
Consequences of the Lack of TNFR1 in Ouabain Response in the Hippocampus of C57BL/6J Mice
 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. Chemicals and Enzyme-Linked Immunosorbent Assay (ELISA) Kits
2.3. Protein Extraction-Membrane Enriched, Cytosolic, and Nuclear Fractions
2.4. Western Blotting
2.5. EMSA
2.6. Open Field
2.7. Elevated Plus-Maze
2.8. Statistical Analysis
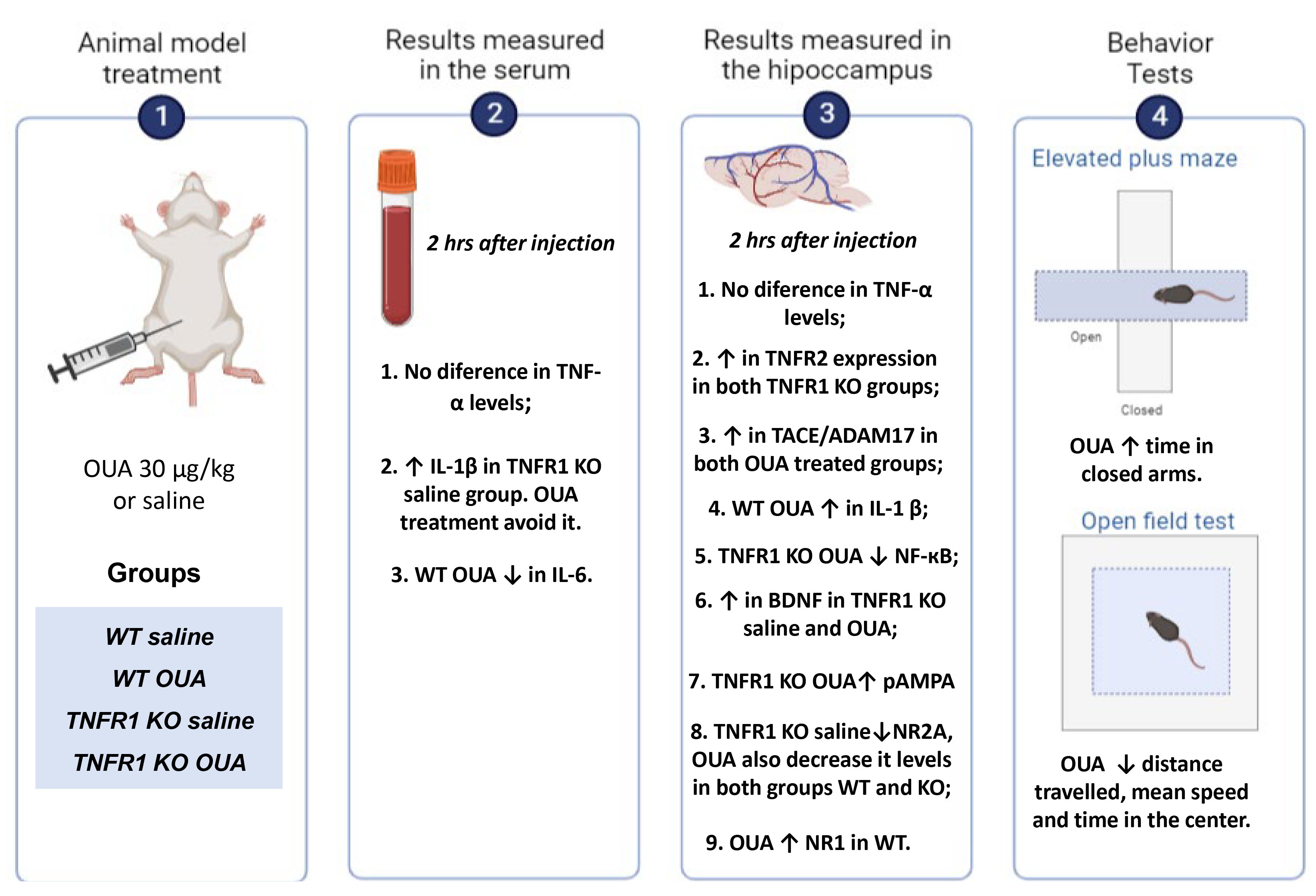
3. Results
3.1. TNFR1 KO Mice Have Altered TNFR2 and Ouabain Increases TACE/ADAM17 Activity
3.2. Ouabain Has an Effect on IL-1β Expression That Is Correlated with NF-кB Activity
3.3. TNFR1 Impacts BDNF Levels in Serum and Ouabain Modulates Glutamate Receptors
3.4. Ouabain Interferes with Behavior Independently of the Presence of TNFR1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamlyn, J.M.; Blaustein, M.P. Endogenous Ouabain: Recent Advances and Controversies. Hypertens 2016, 68, 526–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, A.; Guo, J.; Itagaki, Y.; Bell, C.; Wang, Y.; Haupert, G.T.J.; Magil, S.; Gallagher, R.T.; Berova, N.; Nakanishi, K. On the structure of endogenous ouabain. Proc. Natl. Acad. Sci. USA 1999, 96, 6654–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, J.R.; Randall, J.D.; Rosoff, J.; Zhao, J.; Jensen, R.V.; Gullans, S.R.; Haupert, G.T.J. Endogenous ouabain: Upregulation of steroidogenic genes in hypertensive hypothalamus but not adrenal. Circulation 2005, 112, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W. Ouabain, a new steroid hormone of adrenal gland and hypothalamus. Exp. Clin. Endocrinol. Diabetes 2000, 108, 449–454. [Google Scholar] [CrossRef]
- Nesher, M.; Shpolansky, U.; Rosen, H.; Lichtstein, D. The digitalis-like steroid hormones: New mechanisms of action and biological significance. Life Sci. 2007, 80, 2093–2107. [Google Scholar] [CrossRef]
- Skou, J.C.; Esmann, M. The Na,K-ATPase. J. Bioenerg. Biomembr. 1992, 24, 249–261. [Google Scholar] [CrossRef]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O. V Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef]
- Liang, M.; Tian, J.; Liu, L.; Pierre, S.; Liu, J.; Shapiro, J.; Xie, Z.-J. Identification of a pool of non-pumping Na/K-ATPase. J. Biol. Chem. 2007, 282, 10585–10593. [Google Scholar] [CrossRef] [Green Version]
- Xiao, A.Y.; Wei, L.; Xia, S.; Rothman, S.; Yu, S.P. Ionic mechanism of ouabain-induced concurrent apoptosis and necrosis in individual cultured cortical neurons. J. Neurosci. 2002, 22, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Burlaka, I.; Liu, X.L.; Rebetz, J.; Arvidsson, I.; Yang, L.; Brismar, H.; Karpman, D.; Aperia, A. Ouabain protects against Shiga toxin-triggered apoptosis by reversing the imbalance between Bax and Bcl-xL. J. Am. Soc. Nephrol. 2013, 24, 1413–1423. [Google Scholar] [CrossRef]
- Desfrere, L.; Karlsson, M.; Hiyoshi, H.; Malmersjo, S.; Nanou, E.; Estrada, M.; Miyakawa, A.; Lagercrantz, H.; El Manira, A.; Lal, M.; et al. Na,K-ATPase signal transduction triggers CREB activation and dendritic growth. Proc. Natl. Acad. Sci. USA 2009, 106, 2212–2217. [Google Scholar] [CrossRef] [Green Version]
