
Surgical Management of Synucleinopathies

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Parkinson’s Disease
1.2. Multiple System Atrophy and Dementia with Lewy Bodies
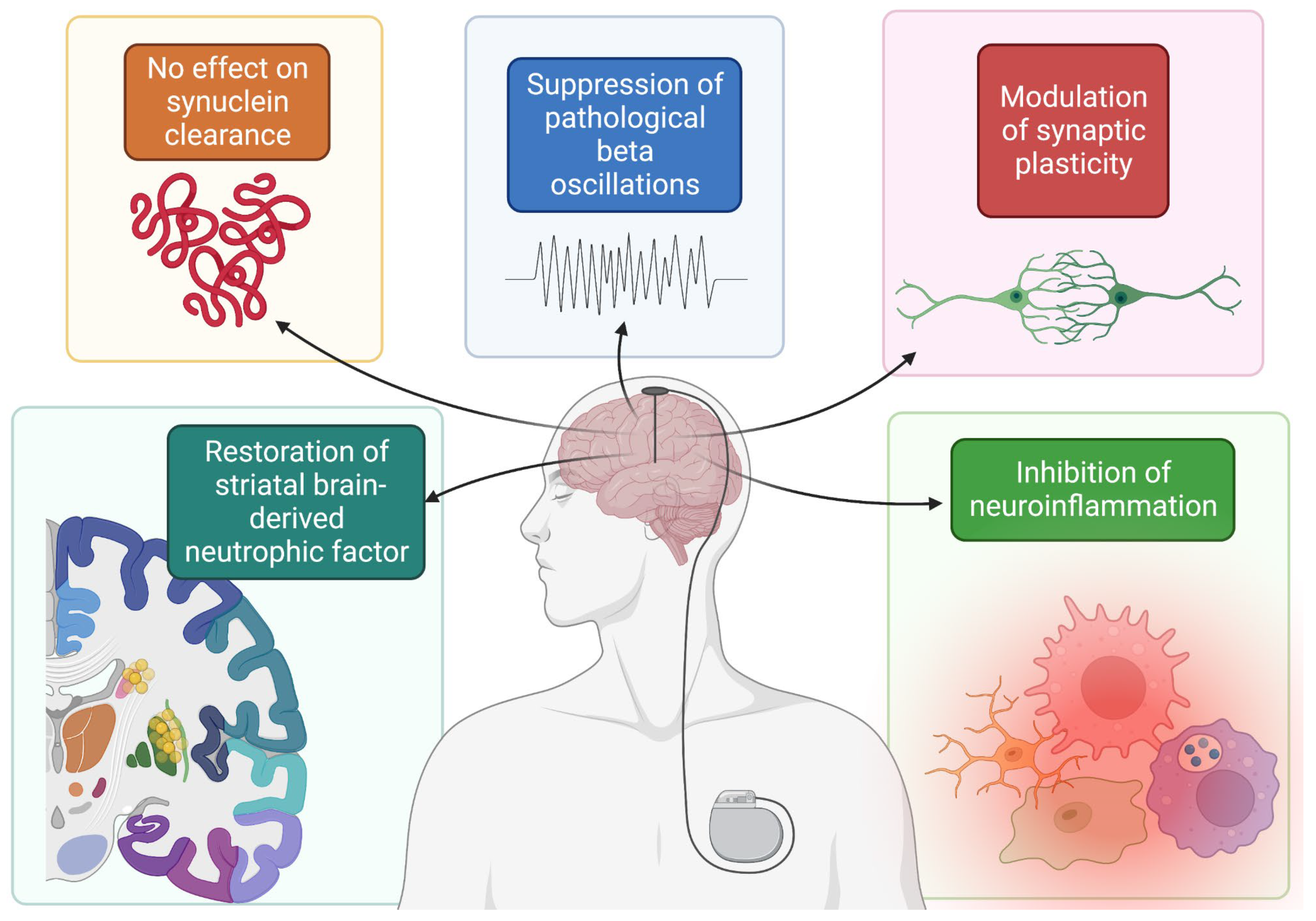
2. Deep Brain Stimulation (DBS)
2.1. DBS and α-Synuclein in PD
2.2. DBS in DLB and MSA
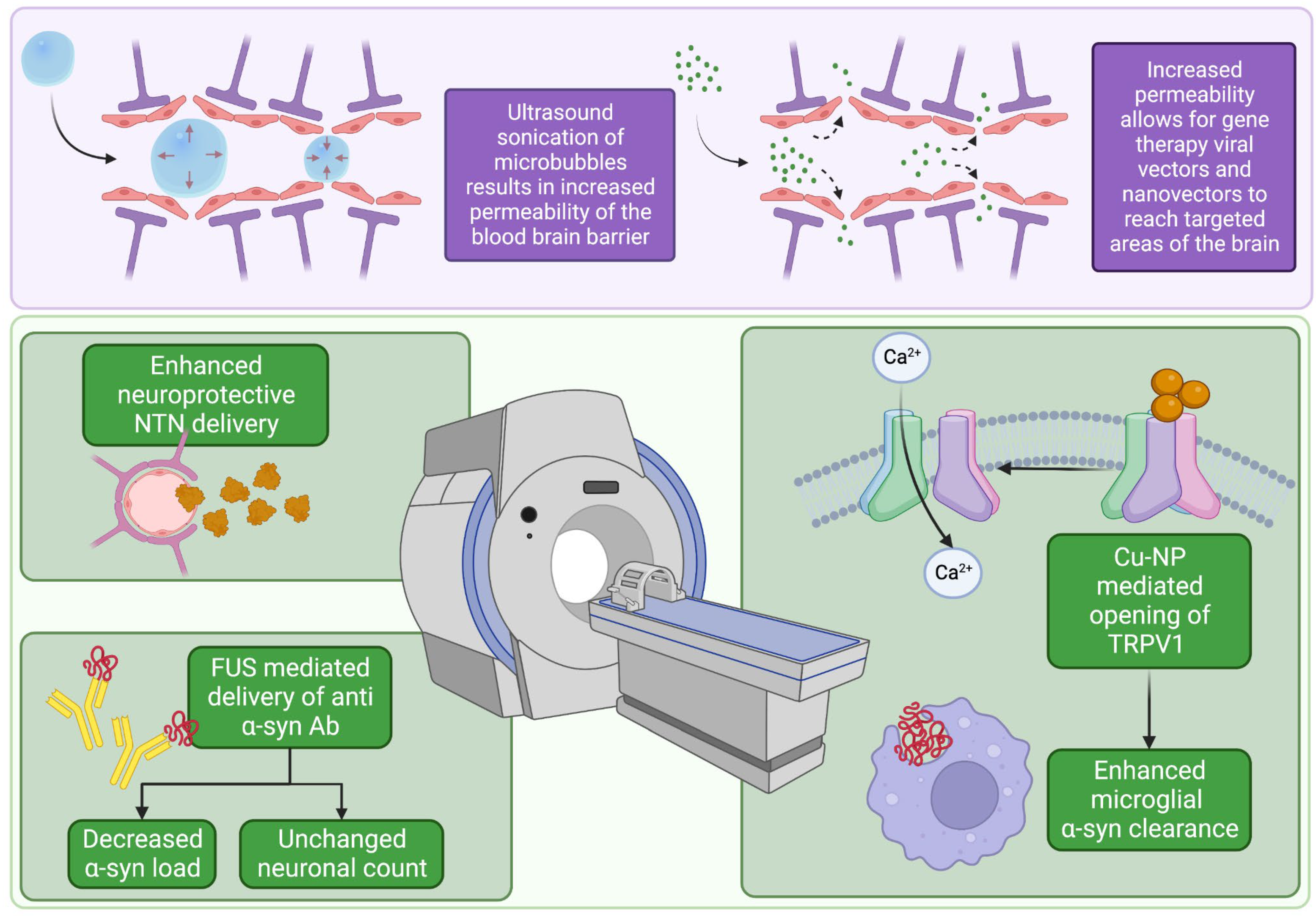
3. Focused Ultrasound
3.1. FUS and Synuclein
3.2. FUS Gene Therapy Approaches
4. Other Approaches in the Management of MSA and DLB
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kianirad, Y.; Simuni, T. Pimavanserin, a novel antipsychotic for management of Parkinson’s disease psychosis. Expert Rev. Clin. Pharmacol. 2017, 10, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Abrantes, A.M.; Friedman, J.H.; Brown, R.A.; Strong, D.R.; Desaulniers, J.; Ing, E.; Saritelli, J.; Riebe, D. Physical Activity and Neuropsychiatric Symptoms of Parkinson Disease. J. Geriatr. Psychiatry Neurol. 2012, 25, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Wickremaratchi, M.M.; Ben-Shlomo, Y.; Morris, H.R. The effect of onset age on the clinical features of Parkinson’s disease. Eur. J. Neurol. 2009, 16, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Ray Dorsey, E.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and projected future economic burden of Parkinson’s disease in the U.S. NPJ Park. Dis. 2020, 6, 1–9. [Google Scholar] [CrossRef]
- De Souza, R.M.; Moro, E.; Lang, A.E.; Schapira, A.H.V. Timing of Deep Brain Stimulation in Parkinson Disease: A Need for Reappraisal? Ann. Neurol. 2013, 73, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. NPJ Park. Dis. 2018, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease: Etiology and Pathogenesis. Mov. Disord. 2011, 26, 1049–1055. [Google Scholar] [CrossRef]
- Schapira, A.H.; Agid, Y.; Barone, P.; Jenner, P.; Lemke, M.R.; Poewe, W.; Rascol, O.; Reichmann, H.; Tolosa, E. Perspectives on recent advances in the understanding and treatment of Parkinson’s disease. Eur. J. Neurol. 2009, 16, 1090–1099. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Yang, R.; Guo, J.C.; Ren, H.M.; Zha, X.L.; Cheng, J.S.; Cai, D.F. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 2007, 18, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, C.; Yin, J.; Li, X.; Cheng, F.; Li, Y.; Yang, H.; Uéda, K.; Chan, P.; Yu, S. alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 2009, 454, 187–192. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.K.; Choi, M.G.; Kim, J.Y.; Yang, Y.; Lai, Y.; Kweon, D.H.; Lee, N.K.; Shin, Y.K. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 2013, 110, 4087–4092. