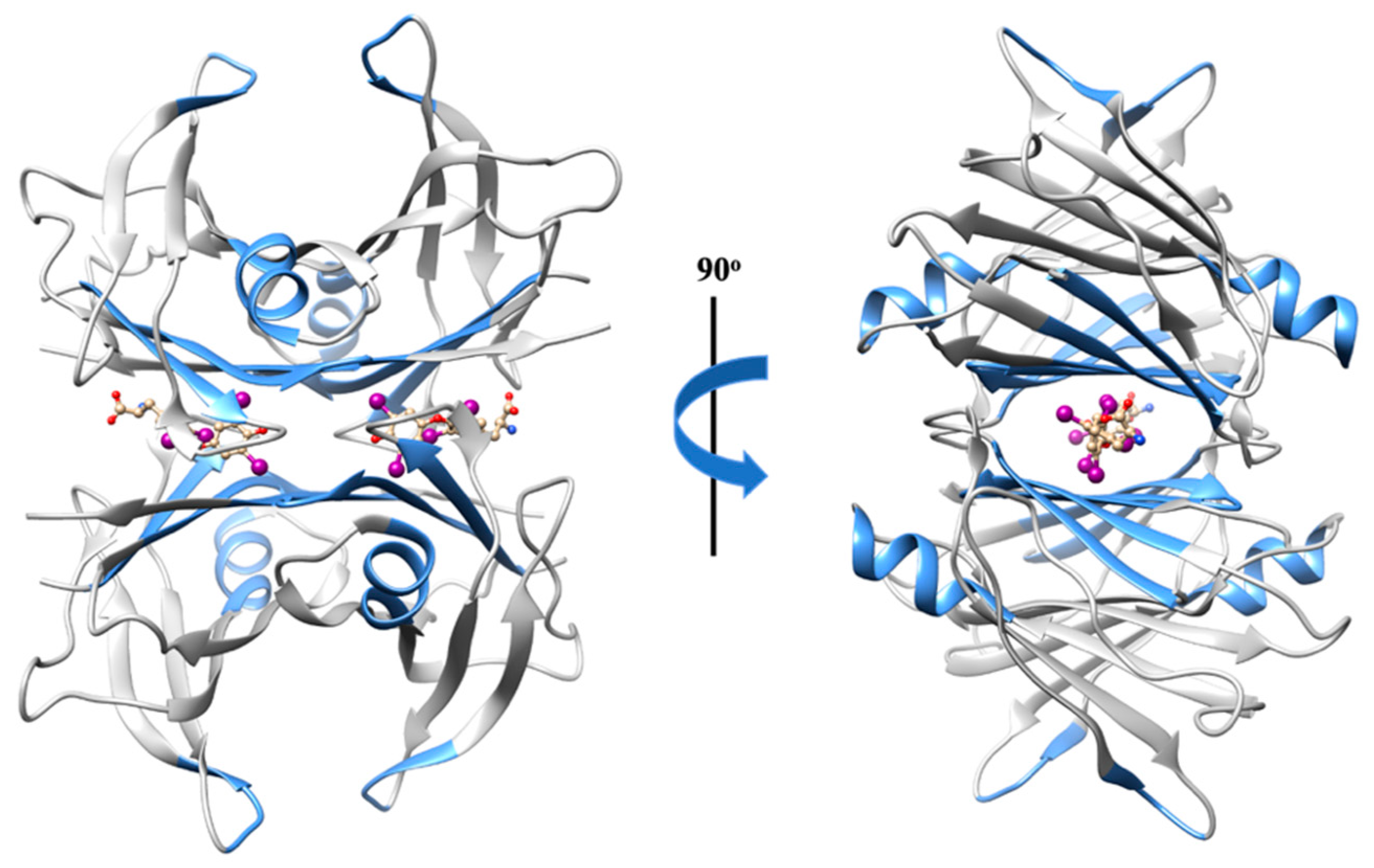
2.1.5. Transthyretin (TTR)
2.1.6. Cyclophilin 40 (CyP40)
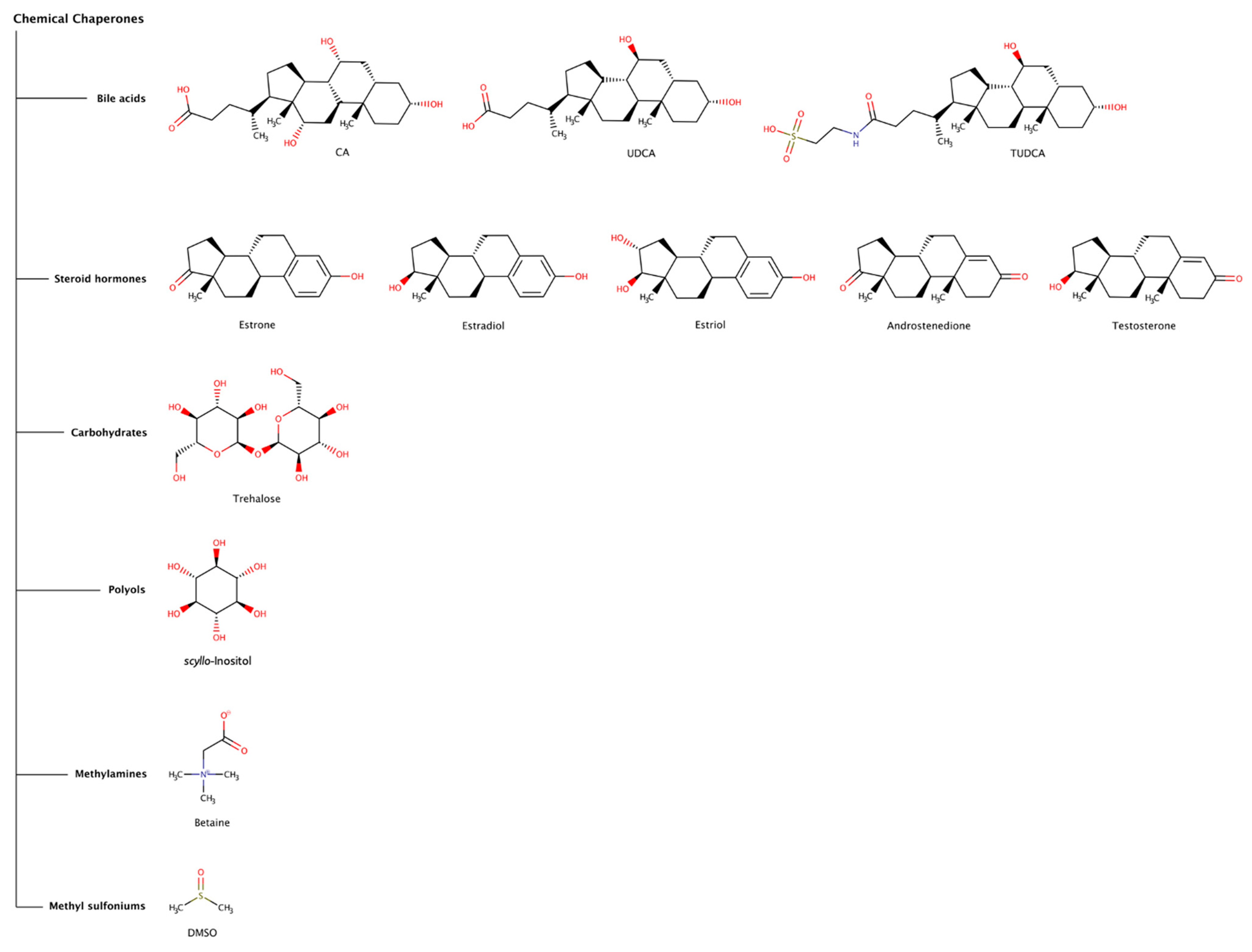
2.2. Chemical Chaperones in Amyloid Disassembly
2.2.1. Bile Acids
2.2.2. Steroid Hormones
2.2.3. Trehalose
2.2.4. Scyllo-Inositol
2.2.5. Betaine
2.2.6. Dimethyl Sulfoxide (DMSO)
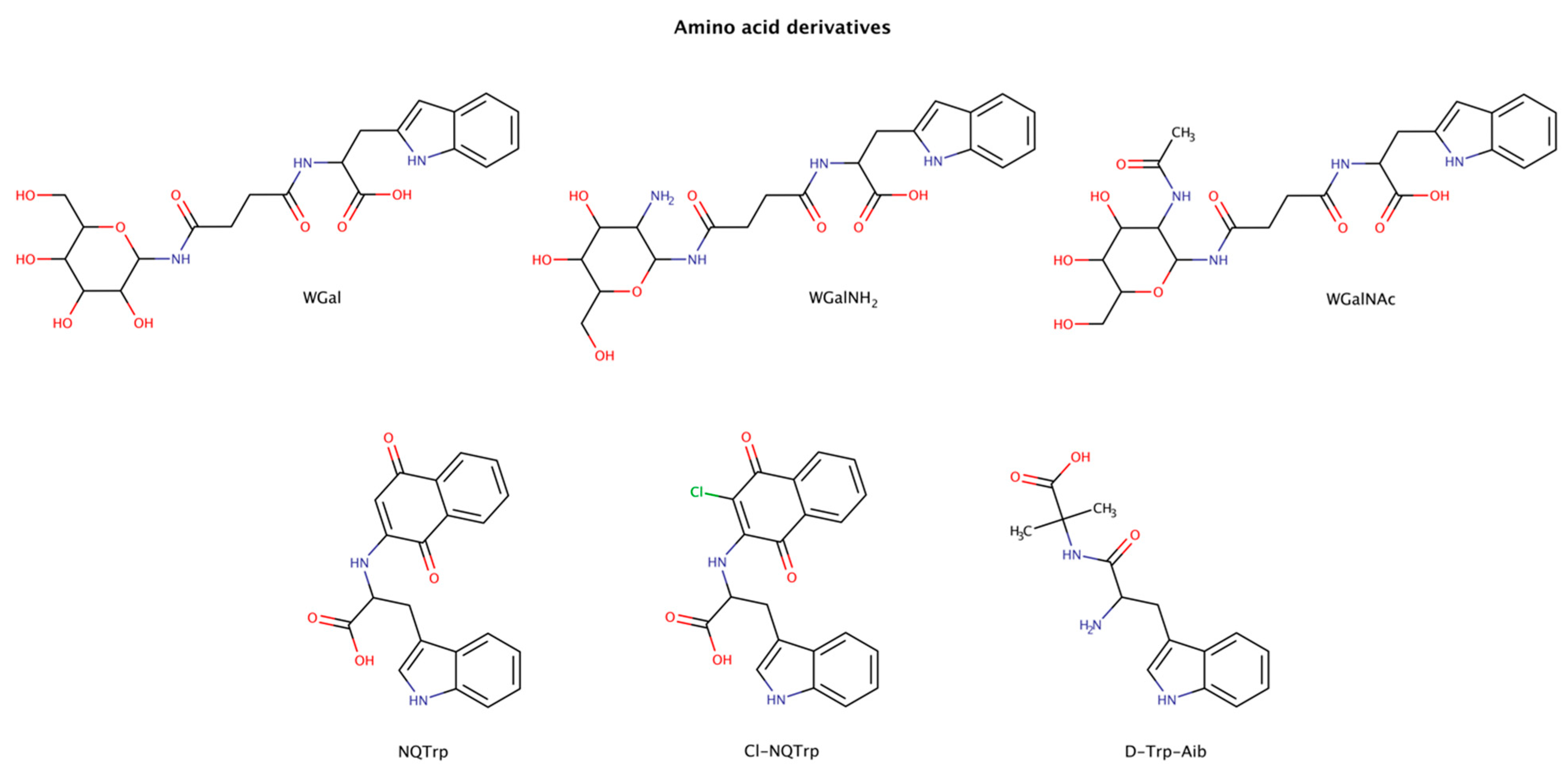
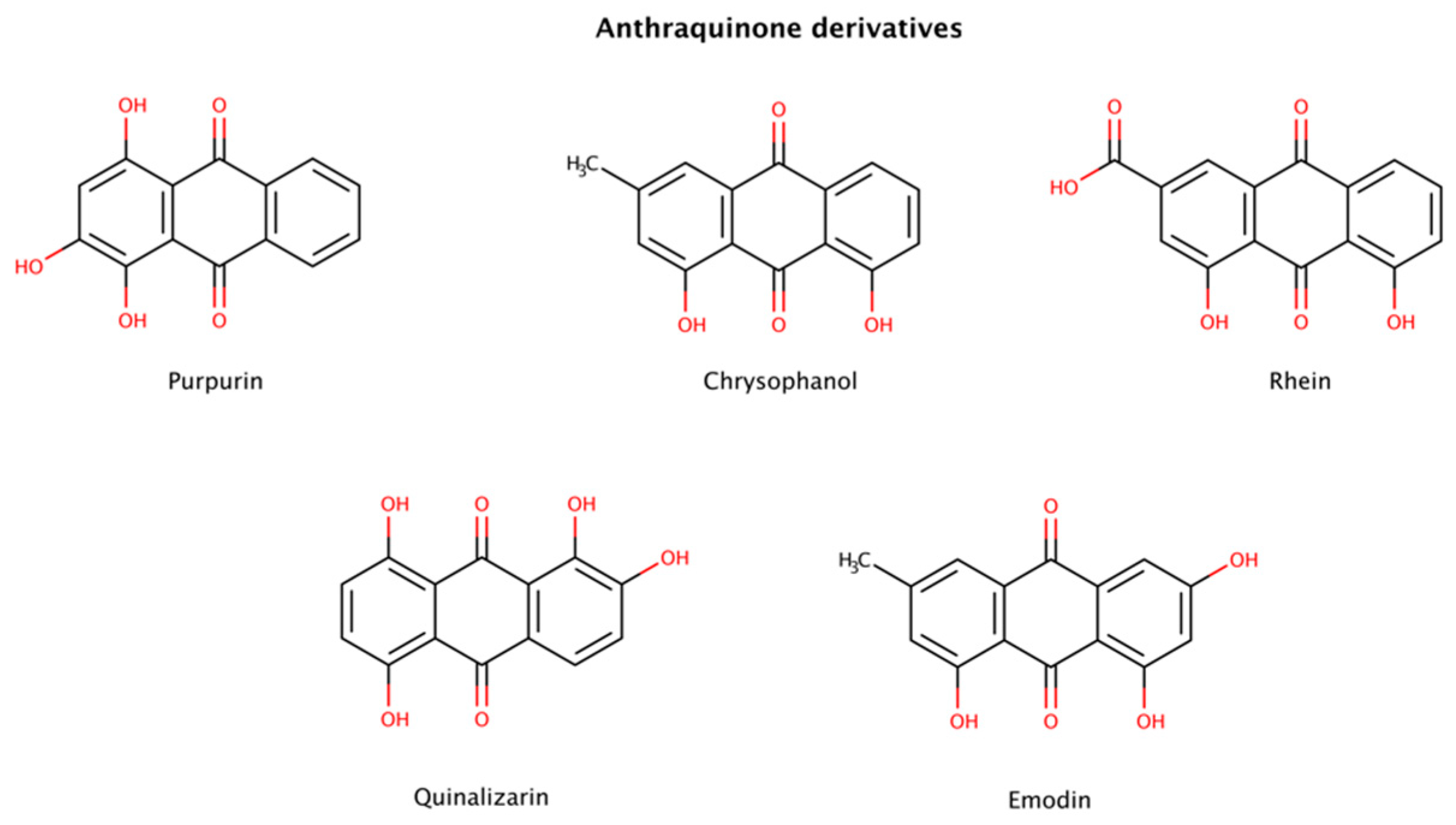
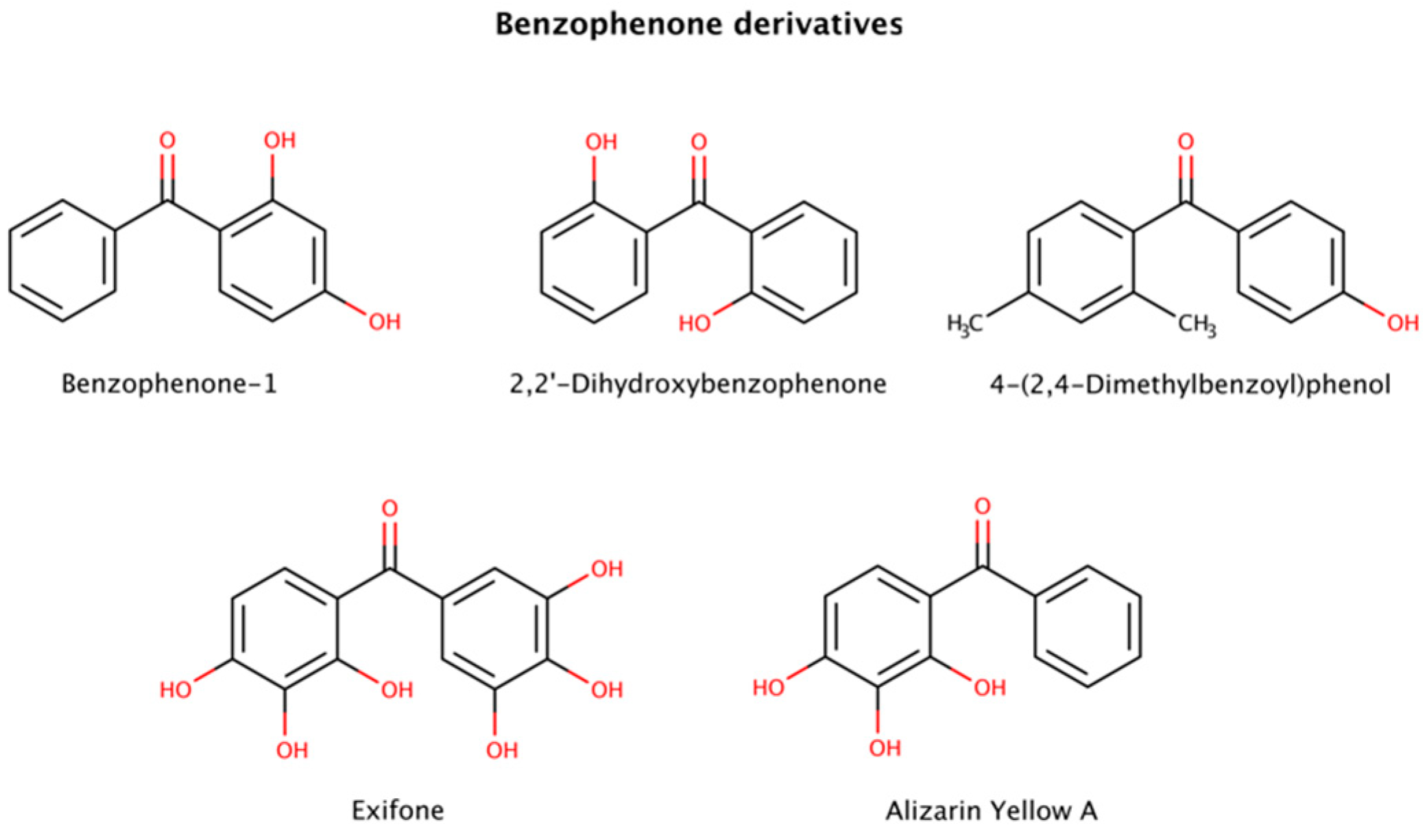
2.3. Pharmacological Chaperones in Amyloid Disassembly
2.3.1. Amino-Acid Derivatives
2.3.2. Anthraquinone Derivatives
2.3.3. Benzophenone Derivatives
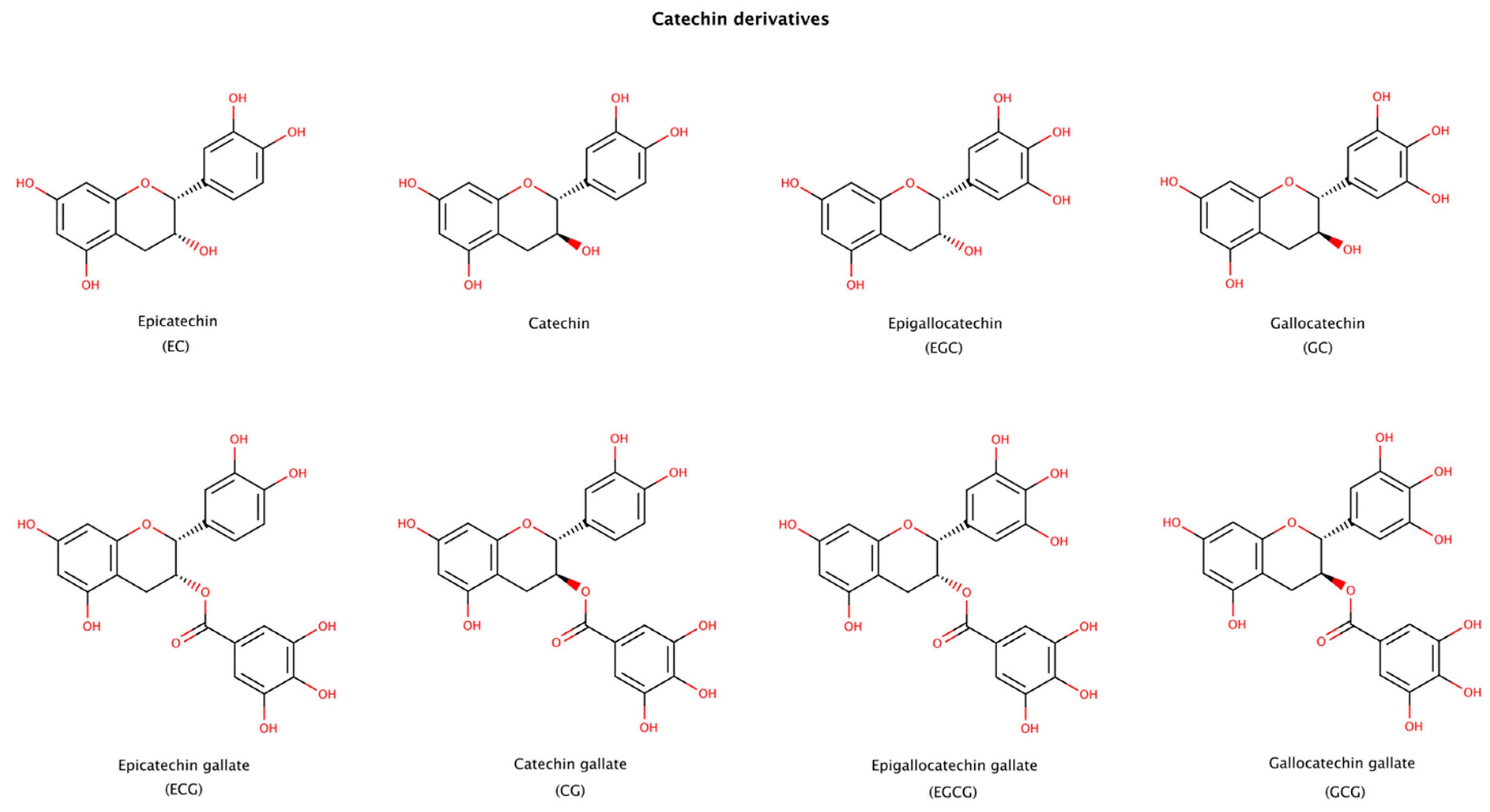
2.3.4. Catechin Derivatives
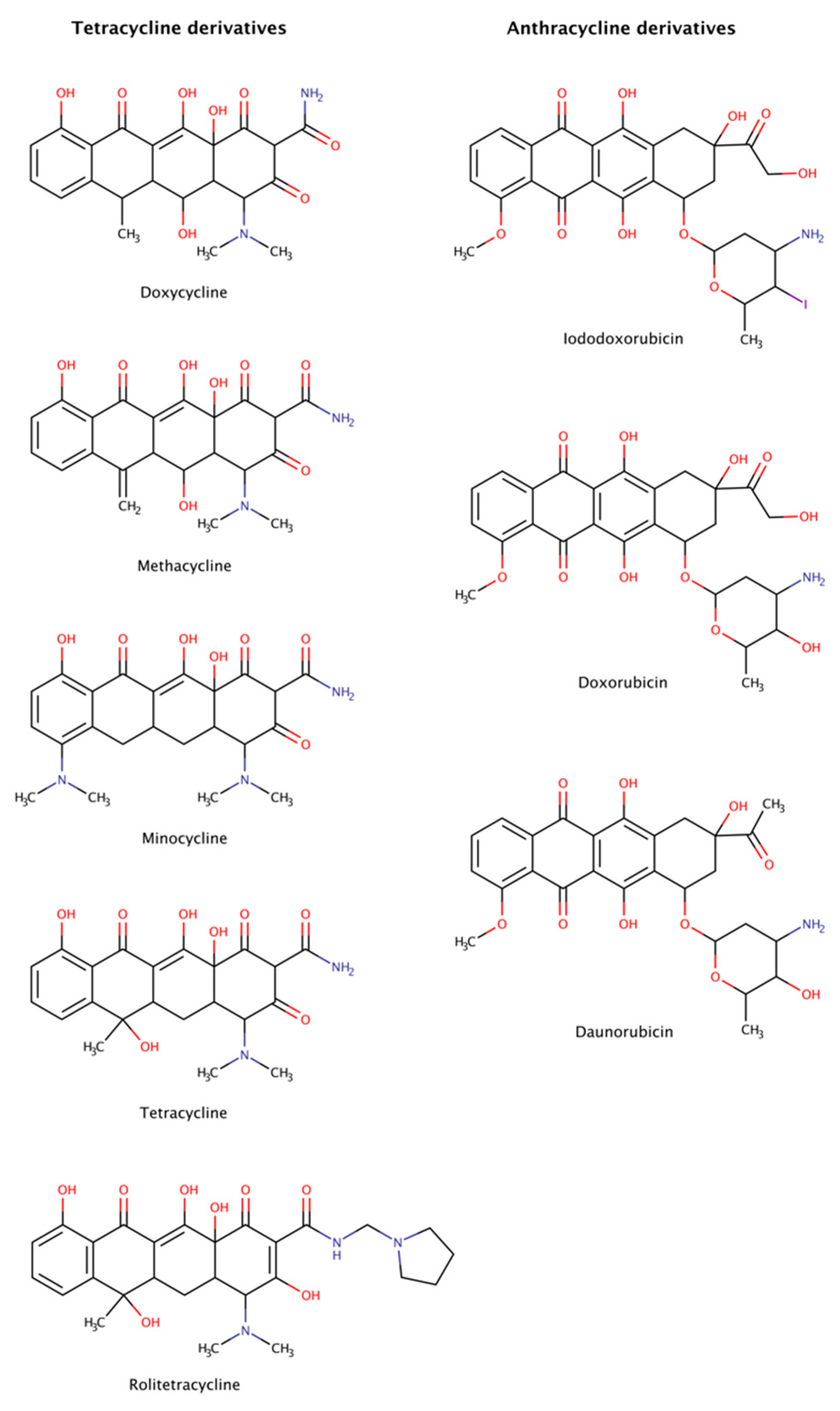
2.3.5. Anthracycline and Tetracycline Derivatives
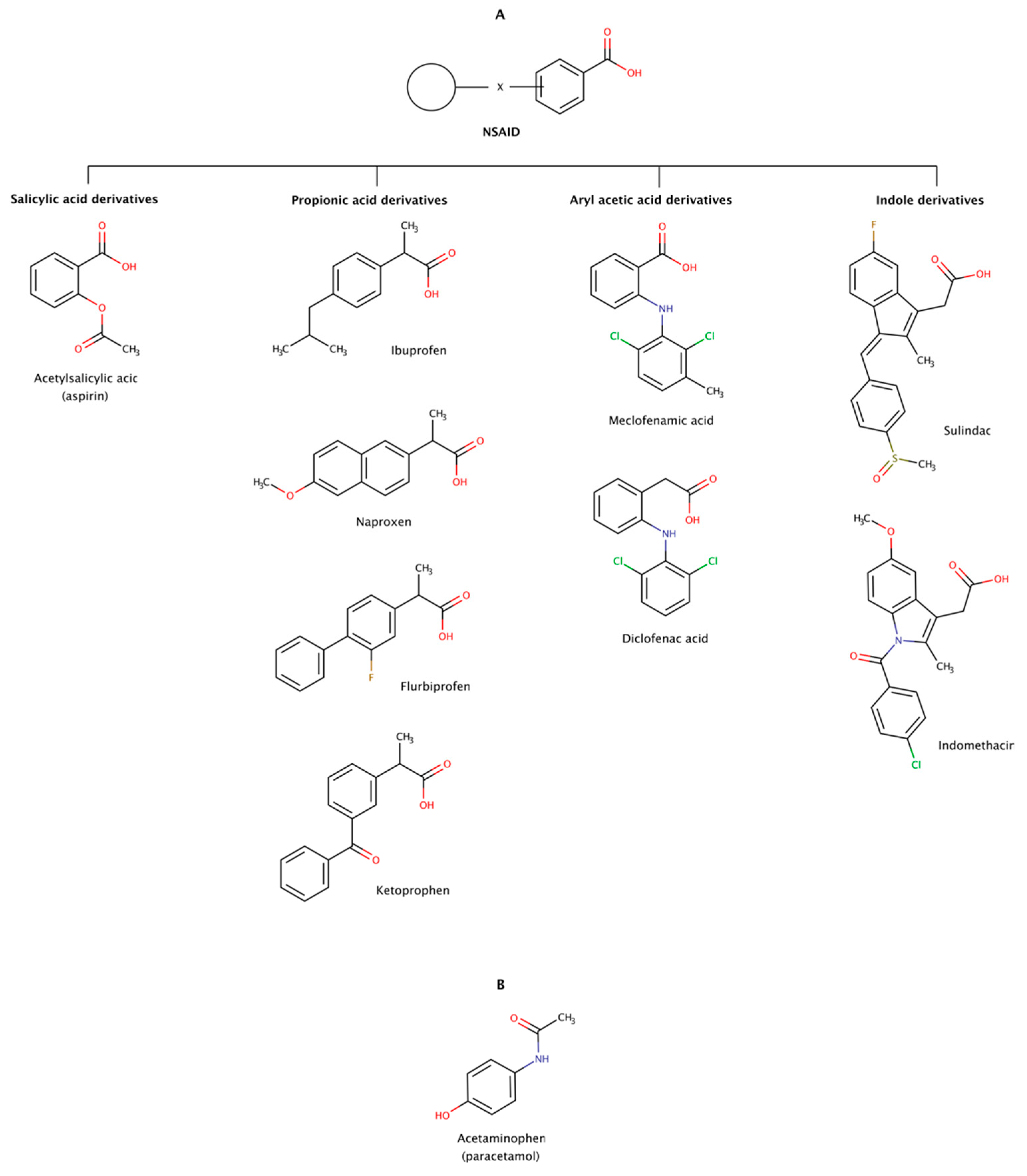
2.3.6. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
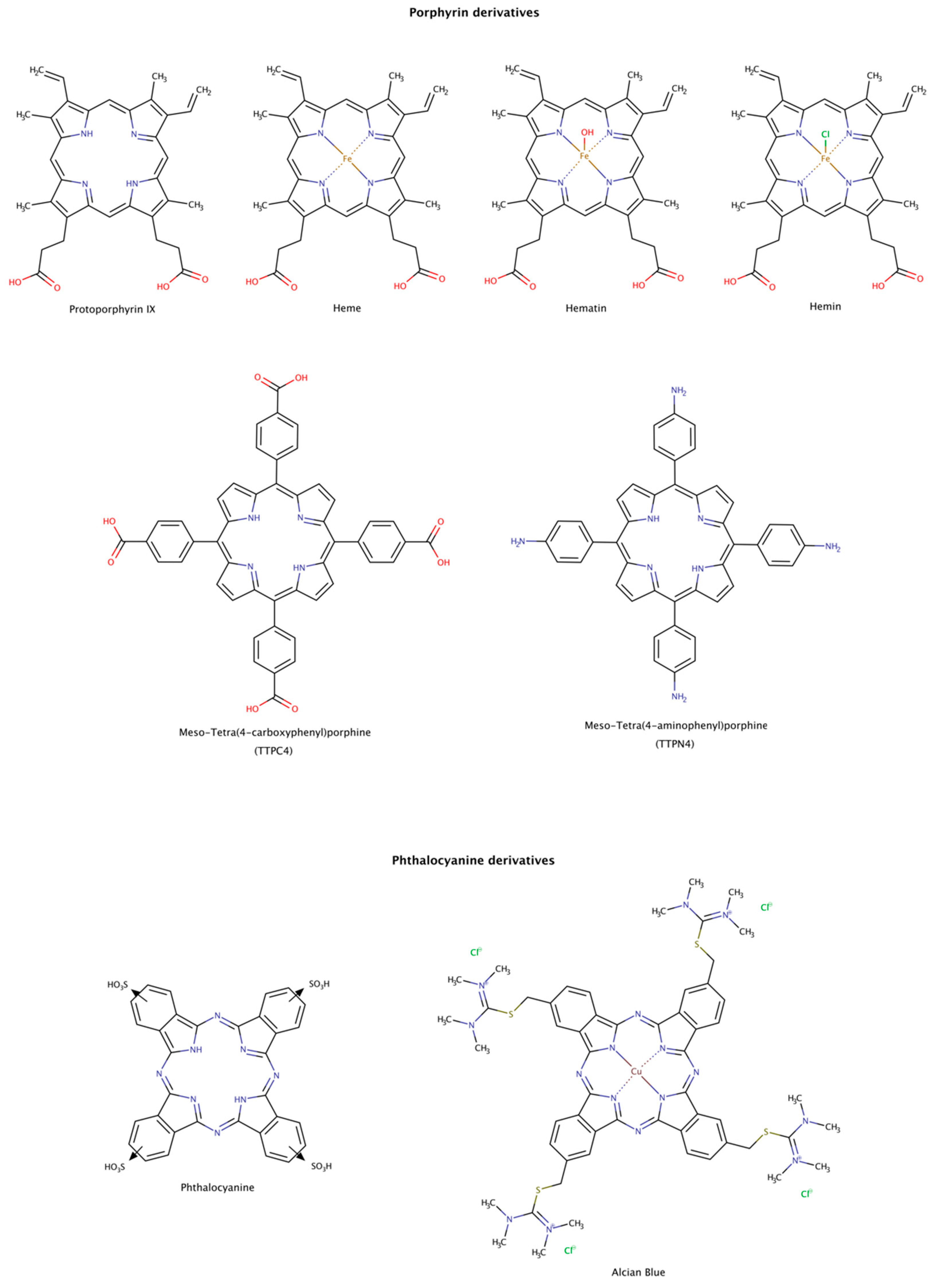
2.3.7. Porphyrin and Phthalocyanine Derivatives
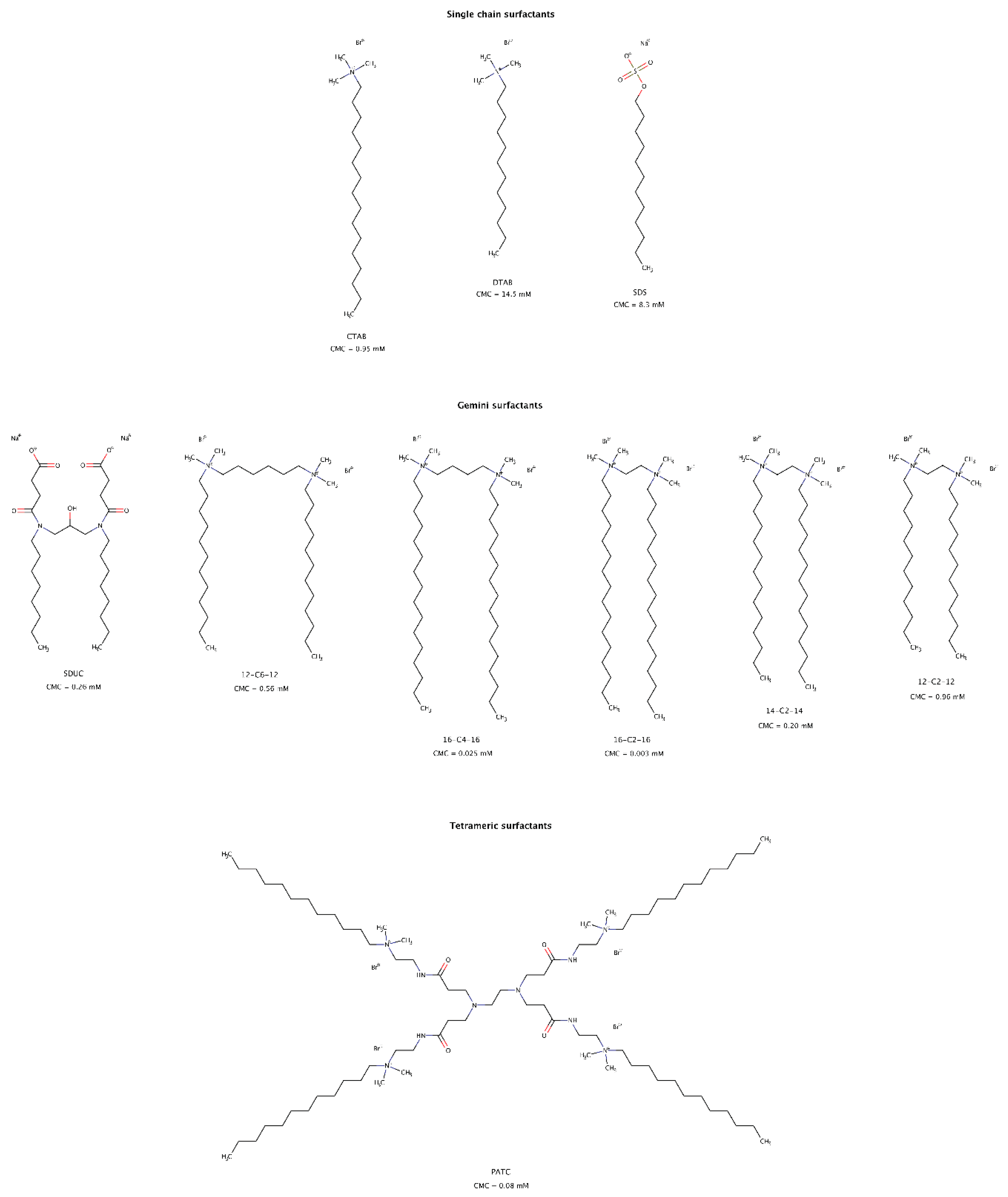
2.3.8. Surfactants
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Almeida, Z.L.; Brito, R.M.M. Structure and Aggregation Mechanisms in Amyloids. Molecules 2020, 25, 1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamcik, J.; Jung, J.M.; Flakowski, J.; De Los Rios, P.; Dietler, G.; Mezzenga, R. Understanding amyloid aggregation by statistical analysis of atomic force microscopy images. Nat. Nanotechnol. 2010, 5, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lövestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; MacDonald, J.; et al. Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 2022, 375, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Wiese, S.; Adak, V.; Engler, J.; Agarwal, S.; Fritz, G.; Westermark, P.; Zacharias, M.; Fändrich, M. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Pujols, J.; Peña-Díaz, S.; Pallarès, I.; Ventura, S. Chemical Chaperones as Novel Drugs for Parkinson’s Disease. Trends Mol. Med. 2020, 26, 408–421. [Google Scholar] [CrossRef]
- Ren, B.; Liu, Y.; Zhang, Y.; Zhang, M.; Sun, Y.; Liang, G.; Xu, J.; Zheng, J. Tanshinones inhibit hIAPP aggregation, disaggregate preformed hIAPP fibrils, and protect cultured cells. J. Mater. Chem. B 2017, 6, 56–67. [Google Scholar] [CrossRef]
- Choi, E.Y.; Kang, S.S.; Lee, S.K.; Han, B.H. Polyphenolic biflavonoids inhibit amyloid-beta fibrillation and disaggregate preformed amyloid-beta fibrils. Biomol. Ther. 2020, 28, 145–151. [Google Scholar] [CrossRef]
- Khan, A.N.; Qureshi, I.A.; Khan, U.K.; Uversky, V.N.; Khan, R.H. Inhibition and disruption of amyloid formation by the antibiotic levofloxacin: A new direction for antibiotics in an era of multi-drug resistance. Arch. Biochem. Biophys. 2021, 714, 109077. [Google Scholar] [CrossRef]
- Abioye, R.O.; Okagu, O.D.; Udenigwe, C.C. Inhibition of Islet Amyloid Polypeptide Fibrillation by Structurally Diverse Phenolic Compounds and Fibril Disaggregation Potential of Rutin and Quercetin. J. Agric. Food Chem. 2022, 70, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Mandal, H.; Basak, A.; Prabhu, T.; Kolli, V.; Sarkar, N. Inhibitory as well as Disaggregation Potential of Selected Hydroxy Benzoic Phytochemicals on Hen Egg-White Lysozyme Amyloidogenesis. Curr. Proteomics 2021, 18, 349–361. [Google Scholar] [CrossRef]
- Huang, C.; Rossi, P.; Saio, T.; Kalodimos, C.G. Structural basis for the antifolding activity of a molecular chaperone. Nature 2016, 537, 202–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; Kwon, Y.T. Protein quality control by molecular chaperones in neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannini, B.; Chiti, F. Chaperones as suppressors of protein misfolded oligomer toxicity. Front. Mol. Neurosci. 2017, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Low, K.J.Y.; Venkatraman, A.; Mehta, J.S.; Pervushin, K. Molecular mechanisms of amyloid disaggregation. J. Adv. Res. 2022, 36, 113–132. [Google Scholar] [CrossRef]
- Ohtsuka, K.; Kawashima, D.; Gu, Y.; Saito, K. Inducers and co-inducers of molecular chaperones. Int. J. Hyperth. 2005, 21, 703–711. [Google Scholar] [CrossRef]
- Shorter, J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 2011, 6, e26319. [Google Scholar] [CrossRef] [Green Version]
- Rüdiger, S.; Germeroth, L.; Schneider-Mergener, J.; Bukau, B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997, 16, 1501–1507. [Google Scholar] [CrossRef] [Green Version]
- Nillegoda, N.B.; Bukau, B. Metazoan Hsp70-based protein disaggregases: Emergence and mechanisms. Front. Mol. Biosci. 2015, 2, 57. [Google Scholar] [CrossRef]
- Nillegoda, N.B.; Stank, A.; Malinverni, D.; Alberts, N.; Szlachcic, A.; Barducci, A.; De Los Rios, P.; Wade, R.C.; Bukau, B. Evolution of an intricate J-protein network driving protein disaggregation in eukaryotes. Elife 2017, 6, e24560. [Google Scholar] [CrossRef] [PubMed]
- Duennwald, M.L.; Echeverria, A.; Shorter, J. Small Heat Shock Proteins Potentiate Amyloid Dissolution by Protein Disaggregases from Yeast and Humans. PLoS Biol. 2012, 10, e1001346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Carroni, M.; Nussbaum-Krammer, C.; Mogk, A.; Nillegoda, N.B.; Szlachcic, A.; Guilbride, D.L.; Saibil, H.R.; Mayer, M.P.; Bukau, B. Human Hsp70 Disaggregase Reverses Parkinson’s-Linked α-Synuclein Amyloid Fibrils. Mol. Cell 2015, 59, 781–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scior, A.; Buntru, A.; Arnsburg, K.; Ast, A.; Iburg, M.; Juenemann, K.; Pigazzini, M.L.; Mlody, B.; Puchkov, D.; Priller, J.; et al. Complete suppression of Htt fibrilization and disaggregation of Htt fibrils by a trimeric chaperone complex. EMBO J. 2018, 37, 282–299. [Google Scholar] [CrossRef]
- Ferrari, L.; Geerts, W.J.C.; van Wezel, M.; Kos, R.; Konstantoulea, A.; van Bezouwen, L.S.; Förster, F.G.; Rüdiger, S.G.D. Human chaperones untangle fibrils of the Alzheimer protein Tau. bioRxiv 2018, 426650. [Google Scholar] [CrossRef]
- Nachman, E.; Wentink, A.S.; Madiona, K.; Bousset, L.; Katsinelos, T.; Allinson, K.; Kampinga, H.; McEwan, W.A.; Jahn, T.R.; Melki, R.; et al. Disassembly of Tau fibrils by the human Hsp70 disaggregation machinery generates small seeding-competent species. J. Biol. Chem. 2020, 295, 9676–9690. [Google Scholar] [CrossRef]