- Orellana, A.M.; Leite, J.A.; Kinoshita, P.F.; Vasconcelos, A.R.; Andreotti, D.Z.; de Sa Lima, L.; Xavier, G.F.; Kawamoto, E.M.; Scavone, C. Ouabain increases neuronal branching in hippocampus and improves spatial memory. Neuropharmacology 2018, 140, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Orellana, A.M.; Kinoshita, P.F.; Leite, J.A.; Kawamoto, E.M.; Scavone, C. Cardiotonic steroids as modulators of neuroinflammation. Front. Endocrinol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, P.F.; Yshii, L.M.; Vasconcelos, A.R.; Orellana, A.M.M.; Lima, L.S.; Davel, A.P.C.; Rossoni, L.V.; Kawamoto, E.M.; Scavone, C. Signaling function of Na,K-ATPase induced by ouabain against LPS as an inflammation model in hippocampus. J. Neuroinflamm. 2014, 11, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, P.F.; Yshii, L.M.; Orellana, A.M.M.; Paixao, A.G.; Vasconcelos, A.R.; Lima, L.d.S.; Kawamoto, E.M.; Scavone, C. Alpha 2 Na(+),K(+)-ATPase silencing induces loss of inflammatory response and ouabain protection in glial cells. Sci. Rep. 2017, 7, 4894. [Google Scholar] [CrossRef]
- De Sá Lima, L.; Kawamoto, E.M.; Munhoz, C.D.; Kinoshita, P.F.; Orellana, A.M.M.; Curi, R.; Rossoni, L.V.; Avellar, M.C.W.; Scavone, C. Ouabain activates NFκB through an NMDA signaling pathway in cultured cerebellar cells. Neuropharmacology 2013, 73, 327–336. [Google Scholar] [CrossRef]
- Kawamoto, E.M.; Lima, L.S.; Munhoz, C.D.; Yshii, L.M.; Kinoshita, P.F.; Amara, F.G.; Pestana, R.R.F.; Orellana, A.M.M.; Cipolla-Neto, J.; Britto, L.R.G.; et al. Influence of N-methyl-D-aspartate receptors on ouabain activation of nuclear factor-kappaB in the rat hippocampus. J. Neurosci. Res. 2012, 90, 213–228. [Google Scholar] [CrossRef]
- de Rezende Correa, G.; Araujo dos Santos, A.; Frederico Leite Fontes, C.; Giestal de Araujo, E. Ouabain induces an increase of retinal ganglion cell survival in vitro: The involvement of protein kinase C. Brain Res. 2005, 1049, 89–94. [Google Scholar] [CrossRef]
- Salles von-Held-Ventura, J.; Mazala-de-Oliveira, T.; Candida da Rocha Oliveira, A.; Granja, M.G.; Goncalves-de-Albuquerque, C.F.; Castro-Faria-Neto, H.C.; Giestal-de-Araujo, E. The trophic effect of ouabain on retinal ganglion cells is mediated by IL-1beta and TNF-alpha. Biochem. Biophys. Res. Commun. 2016, 478, 378–384. [Google Scholar] [CrossRef]
- Tracey, K.J.; Cerami, A. Tumor necrosis factor: A pleiotropic cytokine and therapeutic target. Annu. Rev. Med. 1994, 45, 491–503. [Google Scholar] [CrossRef]
- Gearing, A.J.; Beckett, P.; Christodoulou, M.; Churchill, M.; Clements, J.; Davidson, A.H.; Drummond, A.H.; Galloway, W.A.; Gilbert, R.; Gordon, J.L. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature 1994, 370, 555–557. [Google Scholar] [CrossRef]