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W. The natural history of Parkinson’s disease. J. Neurol. 2006, 253 (Suppl. S7), VII2–VII6. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovic, J.; Stacy, M. Medical management of levodopa-associated motor complications in patients with Parkinson’s disease. CNS Drugs 2007, 21, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Jost, W.H.; Augustis, S. Severity of orthostatic hypotension in the course of Parkinson’s disease: No correlation with the duration of the disease. Park. Relat. Disord. 2015, 21, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Riedel, O.; Klotsche, J.; Spottke, A.; Deuschl, G.; Förstl, H.; Henn, F.; Heuser, I.; Oertel, W.; Reichmann, H.; Riederer, P.; et al. Cognitive impairment in 873 patients with idiopathic Parkinson’s disease. Results from the German Study on Epidemiology of Parkinson’s Disease with Dementia (GEPAD). J. Neurol. 2008, 255, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knie, B.; Mitra, M.T.; Logishetty, K.; Chaudhuri, K.R. Excessive Daytime Sleepiness in Patients with Parkinson’s Disease. CNS Drugs 2011, 25, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.E.; Hess, C.W.; Fox, S.H. Motor Complications of Dopaminergic Medications in Parkinson’s Disease. Semin. Neurol. 2017, 37, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Rocca, W.A. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology 1997, 49, 1284–1288. [Google Scholar] [CrossRef] [PubMed]
- Vann Jones, S.A.; O’Brien, J.T. The prevalence and incidence of dementia with Lewy bodies: A systematic review of population and clinical studies. Psychol. Med. 2014, 44, 673–683. [Google Scholar] [CrossRef]
- Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N. Engl. J. Med. 2013, 369, 233–244. [Google Scholar] [CrossRef]
- Wenning, G.K.; Krismer, F. Multiple system atrophy. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 117, pp. 229–241. [Google Scholar] [CrossRef]
- Boot, B.P.; Orr, C.F.; Ahlskog, J.E.; Ferman, T.J.; Roberts, R.; Pankratz, V.S.; Dickson, D.W.; Parisi, J.; Aakre, J.A.; Geda, Y.E.; et al. Risk factors for dementia with Lewy bodies: A case-control study. Neurology 2013, 81, 833–840. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.S.; Kågedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef]
- Gilman, S.; Wenning, G.K.; Low, P.A.; Brooks, D.J.; Mathias, C.J.; Trojanowski, J.Q.; Wood, N.W.; Colosimo, C.; Dürr, A.; Fowler, C.J.; et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008, 71, 670–676. [Google Scholar] [CrossRef]