- Nillegoda, N.B.; Kirstein, J.; Szlachcic, A.; Berynskyy, M.; Stank, A.; Stengel, F.; Arnsburg, K.; Gao, X.; Scior, A.; Aebersold, R.; et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 2015, 524, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Mok, S.A.; Condello, C.; Freilich, R.; Gillies, A.; Arhar, T.; Oroz, J.; Kadavath, H.; Julien, O.; Assimon, V.A.; Rauch, J.N.; et al. Mapping interactions with the chaperone network reveals factors that protect against tau aggregation. Nat. Struct. Mol. Biol. 2018, 25, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Tittelmeier, J.; Sandhof, C.A.; Ries, H.M.; Druffel-Augustin, S.; Mogk, A.; Bukau, B.; Nussbaum-Krammer, C. The HSP110/HSP70 disaggregation system generates spreading-competent toxic α-synuclein species. EMBO J. 2020, 39, e103954. [Google Scholar] [CrossRef]
- Carlomagno, Y.; Zhang, Y.; Davis, M.; Lin, W.L.; Cook, C.; Dunmore, J.; Tay, W.; Menkosky, K.; Cao, X.; Petrucelli, L.; et al. Casein kinase II induced polymerization of soluble TDP-43 into filaments is inhibited by heat shock proteins. PLoS ONE 2014, 9, e90452. [Google Scholar] [CrossRef]
- Ali, Y.O.; Allen, H.M.; Yu, L.; Li-Kroeger, D.; Bakhshizadehmahmoudi, D.; Hatcher, A.; McCabe, C.; Xu, J.; Bjorklund, N.; Taglialatela, G.; et al. NMNAT2:HSP90 Complex Mediates Proteostasis in Proteinopathies. PLOS Biol. 2016, 14, e1002472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wu, H.; Wu, W.; Zhuo, W.; Liu, W.; Zhang, Y.; Cheng, M.; Chen, Y.G.; Gao, N.; Yu, H.; et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014, 24, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, F.P.; Haubrich, K.; Perez-Borrajero, C.; Hennig, J. Emerging RNA-binding roles in the TRIM family of ubiquitin ligases. Biol. Chem. 2019, 400, 1443–1464. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Harischandra, D.S.; Ghaisas, S.; Zhang, P.; Prall, W.; Huang, L.; Maghames, C.; Guo, L.; Luna, E.; Mack, K.L.; et al. TRIM11 Prevents and Reverses Protein Aggregation and Rescues a Mouse Model of Parkinson’s Disease. Cell Rep. 2020, 33, 108418. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Giasson, B.I.; Glavis-Bloom, A.; Brewer, M.D.; Shorter, J.; Gitler, A.D.; Yang, X. A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol. Cell 2014, 55, 15–30. [Google Scholar] [CrossRef] [Green Version]
- Tumani, H.; Huss, A.; Bachhuber, F. The cerebrospinal fluid and barriers—Anatomic and physiologic considerations. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 146, pp. 3–20. [Google Scholar]
- Lescuyer, P.; Gandini, A.; Burkhard, P.R.; Hochstrasser, D.F.; Sanchez, J.-C. Prostaglandin D2 synthase and its post-translational modifications in neurological disorders. Electrophoresis 2005, 26, 4563–4570. [Google Scholar] [CrossRef]
- Qu, W.M.; Huang, Z.L.; Xu, X.H.; Aritake, K.; Eguchi, N.; Nambu, F.; Narumiya, S.; Urade, Y.; Hayaishi, O. Lipocalin-type prostaglandin D syntase produces prostaglandin D2 involved in regulation of physiological sleep. Proc. Natl. Acad. Sci. USA 2006, 103, 17949–17954. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, T.; Ban, T.; Aritake, K.; Huang, Z.L.; Qu, W.M.; Okazaki, I.; Mohri, I.; Murayama, S.; Ozono, K.; Taniike, M.; et al. Lipocalin-type prostaglandin D synthase/β-trace is a major amyloid β-chaperone in human cerebrospinal fluid. Proc. Natl. Acad. Sci. USA 2007, 104, 6412–6417. [Google Scholar] [CrossRef] [Green Version]
- Kannaian, B.; Sharma, B.; Phillips, M.; Chowdhury, A.; Manimekalai, M.S.S.; Adav, S.S.; Ng, J.T.Y.; Kumar, A.; Lim, S.; Mu, Y.; et al. Abundant neuroprotective chaperone Lipocalin-type prostaglandin D synthase (L-PGDS) disassembles the Amyloid-β fibrils. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Esler, W.P.; Stimson, E.R.; Ghilardi, J.R.; Lu, Y.-A.; Felix, A.M.; Vinters, H.V.; Mantyh, P.W.; Lee, J.P.; Maggio, J.E. Point Substitution in the Central Hydrophobic Cluster of a Human-Amyloid Congener Disrupts Peptide Folding and Abolishes Plaque Competence. Biochemistry 1996, 35, 13914–13921. [Google Scholar] [CrossRef]
- Fukuyama, R.; Mizuno, T.; Mizuno, T.; Mori, S.; Nakajima, K.; Fushiki, S.; Yanagisawa, K. Age-dependent change in the levels of Aβ40 and Aβ42 in cerebrospinal fluid from control subjects, and a decrease in the ratio of Aβ42 to Aβ40 level in cerebrospinal fluid from Alzheimer’s disease patients. Eur. Neurol. 2000, 43, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Puchades, M.; Hansson, S.F.; Nilsson, C.L.; Andreasen, N.; Blennow, K.; Davidsson, P. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer’s disease. Mol. Brain Res. 2003, 118, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Hansson, S.F.; Andréasson, U.; Wall, M.; Skoog, I.; Andreasen, N.; Wallin, A.; Zetterberg, H.; Blennow, K. Reduced levels of amyloid-β-binding proteins in cerebrospinal fluid from Alzheimer’s disease patients. J. Alzheimer’s Dis. 2009, 16, 389–397. [Google Scholar] [CrossRef]
- Frangolho, A.; Correia, B.E.; Vaz, D.C.; Almeida, Z.L.; Brito, R.M.M. Oligomerization Profile of Human Transthyretin Variants with Distinct Amyloidogenicity. Molecules 2020, 25, 5698. [Google Scholar] [CrossRef]
- Ferreira, E.; Almeida, Z.L.; Cruz, P.F.; Silva e Sousa, M.; Veríssimo, P.; Brito, R.M.M. Searching for the Best Transthyretin Aggregation Protocol to Study Amyloid Fibril Disruption. Int. J. Mol. Sci. 2021, 23, 391. [Google Scholar] [CrossRef] [PubMed]
- Quintas, A.; Vaz, D.C.; Cardoso, I.; Saraiva, M.J.M.; Brito, R.M.M. Tetramer Dissociation and Monomer Partial Unfolding Precedes Protofibril Formation in Amyloidogenic Transthyretin Variants. J. Biol. Chem. 2001, 276, 27207–27213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurshman, A.R.; White, J.T.; Powers, E.T.; Kelly, J.W. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 2004, 43, 7365–7381. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Murphy, R.M. Characterization of the interaction of β-Amyloid with Transthyretin monomers and tetramers. Biochemistry 2010, 49, 8276–8289. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, X.; Ladiwala, A.R.A.; Du, D.; Yadav, J.K.; Tessier, P.M.; Wright, P.E.; Kelly, J.W.; Buxbaum, J.N. Mechanisms of transthyretin inhibition of β-amyloid aggregation in vitro. J. Neurosci. 2013, 33, 19423–19433. [Google Scholar] [CrossRef] [Green Version]