- Smookler, D.S.; Mohammed, F.F.; Kassiri, Z.; Duncan, G.S.; Mak, T.W.; Khokha, R. Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J. Immunol. 2006, 176, 721–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brás, J.P.; Bravo, J.; Freitas, J.; Barbosa, M.A.; Santos, S.G.; Summavielle, T.; Almeida, M.I. TNF-alpha-induced microglia activation requires miR-342: Impact on NF-kB signaling and neurotoxicity. Cell Death Dis. 2020, 11, 415. [Google Scholar] [CrossRef] [PubMed]
- Benson, C.A.; Powell, H.R.; Liput, M.; Dinham, S.; Freedman, D.A.; Ignatowski, T.A.; Stachowiak, E.K.; Stachowiak, M.K. Immune Factor, TNFα, Disrupts Human Brain Organoid Development Similar to Schizophrenia-Schizophrenia Increases Developmental Vulnerability to TNFα. Front. Cell. Neurosci. 2020, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, A.; Htike, T.T.; James, R.; Picon, C.; Reynolds, R. TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocampus. Acta Neuropathol. Commun. 2021, 9, 159. [Google Scholar] [CrossRef]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body Fluid Cytokine Levels in Mild Cognitive Impairment and Alzheimer’s Disease: A Comparative Overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Rossi, S.; Motta, C.; Studer, V.; Barbieri, F.; Buttari, F.; Bergami, A.; Sancesario, G.; Bernardini, S.; De Angelis, G.; Martino, G.; et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult. Scler. 2014, 20, 304–312. [Google Scholar] [CrossRef]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.S.; Beiser, A.S.; Vasan, R.S.; Roubenoff, R.; Dinarello, C.A.; Harris, T.B.; Benjamin, E.J.; Au, R.; Kiel, D.P.; Wolf, P.A.; et al. Inflammatory markers and the risk of Alzheimer disease: The Framingham Study. Neurology 2007, 68, 1902–1908. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, R.; Kaelber, D.C.; Gurney, M.E. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer’s disease in patients with rheumatoid arthritis and psoriasis. PLoS ONE 2020, 15, e0229819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobinick, E.; Gross, H.; Weinberger, A.; Cohen, H. TNF-alpha modulation for treatment of Alzheimer’s disease: A 6-month pilot study. MedGenMed 2006, 8, 25. [Google Scholar] [PubMed]
- Chang, R.; Yee, K.-L.; Sumbria, R.K. Tumor necrosis factor α Inhibition for Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2017, 9, 9278. [Google Scholar] [CrossRef] [PubMed]
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. Therapeutic Potential of TNF-α Inhibition for Alzheimer’s Disease Prevention. J. Alzheimer’s Dis. 2020, 78, 619–626. [Google Scholar] [CrossRef]
- Kemanetzoglou, E.; Andreadou, E. CNS Demyelination with TNF-alpha Blockers. Curr. Neurol. Neurosci. Rep. 2017, 17, 36. [Google Scholar] [CrossRef] [Green Version]
- Pegoretti, V.; Baron, W.; Laman, J.D.; Eisel, U.L.M. Selective Modulation of TNF-TNFRs Signaling: Insights for Multiple Sclerosis Treatment. Front. Immunol. 2018, 9, 925. [Google Scholar] [CrossRef] [Green Version]