- Ozawa, T.; Paviour, D.; Quinn, N.P.; Josephs, K.A.; Sangha, H.; Kilford, L.; Healy, D.G.; Wood, N.W.; Lees, A.J.; Holton, J.L.; et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: Clinicopathological correlations. Brain J. Neurol. 2004, 127 Pt 12, 2657–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Esparcia, P.; López-González, I.; Grau-Rivera, O.; García-Garrido, M.F.; Konetti, A.; Llorens, F.; Zafar, S.; Carmona, M.; del Rio, J.A.; Zerr, I.; et al. Dementia with Lewy Bodies: Molecular Pathology in the Frontal Cortex in Typical and Rapidly Progressive Forms. Front. Neurol. 2017, 8, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, G.M.; Holton, J.L.; Revesz, T.; Dickson, D.W. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011, 122, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Jecmenica-Lukic, M.; Poewe, W.; Tolosa, E.; Wenning, G.K. Premotor signs and symptoms of multiple system atrophy. Lancet Neurol. 2012, 11, 361–368. [Google Scholar] [CrossRef]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [Green Version]
- Burn, D.J.; Rowan, E.N.; Minett, T.; Sanders, J.; Myint, P.; Richardson, J.; Thomas, A.; Newby, J.; Reid, J.; O’Brien, J.T.; et al. Extrapyramidal features in Parkinson’s disease with and without dementia and dementia with Lewy bodies: A cross-sectional comparative study. Mov. Disord. Off. J. Mov. Disord. Soc. 2003, 18, 884–889. [Google Scholar] [CrossRef]
- Köllensperger, M.; Geser, F.; Ndayisaba, J.P.; Boesch, S.; Seppi, K.; Ostergaard, K.; Dupont, E.; Cardozo, A.; Tolosa, E.; Abele, M.; et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: Final analysis of the European multiple system atrophy registry. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 2604–2612. [Google Scholar] [CrossRef]
- Hamilton, J.M.; Salmon, D.P.; Galasko, D.; Raman, R.; Emond, J.; Hansen, L.A.; Masliah Eliezer Thal, L.J. Visuospatial Deficits Predict Rate of Cognitive Decline in Autopsy-Verified Dementia with Lewy Bodies. Neuropsychology 2008, 22, 729–737. [Google Scholar] [CrossRef] [Green Version]
- Abbott, A. Levodopa: The story so far. Nature 2010, 466, S6–S7. [Google Scholar] [CrossRef]
- Brodell, D.W.; Stanford, N.T.; Jacobson, C.E.; Schmidt, P.; Okun, M.S. Carbidopa/levodopa dose elevation and safety concerns in Parkinson’s patients: A cross-sectional and cohort design. BMJ Open 2012, 2, e001971. [Google Scholar] [CrossRef] [Green Version]