- Cotrina, E.Y.; Gimeno, A.; Llop, J.; Jiménez-Barbero, J.; Quintana, J.; Valencia, G.; Cardoso, I.; Prohens, R.; Arsequell, G. Calorimetric Studies of Binary and Ternary Molecular Interactions between Transthyretin, Aβ Peptides, and Small-Molecule Chaperones toward an Alternative Strategy for Alzheimer’s Disease Drug Discovery. J. Med. Chem. 2020, 63, 3205–3214. [Google Scholar] [CrossRef]
- Nilsson, L.; Pamrén, A.; Islam, T.; Brännström, K.; Golchin, S.A.; Pettersson, N.; Iakovleva, I.; Sandblad, L.; Gharibyan, A.L.; Olofsson, A. Transthyretin Interferes with Aβ Amyloid Formation by Redirecting Oligomeric Nuclei into Non-Amyloid Aggregates. J. Mol. Biol. 2018, 430, 2722–2733. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Gonçalves, A.; Saraiva, M.J.; Cardoso, I. Transthyretin binding to A-Beta peptide - Impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008, 582, 936–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Buxbaum, J.N. Transthyretin and the brain re-visited: Is neuronal synthesis of transthyretin protective in Alzheimer’s disease? Mol. Neurodegener. 2011, 6, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascella, R.; Conti, S.; Mannini, B.; Li, X.; Buxbaum, J.N.; Tiribilli, B.; Chiti, F.; Cecchi, C. Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins in vitro. Biochim. Biophys. Acta—Mol. Basis Dis. 2013, 1832, 2302–2314. [Google Scholar] [CrossRef] [Green Version]
- Garai, K.; Posey, A.E.; Li, X.; Buxbaum, J.N.; Pappu, R.V. Inhibition of amyloid beta fibril formation by monomeric human transthyretin. Protein Sci. 2018, 27, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Vatassery, G.T.; Quach, H.T.; Smith, W.E.; Benson, B.A.; Eckfeldt, J.H. A sensitive assay of transthyretin (prealbumin) in human cerebrospinal fluid in nanogram amounts by ELISA. Clin. Chim. Acta 1991, 197, 19–25. [Google Scholar] [CrossRef]
- Smith, F.R.; Goodman, D.S. The effects of diseases of the liver, thyroid, and kidneys on the transport of vitamin A in human plasma. J. Clin. Invest. 1971, 50, 2426–2436. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Serot, J.M.; Christmann, D.; Dubost, T.; Couturier, M. Cerebrospinal fluid transthyretin: Aging and late onset Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 63, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Han, S.H.; Jung, E.S.; Sohn, J.H.; Hong, H.J.; Hong, H.S.; Kim, J.W.; Na, D.L.; Kim, M.; Kim, H.; Ha, H.J.; et al. Human serum transthyretin levels correlate inversely with Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 25, 77–84. [Google Scholar] [CrossRef]
- Gimeno, A.; Santos, L.M.; Alemi, M.; Rivas, J.; Blasi, D.; Cotrina, E.Y.; Llop, J.; Valencia, G.; Cardoso, I.; Quintana, J.; et al. Insights on the Interaction between Transthyretin and Aβ in Solution. A Saturation Transfer Difference (STD) NMR Analysis of the Role of Iododiflunisal. J. Med. Chem. 2017, 60, 5749–5758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzman, A.L.; Goldgaber, D. Interaction of transthyretin with amyloid β-protein: Binding and inhibition of amyloid formation. CIBA Found. Symp. 1996, 199, 146–164. [Google Scholar] [PubMed]
- Du, J.; Cho, P.Y.; Yang, D.T.; Murphy, R.M. Identification of beta-amyloid-binding sites on transthyretin. Protein Eng. Des. Sel. 2012, 25, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, J.D.; Shelton, L.B.; Zheng, D.; Favretto, F.; Nordhues, B.A.; Darling, A.; Sullivan, L.E.; Sun, Z.; Solanki, P.K.; Martin, M.D.; et al. Human cyclophilin 40 unravels neurotoxic amyloids. PLOS Biol. 2017, 15, e2001336. [Google Scholar] [CrossRef] [Green Version]
- Gur, M.; Blackburn, E.A.; Ning, J.; Narayan, V.; Ball, K.L.; Walkinshaw, M.D.; Erman, B. Molecular dynamics simulations of site point mutations in the TPR domain of cyclophilin 40 identify conformational states with distinct dynamic and enzymatic properties. J. Chem. Phys. 2018, 148, 145101. [Google Scholar] [CrossRef] [Green Version]
- Papp, E.; Csermely, P. Chemical chaperones: Mechanisms of action and potential use. Handb. Exp. Pharmacol. 2006, 172, 405–416. [Google Scholar]
- Kushwah, N.; Jain, V.; Yadav, D. Osmolytes: A possible therapeutic molecule for ameliorating the neurodegeneration caused by protein misfolding and aggregation. Biomolecules 2020, 10, 132. [Google Scholar] [CrossRef] [Green Version]
- Schonewille, M.; de Boer, J.F.; Groen, A.K. Bile salts in control of lipid metabolism. Curr. Opin. Lipidol. 2016, 27, 295–301. [Google Scholar] [CrossRef]
- Ackerman, H.D.; Gerhard, G.S. Bile acids in neurodegenerative disorders. Front. Aging Neurosci. 2016, 8, 263. [Google Scholar] [CrossRef] [Green Version]
- Majid, N.; Siddiqi, M.K.; Khan, A.N.; Shabnam, S.; Malik, S.; Alam, A.; Uversky, V.N.; Khan, R.H. Biophysical Elucidation of Amyloid Fibrillation Inhibition and Prevention of Secondary Nucleation by Cholic Acid: An Unexplored Function of Cholic Acid. ACS Chem. Neurosci. 2019, 10, 4704–4715. [Google Scholar] [CrossRef]
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.S.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M.P. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.C.; Callaerts-Vegh, Z.; Nunes, A.F.; Rodrigues, C.M.P.; D’Hooge, R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 2013, 50, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Macedo, B.; Batista, A.R.; Ferreira, N.; Almeida, M.R.; Saraiva, M.J. Anti-apoptotic treatment reduces transthyretin deposition in a transgenic mouse model of Familial Amyloidotic Polyneuropathy. Biochim. Biophys. Acta—Mol. Basis Dis. 2008, 1782, 517–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, I.; Martins, D.; Ribeiro, T.; Merlini, G.; Saraiva, M.J. Synergy of combined Doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: Studies in FAP mouse models. J. Transl. Med. 2010, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obici, L.; Cortese, A.; Lozza, A.; Lucchetti, J.; Gobbi, M.; Palladini, G.; Perlini, S.; Saraiva, M.J.; Merlini, G. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 2012, 19, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Wixner, J.; Pilebro, B.; Lundgren, H.-E.; Olsson, M.; Anan, I. Effect of doxycycline and ursodeoxycholic acid on transthyretin amyloidosis. Amyloid 2017, 24, 78–79. [Google Scholar] [CrossRef]
- Karlstedt, E.; Jimenez-Zepeda, V.; Howlett, J.G.; White, J.A.; Fine, N.M. Clinical Experience With the Use of Doxycycline and Ursodeoxycholic Acid for the Treatment of Transthyretin Cardiac Amyloidosis. J. Card. Fail. 2019, 25, 147–153. [Google Scholar] [CrossRef]