- Titelbaum, D.S.; Degenhardt, A.; Kinkel, R.P. Anti-tumor necrosis factor alpha-associated multiple sclerosis. AJNR. Am. J. Neuroradiol. 2005, 26, 1548–1550. [Google Scholar]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Dolga, A.M.; Nijholt, I.M.; Ostroveanu, A.; Ten Bosch, Q.; Luiten, P.G.M.; Eisel, U.L.M. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J. Alzheimer’s Dis. 2008, 13, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L.M. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef] [Green Version]
- Nophar, Y.; Kemper, O.; Brakebusch, C.; Englemann, H.; Zwang, R.; Aderka, D.; Holtmann, H.; Wallach, D. Soluble forms of tumor necrosis factor receptors (TNF-Rs). The cDNA for the type I TNF-R, cloned using amino acid sequence data of its soluble form, encodes both the cell surface and a soluble form of the receptor. EMBO J. 1990, 9, 3269–3278. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Libert, C.; Vandenbroucke, R.E. A New Venue of TNF Targeting. Int. J. Mol. Sci. 2018, 19, 1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallach, D.; Engelmann, H.; Nophar, Y.; Aderka, D.; Kemper, O.; Hornik, V.; Holtmann, H.; Brakebusch, C. Soluble and cell surface receptors for tumor necrosis factor. Agents Actions Suppl. 1991, 35, 51–57. [Google Scholar] [PubMed]
- Chowdhury, D.; Hell, J.W. Homeostatic synaptic scaling: Molecular regulators of synaptic AMPA-type glutamate receptors. F1000Research 2018, 7, 234. [Google Scholar] [CrossRef] [Green Version]
- Heir, R.; Stellwagen, D. TNF-Mediated Homeostatic Synaptic Plasticity: From in vitro to in vivo Models. Front. Cell. Neurosci. 2020, 14, 565841. [Google Scholar] [CrossRef] [PubMed]
- Siddoway, B.; Hou, H.; Xia, H. Molecular mechanisms of homeostatic synaptic downscaling. Neuropharmacology 2014, 78, 38–44. [Google Scholar] [CrossRef]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-alpha. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [Green Version]
- Dostanic, I.; Paul, R.J.; Lorenz, J.N.; Theriault, S.; Van Huysse, J.W.; Lingrel, J.B. The alpha2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H477–H485. [Google Scholar] [CrossRef] [Green Version]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides: Their roles in hypertension, salt metabolism, and cell growth. Am. J. Physiol. Cell Physiol. 2007, 293, C509–C536. [Google Scholar] [CrossRef]
- Sajeevadathan, M.; Pettitt, M.J.; Buhr, M. Interaction of ouabain and progesterone on induction of bull sperm capacitation. Theriogenology 2019, 126, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Kobayashi, M.; Usui-Kawanishi, F.; Karasawa, T.; Kimura, H.; Watanabe, S.; Mise, N.; Kayama, F.; Kasahara, T.; Hasebe, N.; Takahashi, M. The cardiac glycoside ouabain activates NLRP3 inflammasomes and promotes cardiac inflammation and dysfunction. PLoS ONE 2017, 12, e0176676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najjari, M.; Vaezi, G.; Hojati, V.; Mousavi, Z.; Bakhtiarian, A.; Nikoui, V. Involvement of IL-1β and IL-6 in antiarrhythmic properties of atorvastatin in ouabain-induced arrhythmia in rats. Immunopharmacol. Immunotoxicol. 2018, 40, 256–261. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Saperstein, S.; Chen, L.; Oakes, D.; Pryhuber, G.; Finkelstein, J. IL-1beta augments TNF-alpha-mediated inflammatory responses from lung epithelial cells. J. Interferon Cytokine Res. 2009, 29, 273–284. [Google Scholar] [CrossRef]