- Honey, C.R.; Hamani, C.; Kalia, S.K.; Sankar, T.; Picillo, M.; Munhoz, R.P.; Fasano, A.; Panisset, M. Deep Brain Stimulation Target Selection for Parkinson’s Disease. Can. J. Neurol. Sci. 2017, 44, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinkhauser, G.; Pogosyan, A.; Tan, H.; Herz, D.M.; Kühn, A.A.; Brown, P. Beta burst dynamics in Parkinson’s disease OFF and ON dopaminergic medication. Brain 2017, 140, 2968–2981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner-Fisman, G.; Herzog, J.; Fisman, D.N.; Tamma, F.; Lyons, K.E.; Pahwa, R.; Lang, A.E.; Deuschl, G. Subthalamic nucleus deep brain stimulation: Summary and meta-analysis of outcomes. Mov. Disord. 2006, 21 (Suppl. S14), S290–S304. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef] [Green Version]
- Fischer, D.L.; Manfredsson, F.P.; Kemp, C.J.; Cole-Strauss, A.; Lipton, J.W.; Duffy, M.F.; Polinski, N.K.; Steece-Collier, K.; Collier, T.J.; Gombash, S.E.; et al. Subthalamic Nucleus Deep Brain Stimulation Does Not Modify the Functional Deficits or Axonopathy Induced by Nigrostriatal α-Synuclein Overexpression. Sci. Rep. 2017, 7, 16356. [Google Scholar] [CrossRef] [Green Version]
- Musacchio, T.; Rebenstorff, M.; Fluri, F.; Brotchie, J.M.; Volkmann, J.; Koprich, J.B.; Ip, C.W. Subthalamic nucleus deep brain stimulation is neuroprotective in the A53T α-synuclein Parkinson’s disease rat model. Ann. Neurol. 2017, 81, 825–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, S.; Brown, P. The functional role of beta oscillations in Parkinson’s disease. Park. Relat. Disord. 2014, 20, S44–S48. [Google Scholar] [CrossRef]
- Chen, J.Z.; Hofman, K.; Koprich, J.B.; Brotchie, J.M.; Volkmann, J.; Ip, C.W. P 15 Long-term subthalamic deep brain stimulation modulates pathological beta oscillations in the AAV-A53T-Synuclein Parkinson’s disease rat model. Clin. Neurophysiol. 2022, 137, e23–e24. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. Available online: https://www.frontiersin.org/articles/10.3389/fncel.2019.00363 (accessed on 18 September 2022). [CrossRef]
- Miller, K.M.; Patterson, J.R.; Kochmanski, J.; Kemp, C.J.; Stoll, A.C.; Onyekpe, C.U.; Cole-Strauss, A.; Steece-Collier, K.; Howe, J.W.; Luk, K.C.; et al. Striatal Afferent BDNF Is Disrupted by Synucleinopathy and Partially Restored by STN DBS. J. Neurosci. 2021, 41, 2039–2052. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, G.; Liu, D.; Zhang, X.; Liu, Y.; Yuan, T.; Du, T.; Zhang, J. Subthalamic nucleus deep brain stimulation suppresses neuroinflammation by Fractalkine pathway in Parkinson’s disease rat model. Brain Behav. Immun. 2020, 90, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Herrington, T.M.; Cheng, J.J.; Eskandar, E.N. Mechanisms of deep brain stimulation. J. Neurophysiol. 2016, 115, 19–38. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. α-Synuclein Is Degraded by Both Autophagy and the Proteasome*. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Li, Z.; Han, D.; Mi, X.; Tian, M.; Liu, T.; Li, Y.; He, J.; Kuang, C.; Cao, Y.; et al. Autophagy prevents hippocampal α-synuclein oligomerization and early cognitive dysfunction after anesthesia/surgery in aged rats. Aging 2020, 12, 7262–7281. [Google Scholar] [CrossRef]
- Witt, K.; Daniels, C.; Volkmann, J. Factors associated with neuropsychiatric side effects after STN-DBS in Parkinson’s disease. Park. Relat. Disord. 2012, 18, S168–S170. [Google Scholar] [CrossRef]
- Gratwicke, J.; Zrinzo, L.; Kahan, J.; Peters, A.; Brechany, U.; McNichol, A.; Beigi, M.; Akram, H.; Hyam, J.; Oswal, A.; et al. Bilateral nucleus basalis of Meynert deep brain stimulation for dementia with Lewy bodies: A randomised clinical trial. Brain Stimulat. 2020, 13, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Maltête, D.; Wallon, D.; Bourilhon, J.; Lefaucheur, R.; Danaila, T.; Thobois, S.; Defebvre, L.; Dujardin, K.; Houeto, J.L.; Godefroy, O.; et al. Nucleus Basalis of Meynert Stimulation for Lewy Body Dementia: A Phase I Randomized Clinical Trial. Neurology 2021, 96, e684–e697. [Google Scholar] [CrossRef] [PubMed]
- Thavanesan, N.; Gillies, M.; Farrell, M.; Green, A.L.; Aziz, T. Deep Brain Stimulation in Multiple System Atrophy Mimicking Idiopathic Parkinson’s Disease. Case Rep. Neurol. 2014, 6, 232–237. [Google Scholar] [CrossRef]
- Lambrecq, V.; Krim, E.; Meissner, W.; Guehl, D.; Tison, F. Deep-brain stimulation of the internal pallidum in multiple system atrophy. Rev. Neurol. 2008, 164, 398–402. [Google Scholar] [CrossRef]
- Artusi, C.A.; Rinaldi, D.; Balestrino, R.; Lopiano, L. Deep brain stimulation for atypical parkinsonism: A systematic review on efficacy and safety. Park. Relat. Disord. 2022, 96, 109–118. [Google Scholar] [CrossRef]
- Stankovic, I.; Krismer, F.; Jesic, A.; Antonini, A.; Benke, T.; Brown, R.G.; Burn, D.J.; Holton, J.L.; Kaufmann, H.; Kostic, V.S.; et al. Cognitive impairment in multiple system atrophy. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Hynynen, K.; Lipsman, N. Applications of focused ultrasound in the brain: From thermoablation to drug delivery. Nat. Rev. Neurol. 2021, 17, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, Z.; Izadifar, Z.; Chapman, D.; Babyn, P. An Introduction to High Intensity Focused Ultrasound: Systematic Review on Principles, Devices, and Clinical Applications. J. Clin. Med. 2020, 9, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, P.P.; Brown, J.R.; Pauly, K.B. Frequency Dependence of Ultrasound Neurostimulation in the Mouse Brain. Ultrasound Med. Biol. 2016, 42, 1512–1530. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.F. High intensity focused ultrasound in clinical tumor ablation. World J. Clin. Oncol. 2011, 2, 8–27. [Google Scholar] [CrossRef]