- Wehling, M. Specific, Nongenomic Actions of Steroid Hormones. Annu. Rev. Physiol. 1997, 59, 365–393. [Google Scholar] [CrossRef]
- Campbell, C.M.; LoRusso, S.; Dispenzieri, A.; Kristen, A.V.; Maurer, M.S.; Rapezzi, C.; Lairez, O.; Drachman, B.; Garcia-Pavia, P.; Grogan, M.; et al. Sex Differences in Wild-Type Transthyretin Amyloidosis: An Analysis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Cardiol. Ther. 2022, 11, 393–405. [Google Scholar] [CrossRef]
- Caponetti, A.G.; Rapezzi, C.; Gagliardi, C.; Milandri, A.; Dispenzieri, A.; Kristen, A.V.; Wixner, J.; Maurer, M.S.; Garcia-Pavia, P.; Tournev, I.; et al. Sex-Related Risk of Cardiac Involvement in Hereditary Transthyretin Amyloidosis: Insights From THAOS. JACC Hear. Fail. 2021, 9, 736–746. [Google Scholar] [CrossRef]
- Van Den Eeden, S.K. Incidence of Parkinson’s Disease: Variation by Age, Gender, and Race/Ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Vest, R.S.; Pike, C.J. Gender, sex steroid hormones, and Alzheimer’s disease. Horm. Behav. 2013, 63, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morinaga, A.; Hirohata, M.; Ono, K.; Yamada, M. Estrogen has anti-amyloidogenic effects on Alzheimer’s β-amyloid fibrils in vitro. Biochem. Biophys. Res. Commun. 2007, 359, 697–702. [Google Scholar] [CrossRef]
- Hirohata, M.; Ono, K.; Morinaga, A.; Ikeda, T.; Yamada, M. Anti-aggregation and fibril-destabilizing effects of sex hormones on α-synuclein fibrils in vitro. Exp. Neurol. 2009, 217, 434–439. [Google Scholar] [CrossRef]
- Song, Y.; Li, S.; Li, X.; Chen, X.; Wei, Z.; Liu, Q.; Cheng, Y. The Effect of Estrogen Replacement Therapy on Alzheimer’s Disease and Parkinson’s Disease in Postmenopausal Women: A Meta-Analysis. Front. Neurosci. 2020, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Goodman, Y.; Bruce, A.J.; Cheng, B.; Mattson, M.P. Estrogens Attenuate and Corticosterone Exacerbates Excitotoxicity, Oxidative Injury, and Amyloid β-Peptide Toxicity in Hippocampal Neurons. J. Neurochem. 1996, 66, 1836–1844. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Shen, Y.; Yang, L.-B.; Lue, L.-F.; Finch, C.; Rogers, J. Estrogen Enhances Uptake of Amyloid β-Protein by Microglia Derived from the Human Cortex. J. Neurochem. 2002, 75, 1447–1454. [Google Scholar] [CrossRef]
- Ragonese, P.; D’Amelio, M.; Savettieri, G. Implications for Estrogens in Parkinson’s Disease: An Epidemiological Approach. Ann. N. Y. Acad. Sci. 2006, 1089, 373–382. [Google Scholar] [CrossRef]
- Pinkerton, J.A.V.; Henderson, V.W. Estrogen and cognition, with a focus on Alzheimer’s disease. Semin. Reprod. Med. 2005, 23, 172–179. [Google Scholar] [CrossRef]
- Yu, W.B.; Jiang, T.; Lan, D.M.; Lu, J.H.; Yue, Z.Y.; Wang, J.; Zhou, P. Trehalose inhibits fibrillation of A53T mutant alpha-synuclein and disaggregates existing fibrils. Arch. Biochem. Biophys. 2012, 523, 144–150. [Google Scholar] [CrossRef]
- Liu, R.; Barkhordarian, H.; Emadi, S.; Chan, B.P.; Sierks, M.R. Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiol. Dis. 2005, 20, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, R.; Kolli, V.; Sarkar, N. Trehalose and Magnesium Chloride Exert a Common Anti-amyloidogenic Effect Towards Hen Egg White Lysozyme. Protein J. 2017, 36, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Lan, C.P.; Hasan, S.; Brown, M.E.; McLaurin, J.A. Scyllo-Inositol promotes Robust Mutant Huntingtin Protein degradation. J. Biol. Chem. 2014, 289, 3666–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, T.; McLaurin, J.A. α-Synuclein aggregation, seeding and inhibition by scyllo-inositol. Biochem. Biophys. Res. Commun. 2016, 469, 529–534. [Google Scholar] [CrossRef]
- McLaurin, J.A.; Kierstead, M.E.; Brown, M.E.; Hawkes, C.A.; Lambermon, M.H.L.; Phinney, A.L.; Darabie, A.A.; Cousins, J.E.; French, J.E.; Lan, M.F.; et al. Cyclohexanehexol inhibitors of Aβ aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat. Med. 2006, 12, 801–808. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Keren, R.; Porsteinsson, A.P.; Van Dyck, C.H.; Tariot, P.N.; Gilman, S.; Arnold, D.; Abushakra, S.; Hernandez, C.; et al. A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology 2011, 77, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Fenili, D.; Brown, M.; Rappaport, R.; McLaurin, J.A. Properties of scyllo-inositol as a therapeutic treatment of AD-like pathology. J. Mol. Med. 2007, 85, 603–611. [Google Scholar] [CrossRef]
- Townsend, M.; Cleary, J.P.; Mehta, T.; Hofmeister, J.; Lesne, S.; O’Hare, E.; Walsh, D.M.; Selkoe, D.J. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-β oligomers. Ann. Neurol. 2006, 60, 668–676. [Google Scholar] [CrossRef]
- Ma, K.; Thomason, L.A.M.; McLaurin, J.A. Scyllo-Inositol, Preclinical, and Clinical Data for Alzheimer’s Disease. In Advances in Pharmacology; Academic Press Inc.: Cambridge, MA, USA, 2012; Volume 64, pp. 177–212. [Google Scholar]
- Natalello, A.; Liu, J.; Ami, D.; Doglia, S.M.; de Marco, A. The osmolyte betaine promotes protein misfolding and disruption of protein aggregates. Proteins Struct. Funct. Bioinforma. 2009, 75, 509–517. [Google Scholar] [CrossRef]
- De Marco, A.; Vigh, L.; Diamant, S.; Goloubinoff, P. Native folding of aggregation-prone recombinant proteins in Escherichia coli by osmolytes, plasmid- or benzyl alcohol-overexpressed molecular chaperones. Cell Stress Chaperones 2005, 10, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Santos, N.C.; Figueira-Coelho, J.; Martins-Silva, J.; Saldanha, C. Multidisciplinary utilization of dimethyl sulfoxide: Pharmacological, cellular, and molecular aspects. Biochem. Pharmacol. 2003, 65, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Hanslick, J.L.; Lau, K.; Noguchi, K.K.; Olney, J.W.; Zorumski, C.F.; Mennerick, S.; Farber, N.B. Dimethyl sulfoxide (DMSO) produces widespread apoptosis in the developing central nervous system. Neurobiol. Dis. 2009, 34, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasrallah, F.A.; Garner, B.; Ball, G.E.; Rae, C. Modulation of brain metabolism by very low concentrations of the commonly used drug delivery vehicle dimethyl sulfoxide (DMSO). J. Neurosci. Res. 2008, 86, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Institóris, Á.; Domoki, F.; Mihály, A.; Luiten, P.G.M.; Bari, F. Diazoxide and dimethyl sulphoxide prevent cerebral hypoperfusion-related learning dysfunction and brain damage after carotid artery occlusion. Brain Res. 2004, 1008, 252–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota-Nakaoka, N. Dissolution of 2-Microglobulin Amyloid Fibrils by Dimethylsulfoxide. J. Biochem. 2003, 134, 159–164. [Google Scholar] [CrossRef]