- Liu, J.S.; John, G.R.; Sikora, A.; Lee, S.C.; Brosnan, C.F. Modulation of interleukin-1beta and tumor necrosis factor alpha signaling by P2 purinergic receptors in human fetal astrocytes. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 5292–5299. [Google Scholar] [CrossRef] [Green Version]
- Leal, M.C.; Casabona, J.C.; Puntel, M.; Pitossi, F.J. Interleukin-1β and tumor necrosis factor-α: Reliable targets for protective therapies in Parkinson’s Disease? Front. Cell. Neurosci. 2013, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Clausen, B.H.; Wirenfeldt, M.; Høgedal, S.S.; Frich, L.H.; Nielsen, H.H.; Schrøder, H.D.; Østergaard, K.; Finsen, B.; Kristensen, B.W.; Lambertsen, K.L. Characterization of the TNF and IL-1 systems in human brain and blood after ischemic stroke. Acta Neuropathol. Commun. 2020, 8, 81. [Google Scholar] [CrossRef]
- Wei, F.; Guo, W.; Zou, S.; Ren, K.; Dubner, R. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J. Neurosci. 2008, 28, 10482–10495. [Google Scholar] [CrossRef] [Green Version]
- He, P.; Liu, Q.; Wu, J.; Shen, Y. Genetic deletion of TNF receptor suppresses excitatory synaptic transmission via reducing AMPA receptor synaptic localization in cortical neurons. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 334–345. [Google Scholar] [CrossRef]
- Maggio, N.; Vlachos, A. Tumor necrosis factor (TNF) modulates synaptic plasticity in a concentration-dependent manner through intracellular calcium stores. J. Mol. Med. 2018, 96, 1039–1047. [Google Scholar] [CrossRef]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albensi, B.C.; Mattson, M.P. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse 2000, 35, 151–159. [Google Scholar] [CrossRef]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [Green Version]
- de Vasconcelos, D.I.B.; Leite, J.A.; Carneiro, L.T.; Piuvezam, M.R.; de Lima, M.R.V.; de Morais, L.C.L.; Rumjanek, V.M.; Rodrigues-Mascarenhas, S. Anti-inflammatory and antinociceptive activity of ouabain in mice. Mediat. Inflamm. 2011, 2011, 912925. [Google Scholar] [CrossRef] [Green Version]
- Aperia, A. New roles for an old enzyme: Na,K-ATPase emerges as an interesting drug target. J. Intern. Med. 2007, 261, 44–52. [Google Scholar] [CrossRef]
- Xie, Z.; Askari, A. Na(+)/K(+)-ATPase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.L.; Miyakawa, A.; Aperia, A.; Krieger, P. Na,K-ATPase generates calcium oscillations in hippocampal astrocytes. Neuroreport 2007, 18, 597–600. [Google Scholar] [CrossRef]
- Brambilla, R.; Ashbaugh, J.J.; Magliozzi, R.; Dellarole, A.; Karmally, S.; Szymkowski, D.E.; Bethea, J.R. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 2011, 134, 2736–2754. [Google Scholar] [CrossRef]