- Stewart, E.A.; Rabinovici, J.; Tempany, C.M.C.; Inbar, Y.; Regan, L.; Gastout, B.; Hesley, G.; Kim, H.S.; Hengst, S.; Gedroye, W.M. Clinical outcomes of focused ultrasound surgery for the treatment of uterine fibroids. Fertil. Steril. 2006, 85, 22–29. [Google Scholar] [CrossRef]
- Wu, F.; Wang, Z.B.; Chen, W.Z.; Bai, J.; Zhu, H.; Qiao, T.Y. Preliminary Experience Using High Intensity Focused Ultrasound for the Treatment of Patients with Advanced Stage Renal Malignancy. J. Urol. 2003, 170, 2237–2240. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Zhou, K. High-intensity focused ultrasound ablation: A new strategy to manage primary bone tumors. Curr. Opin. Orthop. 2005, 16, 494–500. [Google Scholar] [CrossRef]
- Prada, F.; Kalani, M.Y.S.; Yagmurlu, K.; Norat, P.; Del Bene, M.; DiMeco, F.; Kassell, N.F. Applications of Focused Ultrasound in Cerebrovascular Diseases and Brain Tumors. Neurotherapeutics 2019, 16, 67–87. [Google Scholar] [CrossRef]
- Samiotaki, G.; Acosta, C.; Wang, S.; Konofagou, E.E. Enhanced delivery and bioactivity of the neurturin neurotrophic factor through focused ultrasound—mediated blood–brain barrier opening in vivo. J. Cereb. Blood Flow Metab. 2015, 35, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Blood-brain barrier drug targeting: The future of brain drug development. Mol. Interv. 2003, 3, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasca-Salas, C.; Fernández-Rodríguez, B.; Pineda-Pardo, J.A.; Rodríguez-Rojas, R.; Obeso, I.; Hernández-Fernández, F.; del Álamo, M.; Mata, D.; Guida, P.; Ordás-Bandera, C.; et al. Blood-brain barrier opening with focused ultrasound in Parkinson’s disease dementia. Nat. Commun. 2021, 12, 779. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sierra, C.; Kwon, N.; Karakatsani, M.E.; Jackson-Lewis, V.R.; Przedborski, S.; Konofagou, E. Focused-ultrasound Mediated Anti-Alpha-Synuclein Antibody Delivery for the Treatment of Parkinson’s Disease. In Proceedings of the 2018 IEEE International Ultrasonics Symposium (IUS), Kobe, Japan, 22–25 October 2018; pp. 1–4. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, H.; Zhang, H.; Wang, T.; Zheng, Q.; Li, Z. Controlled Activation of TRPV1 Channels on Microglia to Boost Their Autophagy for Clearance of Alpha-Synuclein and Enhance Therapy of Parkinson’s Disease. Adv. Mater. 2022, 34, 2108435. [Google Scholar] [CrossRef] [PubMed]