- Loksztejn, A.; Dzwolak, W. Noncooperative dimethyl sulfoxide-induced dissection of insulin fibrils: Toward soluble building blocks of amyloid. Biochemistry 2009, 48, 4846–4851. [Google Scholar] [CrossRef]
- Roy, S.; Bagchi, B. Comparative study of protein unfolding in aqueous urea and dimethyl sulfoxide solutions: Surface polarity, solvent specificity, and sequence of secondary structure melting. J. Phys. Chem. B 2014, 118, 5691–5697. [Google Scholar] [CrossRef]
- Coelho, T.; Merlini, G.; Bulawa, C.E.; Fleming, J.A.; Judge, D.P.; Kelly, J.W.; Maurer, M.S.; Planté-Bordeneuve, V.; Labaudinière, R.; Mundayat, R.; et al. Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis. Neurol. Ther. 2016, 5, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Faria, T.Q.; Almeida, Z.L.; Cruz, P.F.; Jesus, C.S.H.; Castanheira, P.; Brito, R.M.M. A look into amyloid formation by transthyretin: Aggregation pathway and a novel kinetic model. Phys. Chem. Chem. Phys. 2015, 17, 7255–7263. [Google Scholar] [CrossRef]
- Jesus, C.S.H.; Almeida, Z.L.; Vaz, D.C.; Faria, T.Q.; Brito, R.M.M. A new folding kinetic mechanism for human transthyretin and the influence of the amyloidogenic V30M mutation. Int. J. Mol. Sci. 2016, 17, 1428. [Google Scholar] [CrossRef] [Green Version]
- KrishnaKumar, V.G.; Paul, A.; Gazit, E.; Segal, D. Mechanistic insights into remodeled Tau-derived PHF6 peptide fibrils by Naphthoquinone-Tryptophan hybrids. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Viswanathan, G.K.; Mahapatra, S.; Balboni, G.; Pacifico, S.; Gazit, E.; Segal, D. Antagonistic Activity of Naphthoquinone-Based Hybrids toward Amyloids Associated with Alzheimer’s Disease and Type-2 Diabetes. ACS Chem. Neurosci. 2019, 10, 3510–3520. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, G.K.; Paul, A.; Gazit, E.; Segal, D. Naphthoquinone Tryptophan Hybrids: A Promising Small Molecule Scaffold for Mitigating Aggregation of Amyloidogenic Proteins and Peptides. Front. Cell Dev. Biol. 2019, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawar, A.P.; DuBay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation-prone” and “aggregation- susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Scherzer-Attali, R.; Pellarin, R.; Convertino, M.; Frydman-Marom, A.; Egoz-Matia, N.; Peled, S.; Levy-Sakin, M.; Shalev, D.E.; Caflisch, A.; Gazit, E.; et al. Complete phenotypic recovery of an Alzheimer’s disease model by a quinone-tryptophan hybrid aggregation inhibitor. PLoS ONE 2010, 5, e11101. [Google Scholar] [CrossRef]
- Paul, A.; Zhang, B.-D.; Mohapatra, S.; Li, G.; Li, Y.-M.; Gazit, E.; Segal, D. Novel Mannitol-Based Small Molecules for Inhibiting Aggregation of α-Synuclein Amyloids in Parkinson’s Disease. Front. Mol. Biosci. 2019, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Scherzer-Attali, R.; Shaltiel-Karyo, R.; Adalist, Y.; Segal, D.; Gazit, E. Generic inhibition of amyloidogenic proteins by two naphthoquinone-tryptophan hybrid molecules. Proteins Struct. Funct. Bioinforma. 2012, 80, 1962–1973. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hudak, J.E.; Bertozzi, C.R. Glycotherapy: New advances inspire a reemergence of glycans in medicine. Chem. Biol. 2014, 21, 16–37. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.; Frenkel-Pinter, M.; Escobar Alvarez, D.; Milordini, G.; Gazit, E.; Zacco, E.; Segal, D. Tryptophan-galactosylamine conjugates inhibit and disaggregate amyloid fibrils of Aβ42 and hIAPP peptides while reducing their toxicity. Commun. Biol. 2020, 3, 1–12. [Google Scholar] [CrossRef]
- Frydman-Marom, A.; Shaltiel-Karyo, R.; Moshe, S.; Gazit, E. The generic amyloid formation inhibition effect of a designed small aromatic β-breaking peptide. Amyloid 2011, 18, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, J.G. Health functions and structure-activity relationships of natural anthraquinones from plants. Food Funct. 2018, 9, 6063–6080. [Google Scholar] [CrossRef] [PubMed]
- Nam, W.; Kim, S.; Nam, S.; Friedman, M. Structure-Antioxidative and Anti-Inflammatory Activity Relationships of Purpurin and Related Anthraquinones in Chemical and Cell Assays. Molecules 2017, 22, 265. [Google Scholar] [CrossRef]
- Viswanathan, G.K.; Shwartz, D.; Losev, Y.; Arad, E.; Shemesh, C.; Pichinuk, E.; Engel, H.; Raveh, A.; Jelinek, R.; Cooper, I.; et al. Purpurin modulates Tau-derived VQIVYK fibrillization and ameliorates Alzheimer’s disease-like symptoms in animal model. Cell. Mol. Life Sci. 2020, 77, 2795–2813. [Google Scholar] [CrossRef]
- Pickhardt, M.; Gazova, Z.; Von Bergen, M.; Khlistunova, I.; Wang, Y.; Hascher, A.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. J. Biol. Chem. 2005, 280, 3628–3635. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; He, Z.; Peng, A.; Zhang, X.; Cheng, B.; Sun, Y.; Zheng, L.; Huang, K. Effects of several quinones on insulin aggregation. Sci. Rep. 2014, 4, 5648. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.B.; Long, C.; Kennelly, E.J. Structural diversity and bioactivities of natural benzophenones. Nat. Prod. Rep. 2014, 31, 1158–1174. [Google Scholar] [CrossRef]
- Ladiwala, A.R.A.; Mora-Pale, M.; Lin, J.C.; Bale, S.S.; Fishman, Z.S.; Dordick, J.S.; Tessier, P.M. Polyphenolic Glycosides and Aglycones Utilize Opposing Pathways To Selectively Remodel and Inactivate Toxic Oligomers of Amyloid β. ChemBioChem 2011, 12, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Castillo, G.M.; Choi, P.Y.; Snow, A.D. Polyhydroxylated Aromatic Compounds for The Treatment of Amyloidosis and Α-Synuclein Fibril Diseases. WO2001049281A2, 28 December 2000. [Google Scholar]
- Bae, J.; Kim, N.; Shin, Y.; Kim, S.-Y.; Kim, Y.-J. Activity of catechins and their applications. Biomed. Dermatology 2020, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Andrich, K.; Bieschke, J. The effect of (-)-epigallo-catechin-(3)-gallate on amyloidogenic proteins suggests a common mechanism. Adv. Exp. Med. Biol. 2015, 863, 139–161. [Google Scholar] [PubMed]