- Gao, H.; Danzi, M.C.; Choi, C.S.; Taherian, M.; Dalby-Hansen, C.; Ellman, D.G.; Madsen, P.M.; Bixby, J.L.; Lemmon, V.P.; Lambertsen, K.L.; et al. Opposing Functions of Microglial and Macrophagic TNFR2 in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Cell Rep. 2017, 18, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Clausen, B.H.; Fenger, C.; Wulf, H.; Owens, T.; Dagnaes-Hansen, F.; Meldgaard, M.; Finsen, B. Microglia and macrophages express tumor necrosis factor receptor p75 following middle cerebral artery occlusion in mice. Neuroscience 2007, 144, 934–949. [Google Scholar] [CrossRef]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, E.M.; Scavone, C.; Mattson, M.P.; Camandola, S. Curcumin requires tumor necrosis factor alpha signaling to alleviate cognitive impairment elicited by lipopolysaccharide. Neurosignals 2013, 21, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Palmer, T.D. Differential roles of TNFR1 and TNFR2 signaling in adult hippocampal neurogenesis. Brain. Behav. Immun. 2013, 30, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Ortí-Casañ, N.; Wu, Y.; Naudé, P.J.W.; De Deyn, P.P.; Zuhorn, I.S.; Eisel, U.L.M. Targeting TNFR2 as a Novel Therapeutic Strategy for Alzheimer’s Disease. Front. Neurosci. 2019, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Cribbs, D.H.; Anderson, A.J.; Cummings, B.J.; Su, J.H.; Wasserman, A.J.; Cotman, C.W. The induction of the TNFalpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem. Res. 2003, 28, 307–318. [Google Scholar] [CrossRef]
- Cheng, X.; Yang, L.; He, P.; Li, R.; Shen, Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J. Alzheimer’s Dis. 2010, 19, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; He, P.; Xie, J.; Staufenbiel, M.; Li, R.; Shen, Y. Genetic deletion of TNFRII gene enhances the Alzheimer-like pathology in an APP transgenic mouse model via reduction of phosphorylated IκBα. Hum. Mol. Genet. 2014, 23, 4906–4918. [Google Scholar] [CrossRef] [Green Version]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef] [Green Version]
- Moss, M.L.; Minond, D. Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediat. Inflamm. 2017, 2017, 9673537. [Google Scholar] [CrossRef] [PubMed]
- Riethmueller, S.; Ehlers, J.C.; Lokau, J.; Düsterhöft, S.; Knittler, K.; Dombrowsky, G.; Grötzinger, J.; Rabe, B.; Rose-John, S.; Garbers, C. Cleavage Site Localization Differentially Controls Interleukin-6 Receptor Proteolysis by ADAM10 and ADAM17. Sci. Rep. 2016, 6, 25550. [Google Scholar] [CrossRef] [Green Version]
- Bolino, A.; Piguet, F.; Alberizzi, V.; Pellegatta, M.; Rivellini, C.; Guerrero-Valero, M.; Noseda, R.; Brombin, C.; Nonis, A.; D’Adamo, P.; et al. Niacin-mediated Tace activation ameliorates CMT neuropathies with focal hypermyelination. EMBO Mol. Med. 2016, 8, 1438–1454. [Google Scholar] [CrossRef] [PubMed]