- Liu, Y.; Liu, Y.; Wang, S.; Dong, S.; Chang, P.; Jiang, Z. Structural characteristics of (-)-epigallocatechin-3-gallate inhibiting amyloid Aβ42 aggregation and remodeling amyloid fibers. RSC Adv. 2015, 5, 62402–62413. [Google Scholar] [CrossRef]
- Cao, P.; Raleigh, D.P. Analysis of the inhibition and remodeling of islet amyloid polypeptide amyloid fibers by flavanols. Biochemistry 2012, 51, 2670–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, H.E.; Xing, Y.F.; Huang, B.O.; Yi-Zheng Zhang, A.; Zeng, C.M. Tea catechins induce the conversion of preformed lysozyme amyloid fibrils to amorphous aggregates. J. Agric. Food Chem. 2009, 57, 11391–11396. [Google Scholar]
- Zhan, C.; Chen, Y.; Tang, Y.; Wei, G. Green Tea Extracts EGCG and EGC Display Distinct Mechanisms in Disrupting Aβ42Protofibril. ACS Chem. Neurosci. 2020, 11, 1841–1851. [Google Scholar] [CrossRef]
- Acharya, A.; Stockmann, J.; Beyer, L.; Rudack, T.; Nabers, A.; Gumbart, J.C.; Gerwert, K.; Batista, V.S. The Effect of (−)-Epigallocatechin-3-Gallate on the Amyloid-β Secondary Structure. Biophys. J. 2020, 119, 349–359. [Google Scholar] [CrossRef]
- Li, F.; Zhan, C.; Dong, X.; Wei, G. Molecular mechanisms of resveratrol and EGCG in the inhibition of Aβ42aggregation and disruption of Aβ42protofibril: Similarities and differences. Phys. Chem. Chem. Phys. 2021, 23, 18843–18854. [Google Scholar] [CrossRef]
- Yao, Y.; Tang, Y.; Wei, G. Epigallocatechin Gallate Destabilizes α-Synuclein Fibril by Disrupting the E46-K80 Salt-Bridge and Inter-protofibril Interface. ACS Chem. Neurosci. 2020, 11, 4351–4361. [Google Scholar] [CrossRef]
- Seidler, P.; Boyer, D.; Sawaya, M.; Ge, P.; Shin, W.; DeTure, M.; Dickson, D.; Jiang, L.; Eisenberg, D. CryoEM reveals how the small molecule EGCG binds to Alzheimer’s brain-derived tau fibrils and initiates fibril disaggregation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Roy, S.; Bhat, R. Suppression, disaggregation, and modulation of γ-Synuclein fibrillation pathway by green tea polyphenol EGCG. Protein Sci. 2019, 28, 382–402. [Google Scholar] [CrossRef] [Green Version]
- Kristen, A.V.; Lehrke, S.; Buss, S.; Mereles, D.; Steen, H.; Ehlermann, P.; Hardt, S.; Giannitsis, E.; Schreiner, R.; Haberkorn, U.; et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin. Res. Cardiol. 2012, 101, 805–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- aus dem Siepen, F.; Buss, S.J.; Andre, F.; Seitz, S.; Giannitsis, E.; Steen, H.; Katus, H.A.; Kristen, A.V. Extracellular remodeling in patients with wild-type amyloidosis consuming epigallocatechin-3-gallate: Preliminary results of T1 mapping by cardiac magnetic resonance imaging in a small single center study. Clin. Res. Cardiol. 2015, 104, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, F.; Martone, R.; Taborchi, G.; Morini, S.; Bartolini, S.; Angelotti, P.; Farsetti, S.; Di Mario, C.; Perfetto, F. Epigallocatechin-3-gallate tolerability and impact on survival in a cohort of patients with transthyretin-related cardiac amyloidosis. A single-center retrospective study. Intern. Emerg. Med. 2018, 13, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Lange, K.W.; Lange, K.M.; Nakamura, Y. Green tea, epigallocatechin gallate and the prevention of Alzheimer’s disease: Clinical evidence. Food Sci. Hum. Wellness 2022, 11, 765–770. [Google Scholar] [CrossRef]
- Narayan, M.; Henríquez, G.; Gomez, A.; Guerrero, E. Potential role of natural polyphenols against protein aggregation toxicity: In vitro, in vivo, and clinical studies. ACS Chem. Neurosci. 2020, 11, 2915–2934. [Google Scholar]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef]
- DI MARCO, A. Adriamycin (NSC-123,127): A new antibiotic with antitumor activity. Cancer Chemother. Rep. 1969, 53, 33–37. [Google Scholar]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaimomycin, a new antitumor antibiotic fromS. Peucetius var.caesius. Biotechnol. Bioeng. 1969, 11, 1101–1110. [Google Scholar] [CrossRef]
- Post, C.; Tagliavini, F.; McArthur, R.A.; Della Vedova, F.; Gerna, M.; Bandiera, T.; Varasi, M.; Molinari, A.; Lansen, J. Anthracyclines and Amyloidosis. In Advances in Behavioral Biology; Springer: Boston, MA, USA, 1998; pp. 197–204. [Google Scholar]
- Stoilova, T.; Colombo, L.; Forloni, G.; Tagliavini, F.; Salmona, M. A new face for old antibiotics: Tetracyclines in treatment of amyloidoses. J. Med. Chem. 2013, 56, 5987–6006. [Google Scholar] [CrossRef]
- Forloni, G.; Colombo, L.; Girola, L.; Tagliavini, F.; Salmona, M. Anti-amyloidogenic activity of tetracyclines: Studies in vitro. FEBS Lett. 2001, 487, 404–407. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Yamada, M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J. Neurochem. 2006, 97, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hirohata, M.; Yamada, M. Anti-fibrillogenic and fibril-destabilizing activities of anti-Parkinsonian agents for α-synuclein fibrils in vitro. J. Neurosci. Res. 2007, 85, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.; Merlini, G.; Saraiva, M.J. 4 ′-iodo-4′-Deoxydoxorubicin and tetracyclines disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic species: Screening for TTR fibril disrupters. FASEB J. 2003, 17, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.; Saraiva, M.J. Doxycycline disrupts transthyretin amyloid: Evidence from studies in a FAP transgenic mice model. FASEB J. 2006, 20, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Giorgetti, S.; Raimondi, S.; Pagano, K.; Relini, A.; Bucciantini, M.; Corazza, A.; Fogolari, F.; Codutti, L.; Salmona, M.; Mangione, P.; et al. Effect of tetracyclines on the dynamics of formation and destructuration of β2-microglobulin amyloid fibrils. J. Biol. Chem. 2011, 286, 2121–2131. [Google Scholar] [CrossRef] [Green Version]
- Aitken, J.F.; Loomes, K.M.; Konarkowska, B.; Cooper, G.J.S. Suppression by polycyclic compounds of the conversion of human amylin into insoluble amyloid. Biochem. J. 2003, 374, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhao, C.; Huang, X.; Du, W. Tetracycline derivatives resist the assembly behavior of human islet amyloid polypeptide. Biochimie 2020, 174, 95–106. [Google Scholar] [CrossRef]