- Lassuthova, P.; Rebelo, A.P.; Ravenscroft, G.; Lamont, P.J.; Davis, M.R.; Manganelli, F.; Feely, S.M.; Bacon, C.; Brožková, D.Š.; Haberlova, J.; et al. Mutations in ATP1A1 Cause Dominant Charcot-Marie-Tooth Type 2. Am. J. Hum. Genet. 2018, 102, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Viviani, B.; Bartesaghi, S.; Gardoni, F.; Vezzani, A.; Behrens, M.M.; Bartfai, T.; Binaglia, M.; Corsini, E.; Di Luca, M.; Galli, C.L.; et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 8692–8700. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-M.; Jeon, S.-M.; Cho, H.-J. Tumor necrosis factor receptor 1 induces interleukin-6 upregulation through NF-kappaB in a rat neuropathic pain model. Eur. J. Pain 2009, 13, 794–806. [Google Scholar] [CrossRef]
- Pirkmajer, S.; Bezjak, K.; Matkovič, U.; Dolinar, K.; Jiang, L.Q.; Miš, K.; Gros, K.; Milovanova, K.; Pirkmajer, K.P.; Marš, T.; et al. Ouabain Suppresses IL-6/STAT3 Signaling and Promotes Cytokine Secretion in Cultured Skeletal Muscle Cells. Front. Physiol. 2020, 11, 566584. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Rauert, H.; Wicovsky, A.; Müller, N.; Siegmund, D.; Spindler, V.; Waschke, J.; Kneitz, C.; Wajant, H. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J. Biol. Chem. 2010, 285, 7394–7404. [Google Scholar] [CrossRef] [Green Version]
- Petzold, A.; Psotta, L.; Brigadski, T.; Endres, T.; Lessmann, V. Chronic BDNF deficiency leads to an age-dependent impairment in spatial learning. Neurobiol. Learn. Mem. 2015, 120, 52–60. [Google Scholar] [CrossRef]
- Saha, R.N.; Liu, X.; Pahan, K. Up-regulation of BDNF in astrocytes by TNF-alpha: A case for the neuroprotective role of cytokine. J. Neuroimmune Pharmacol. 2006, 1, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, H.; Altar, C.A.; Chen, R.; Nakamura, T.; Nakahashi, T.; Kambayashi, J.; Sun, B.; Tandon, N.N. Brain-derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb. Haemost. 2002, 87, 728–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra-Millàs, M. Are the changes in the peripheral brain-derived neurotrophic factor levels due to platelet activation? World J. Psychiatry 2016, 6, 84–101. [Google Scholar] [CrossRef] [PubMed]
- Bersier, M.G.; Rodríguez de Lores Arnaiz, G. Intracerebroventricular administration of ouabain to rats changes the expression of NMDA receptor subunits in cerebral cortex and hippocampus. Neurochem. Res. 2009, 34, 1650–1657. [Google Scholar] [CrossRef]
- Akkuratov, E.E.; Westin, L.; Vazquez-Juarez, E.; de Marothy, M.; Melnikova, A.K.; Blom, H.; Lindskog, M.; Brismar, H.; Aperia, A. Ouabain Modulates the Functional Interaction Between Na,K-ATPase and NMDA Receptor. Mol. Neurobiol. 2020, 57, 4018–4030. [Google Scholar] [CrossRef]
- Zhang, D.; Hou, Q.; Wang, M.; Lin, A.; Jarzylo, L.; Navis, A.; Raissi, A.; Liu, F.; Man, H.-Y. Na,K-ATPase activity regulates AMPA receptor turnover through proteasome-mediated proteolysis. J. Neurosci. 2009, 29, 4498–4511. [Google Scholar] [CrossRef] [Green Version]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef] [Green Version]
- Bernardino, L.; Xapelli, S.; Silva, A.P.; Jakobsen, B.; Poulsen, F.R.; Oliveira, C.R.; Vezzani, A.; Malva, J.O.; Zimmer, J. Modulator effects of interleukin-1beta and tumor necrosis factor-alpha on AMPA-induced excitotoxicity in mouse organotypic hippocampal slice cultures. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 6734–6744. [Google Scholar] [CrossRef] [Green Version]