- Ward, J.E.; Ren, R.; Toraldo, G.; SooHoo, P.; Guan, J.; O’Hara, C.; Jasuja, R.; Trinkaus-Randall, V.; Liao, R.; Connors, L.H.; et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood 2011, 118, 6610–6617. [Google Scholar] [CrossRef] [Green Version]
- Tagliavini, F.; Forloni, G.; Colombo, L.; Rossi, G.; Girola, L.; Canciani, B.; Angeretti, N.; Giampaolo, L.; Peressini, E.; Awan, T.; et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrP(Sc) in vitro. J. Mol. Biol. 2000, 300, 1309–1322. [Google Scholar] [CrossRef]
- Smith, D.L.; Woodman, B.; Mahal, A.; Sathasivam, K.; Ghazi-Noori, S.; Lowden, P.A.S.; Bates, G.P.; Hockly, E. Minocycline and doxycycline are not beneficial in a model of Huntington’s disease. Ann. Neurol. 2003, 54, 186–196. [Google Scholar] [CrossRef]
- Gertz, M.A.; Zeldenrust, S.R. Treatment of immunoglobulin light chain amyloidosis. Curr. Hematol. Malig. Rep. 2009, 4, 91–98. [Google Scholar] [CrossRef] [PubMed]
- SEBASTIÃO, M.P.; MERLINI, G.; SARAIVA, M.J.; DAMAS, A.M. The molecular interaction of 4′-iodo-4′-deoxydoxorubicin with Leu-55Pro transthyretin ‘amyloid-like’ oligomer leading to disaggregation. Biochem. J. 2000, 351, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Bellotti, V.; Gianni, A.M.; Merlini, G. New drug therapy of amyloidoses: Resorption of AL-type deposits with 4’- iodo-4’-deoxydoxorubicin. Blood 1995, 86, 855–861. [Google Scholar] [CrossRef] [Green Version]
- Merlini, G.; Ascari, E.; Amboldi, N.; Bellotti, V.; Arbustini, E.; Perfetti, V.; Ferrari, M.; Zorzoli, I.; Marinone, M.G.; Garini, P.; et al. Interaction of the anthracycline 4’-iodo-4’-deoxydoxorubicin with amyloid fibrils: Inhibition of amyloidogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 2959–2963. [Google Scholar] [CrossRef]
- Vivenza, D.; Feola, M.; Garrone, O.; Monteverde, M.; Merlano, M.; Lo Nigro, C. Role of the renin-angiotensin-aldosterone system and the glutathione S-transferase Mu, Pi and Theta gene polymorphisms in cardiotoxicity after anthracycline chemotherapy for breast carcinoma. Int. J. Biol. Markers 2013, 28, e336–e347. [Google Scholar] [CrossRef]
- Narezkina, A.; Narayan, H.K.; Zemljic-Harpf, A.E. Molecular mechanisms of anthracycline cardiovascular toxicity. Clin. Sci. 2021, 135, 1311–1332. [Google Scholar] [CrossRef]
- Gautieri, A.; Beeg, M.; Gobbi, M.; Rigoldi, F.; Colombo, L.; Salmona, M. The Anti-Amyloidogenic Action of Doxycycline: A Molecular Dynamics Study on the Interaction with Aβ42. Int. J. Mol. Sci. 2019, 20, 4641. [Google Scholar] [CrossRef] [Green Version]
- Andes, D.; Craig, W.A. Pharmacokinetics and Pharmacodynamics of Tetracyclines. In Antimicrobial Pharmacodynamics in Theory and Clinical Practice, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 279–290. [Google Scholar]
- Danesi, R.; Fogli, S.; Gennari, A.; Conte, P.; Del Tacca, M. Pharmacokinetic-pharmacodynamic relationships of the anthracycline anticancer drugs. Clin. Pharmacokinet. 2002, 41, 431–444. [Google Scholar] [CrossRef]
- Lees, P.; Landoni, M.F.; Giraudel, J.; Toutain, P.L. Pharmacodynamics and pharmacokinetics of nonsteroidal anti-inflammatory drugs in species of veterinary interest. J. Vet. Pharmacol. Ther. 2004, 27, 479–490. [Google Scholar] [CrossRef]
- Miller, S.R.; Sekijima, Y.; Kelly, J.W. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Investig. 2004, 84, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA - J. Am. Med. Assoc. 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekijima, Y.; Tojo, K.; Morita, H.; Koyama, J.; Ikeda, S.I. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid 2015, 22, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Ikram, A.; Donnelly, J.P.; Sperry, B.W.; Samaras, C.; Valent, J.; Hanna, M. Diflunisal tolerability in transthyretin cardiac amyloidosis: A single center’s experience. Amyloid 2018, 25, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Ono, K.; Shibata, S.; Nakamura, K.; Komatsu, J.; Ikeda, Y.; Ikeda, T.; Samuraki, M.; Sakai, K.; Iwasa, K.; et al. Efficacy of diflunisal on autonomic dysfunction of late-onset familial amyloid polyneuropathy (TTR Val30Met) in a Japanese endemic area. J. Neurol. Sci. 2014, 345, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Hirohata, M.; Ono, K.; Naiki, H.; Yamada, M. Non-steroidal anti-inflammatory drugs have anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. Neuropharmacology 2005, 49, 1088–1099. [Google Scholar] [CrossRef] [Green Version]
- Hirohata, M.; Ono, K.; Morinaga, A.; Yamada, M. Non-steroidal anti-inflammatory drugs have potent anti-fibrillogenic and fibril-destabilizing effects for α-synuclein fibrils in vitro. Neuropharmacology 2008, 54, 620–627. [Google Scholar] [CrossRef]
- Azam, F.; Alabdullah, N.H.; Ehmedat, H.M.; Abulifa, A.R.; Taban, I.; Upadhyayula, S. NSAIDs as potential treatment option for preventing amyloid β toxicity in Alzheimer’s disease: An investigation by docking, molecular dynamics, and DFT studies. J. Biomol. Struct. Dyn. 2018, 36, 2099–2117. [Google Scholar] [CrossRef]
- Wichmann, M.A.; Cruickshanks, K.J.; Carlsson, C.M.; Chappell, R.; Fischer, M.E.; Klein, B.E.K.; Klein, R.; Schubert, C.R. NSAID Use and Incident Cognitive Impairment in a Population-based Cohort. Alzheimer Dis. Assoc. Disord. 2016, 30, 105–112. [Google Scholar] [CrossRef]
- Breitner, J.C.S.; Haneuse, S.J.P.A.; Walker, R.; Dublin, S.; Crane, P.K.; Gray, S.L.; Larson, E.B. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 2009, 72, 1899–1905. [Google Scholar] [CrossRef] [Green Version]
- Thal, L.J.; Ferris, S.H.; Kirby, L.; Block, G.A.; Lines, C.R.; Yuen, E.; Assaid, C.; Nessly, M.L.; Norman, B.A.; Baranak, C.C.; et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 2005, 30, 1204–1215. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.; Skelly, D.T. Non-steroidal anti-inflammatory drugs and cognitive function: Are prostaglandins at the heart of cognitive impairment in dementia and delirium? J. Neuroimmune Pharmacol. 2012, 7, 60–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiente-Gabioud, A.A.; Miotto, M.C.; Chesta, M.E.; Lombardo, V.; Binolfi, A.; Fernández, C.O. Phthalocyanines as Molecular Scaffolds to Block Disease-Associated Protein Aggregation. Acc. Chem. Res. 2016, 49, 801–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.; Ghosh, K.S. Inhibition of Amyloid Fibrillation by Small Molecules and Nanomaterials: Strategic Development of Pharmaceuticals Against Amyloidosis. Protein Pept. Lett. 2019, 26, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Gour, N.; Koshti, B. A Chemical Perspective to the Anti-Amyloid Action of Compounds and a Nanoparticle Based Assay for Screening Amyloid Inhibitors. ChemRxiv 2019. [Google Scholar] [CrossRef]