- Selden, R.; Smith, T.W. Ouabain pharmacokinetics in dog and man. Determination by radioimmunoassay. Circulation 1972, 45, 1176–1182. [Google Scholar] [CrossRef] [Green Version]
- Strobach, H.; Wirth, K.E.; Rojsathaporn, K. Absorption, metabolism and elimination of strophanthus glycosides in man. Naunyn. Schmiedebergs. Arch. Pharmacol. 1986, 334, 496–500. [Google Scholar] [CrossRef]
- Picton, L.D.; Nascimento, F.; Broadhead, M.J.; Sillar, K.T.; Miles, G.B. Sodium Pumps Mediate Activity-Dependent Changes in Mammalian Motor Networks. J. Neurosci. 2017, 37, 906–921. [Google Scholar] [CrossRef] [PubMed]
- Kurauchi, Y.; Yoshimaru, Y.; Kajiwara, Y.; Yamada, T.; Matsuda, K.; Hisatsune, A.; Seki, T.; Katsuki, H. Na(+), K(+)-ATPase inhibition causes hyperactivity and impulsivity in mice via dopamine D2 receptor-mediated mechanism. Neurosci. Res. 2019, 146, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Varela, R.B.; Valvassori, S.S.; Lopes-Borges, J.; Mariot, E.; Dal-Pont, G.C.; Amboni, R.T.; Bianchini, G.; Quevedo, J. Sodium butyrate and mood stabilizers block ouabain-induced hyperlocomotion and increase BDNF, NGF and GDNF levels in brain of Wistar rats. J. Psychiatr. Res. 2015, 61, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Decker, S.; Grider, G.; Cobb, M.; Li, X.P.; Huff, M.O.; El-Mallakh, R.S.; Levy, R.S. Open field is more sensitive than automated activity monitor in documenting ouabain-induced hyperlocomotion in the development of an animal model for bipolar illness. Prog. Neuropsychopharmacol. Biol. Psychiatry 2000, 24, 455–462. [Google Scholar] [CrossRef]
- Lopachev, A.; Volnova, A.; Evdokimenko, A.; Abaimov, D.; Timoshina, Y.; Kazanskaya, R.; Lopacheva, O.; Deal, A.; Budygin, E.; Fedorova, T.; et al. Intracerebroventricular injection of ouabain causes mania-like behavior in mice through D2 receptor activation. Sci. Rep. 2019, 9, 15627. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinoshita, P.F.; Orellana, A.M.; Andreotti, D.Z.; de Souza, G.A.; de Mello, N.P.; de Sá Lima, L.; Kawamoto, E.M.; Scavone, C. Consequences of the Lack of TNFR1 in Ouabain Response in the Hippocampus of C57BL/6J Mice. Biomedicines 2022, 10, 2937. https://doi.org/10.3390/biomedicines10112937
Kinoshita PF, Orellana AM, Andreotti DZ, de Souza GA, de Mello NP, de Sá Lima L, Kawamoto EM, Scavone C. Consequences of the Lack of TNFR1 in Ouabain Response in the Hippocampus of C57BL/6J Mice. Biomedicines. 2022; 10(11):2937. https://doi.org/10.3390/biomedicines10112937
Chicago/Turabian StyleKinoshita, Paula Fernanda, Ana Maria Orellana, Diana Zukas Andreotti, Giovanna Araujo de Souza, Natalia Prudente de Mello, Larissa de Sá Lima, Elisa Mitiko Kawamoto, and Cristoforo Scavone. 2022. "Consequences of the Lack of TNFR1 in Ouabain Response in the Hippocampus of C57BL/6J Mice" Biomedicines 10, no. 11: 2937. https://doi.org/10.3390/biomedicines10112937