- Liu, Y.; Carver, J.A.; Ho, L.H.; Elias, A.K.; Musgrave, I.F.; Pukala, T.L. Hemin as a generic and potent protein misfolding inhibitor. Biochem. Biophys. Res. Commun. 2014, 454, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Sonavane, S.; Haider, S.Z.; Kumar, A.; Ahmad, B. Hemin is able to disaggregate lysozyme amyloid fibrils into monomers. Biochim. Biophys. Acta—Proteins Proteomics 2017, 1865, 1315–1325. [Google Scholar] [CrossRef]
- Yuan, C.; Gao, Z. Aβ interacts with both the iron center and the porphyrin ring of heme: Mechanism of heme’s action on Aβ aggregation and disaggregation. Chem. Res. Toxicol. 2013, 26, 262–269. [Google Scholar] [CrossRef]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.I.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005, 280, 7614–7623. [Google Scholar] [CrossRef] [Green Version]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.K.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [Green Version]
- Chernii, S.; Losytskyy, M.; Kelm, A.; Gorski, A.; Tretyakova, I.; Yarmoluk, S.; Chernii, V.; Kovalska, V. Study of tetraphenylporphyrins as modifiers of insulin amyloid aggregation. J. Mol. Recognit. 2020, 33, e2811. [Google Scholar] [CrossRef]
- Abelein, A.; Kaspersen, J.D.; Nielsen, S.B.; Jensen, G.V.; Christiansen, G.; Pedersen, J.S.; Danielsson, J.; Otzen, D.E.; Gräslund, A. Formation of dynamic soluble surfactant-induced amyloid β peptide aggregation intermediates. J. Biol. Chem. 2013, 288, 23518–23528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabaté, R.; Estelrich, J. Stimulatory and inhibitory effects of alkyl bromide surfactants on β-amyloid fibrillogenesis. Langmuir 2005, 21, 6944–6949. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.S.; Chen, Y.T.; Chou, S.W. Inhibition of amyloid fibril formation of β-amyloid peptides via the amphiphilic surfactants. Biochim. Biophys. Acta—Mol. Basis Dis. 2005, 1741, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, N.K.; Ghosh, S.; Dasgupta, S. Effect of surfactants on preformed fibrils of human serum albumin. Int. J. Biol. Macromol. 2013, 59, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, Z.; Rehman, S.U.; Tabish, M.; Shalla, A.H.; Kabir-ud-Din, K.-D. Modulation of bovine serum albumin fibrillation by ester bonded and conventional gemini surfactants. RSC Adv. 2015, 5, 58616–58624. [Google Scholar] [CrossRef]
- Bhat, W.F.; Bhat, I.A.; Bhat, S.A.; Bano, B. In vitro disintegration of goat brain cystatin fibrils using conventional and gemini surfactants: Putative therapeutic intervention in amyloidoses. Int. J. Biol. Macromol. 2016, 93, 493–500. [Google Scholar] [CrossRef]
- Han, Y.; He, C.; Cao, M.; Huango, X.; Wang, Y.; Li, Z. Facile disassembly of amyloid fibrils using gemini surfactant micelles. Langmuir 2010, 26, 1583–1587. [Google Scholar] [CrossRef]
- He, C.; Hou, Y.; Han, Y.; Wang, Y. Disassembly of amyloid fibrils by premicellar and micellar aggregates of a tetrameric cationic surfactant in aqueous solution. Langmuir 2011, 27, 4551–4556. [Google Scholar] [CrossRef]
- Zhu, L.; Han, Y.; He, C.; Huang, X.; Wang, Y. Disaggregation ability of different chelating molecules on copper ion-triggered amyloid fibers. J. Phys. Chem. B 2014, 118, 9298–9305. [Google Scholar] [CrossRef]
- Menger, F.M.; Keiper, J.S. Gemini Surfactants. Angew. Chemie Int. Ed. 2000, 39, 1906–1920. [Google Scholar] [CrossRef]
- Miura, M.; Kodama, M. The Second CMC of the Aqueous Solution of Sodium Dodecyl Sulfate. I. Conductivity. Bull. Chem. Soc. Jpn. 1972, 45, 428–431. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Han, Y.; Deng, M.; Xiang, J.; Wang, Y. Aggregation behavior of a tetrameric cationic surfactant in aqueous solution. Langmuir 2010, 26, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.K.; Chatterjee, S.K.; Bhattarai, A. Micellization of cationic surfactants in alcohol — water mixed solvent media. J. Mol. Liq. 2016, 222, 906–914. [Google Scholar] [CrossRef]
- Bombelli, C.; Giansanti, L.; Luciani, P.; Mancini, G. Gemini Surfactant Based Carriers in Gene and Drug Delivery. Curr. Med. Chem. 2008, 16, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.J.; Camilleri, P.; Engberts, J.B.F.N.; Feiters, M.C.; Nolte, R.J.M.; Söderman, O.; Bergsma, M.; Bell, P.C.; Fielden, M.L.; García Rodríguez, C.L.; et al. Gemini Surfactants: New Synthetic Vectors for Gene Transfection. Angew. Chemie Int. Ed. 2003, 42, 1448–1457. [Google Scholar] [CrossRef]
- Chatani, E.; Lee, Y.H.; Yagi, H.; Yoshimura, Y.; Naiki, H.; Goto, Y. Ultrasonication-dependent production and breakdown lead to minimum-sized amyloid fibrils. Proc. Natl. Acad. Sci. USA 2009, 106, 11119–11124. [Google Scholar] [CrossRef] [Green Version]
- Ikenoue, T.; Lee, Y.-H.; Kardos, J.; Saiki, M.; Yagi, H.; Kawata, Y.; Goto, Y. Cold Denaturation of α-Synuclein Amyloid Fibrils. Angew. Chemie Int. Ed. 2014, 53, 7799–7804. [Google Scholar] [CrossRef]
- Arora, A.; Ha, C.; Park, C.B. Insulin amyloid fibrillation at above 100 °C: New insights into protein folding under extreme temperatures. Protein Sci. 2004, 13, 2429–2436. [Google Scholar] [CrossRef] [Green Version]
- Dubois, J.; Ismail, A.A.; Chan, S.L.; Ali-Khan, Z. Fourier Transform Infrared Spectroscopic Investigation of Temperature- and Pressure-Induced Disaggregation of Amyloid A. Scand. J. Immunol. 1999, 49, 376–380. [Google Scholar] [CrossRef]
- Kim, Y.S.; Randolph, T.W.; Seefeldt, M.B.; Carpenter, J.F. High-Pressure Studies on Protein Aggregates and Amyloid Fibrils. Methods Enzymol. 2006, 413, 237–253. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, Z.L.; Brito, R.M.M. Amyloid Disassembly: What Can We Learn from Chaperones? Biomedicines 2022, 10, 3276. https://doi.org/10.3390/biomedicines10123276
Almeida ZL, Brito RMM. Amyloid Disassembly: What Can We Learn from Chaperones? Biomedicines. 2022; 10(12):3276. https://doi.org/10.3390/biomedicines10123276
Chicago/Turabian StyleAlmeida, Zaida L., and Rui M. M. Brito. 2022. "Amyloid Disassembly: What Can We Learn from Chaperones?" Biomedicines 10, no. 12: 3276. https://doi.org/10.3390/biomedicines10123276