- Moosa, S.; Martínez-Fernández, R.; Elias, W.J.; del Alamo, M.; Eisenberg, H.M.; Fishman, P.S. The role of high-intensity focused ultrasound as a symptomatic treatment for Parkinson’s disease. Mov. Disord. 2019, 34, 1243–1251. [Google Scholar] [CrossRef]
- Schlesinger, I.; Sinai, A.; Zaaroor, M. MRI-Guided Focused Ultrasound in Parkinson’s Disease: A Review. Park. Dis. 2017, 2017, 8124624. [Google Scholar] [CrossRef]
- Martínez-Fernández, R.; Máñez-Miró, J.U.; Rodríguez-Rojas, R.; del Álamo, M.; Shah, B.B.; Hernández-Fernández, F.; Pineda-Pardo, J.A.; Monje, M.H.G.; Fernández-Rodríguez, B.; Sperling, S.A.; et al. Randomized Trial of Focused Ultrasound Subthalamotomy for Parkinson’s Disease. N. Engl. J. Med. 2020, 383, 2501–2513. [Google Scholar] [CrossRef]
- Jung, N.Y.; Park, C.K.; Kim, M.; Lee, P.H.; Sohn, Y.H.; Chang, J.W. The efficacy and limits of magnetic resonance-guided focused ultrasound pallidotomy for Parkinson’s disease: A Phase I clinical trial. J. Neurosurg. 2018, 1853–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef]
- Lin, C.Y.; Hsieh, H.Y.; Chen, C.M.; Wu, S.R.; Tsai, C.H.; Huang, C.Y.; Hua, M.Y.; Wei, K.C.; Yeh, C.K.; Liu, H.L. Non-invasive, neuron-specific gene therapy by focused ultrasound-induced blood-brain barrier opening in Parkinson’s disease mouse model. J. Control. Release 2016, 235, 72–81. [Google Scholar] [CrossRef]
- Marks, W.J.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef]
- Kordower, J.H.; Emborg, M.E.; Bloch, J.; Ma, S.Y.; Chu, Y.; Leventhal, L.; McBride, J.; Chen, E.Y.; Palfi, S.; Roitberg, B.Z.; et al. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 2000, 290, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Johnson, E.M. Clinical tests of neurotrophic factors for human neurodegenerative diseases, part 2: Where do we stand and where must we go next? Neurobiol. Dis. 2017, 97 Pt B, 169–178. [Google Scholar] [CrossRef]
- Price, R.J.; Fisher, D.G.; Suk, J.S.; Hanes, J.; Ko, H.S.; Kordower, J.H. Parkinson’s Disease Gene Therapy: Will Focused Ultrasound and Nanovectors Be the Next Frontier? Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 1279–1282. [Google Scholar] [CrossRef]
- Xhima, K.; Nabbouh, F.; Hynynen, K.; Aubert, I.; Tandon, A. Noninvasive delivery of an α-synuclein gene silencing vector with magnetic resonance–guided focused ultrasound. Mov. Disord. 2018, 33, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Olumolade, O.O.; Sun, T.; Samiotaki, G.; Konofagou, E.E. Noninvasive, neuron-specific gene therapy can be facilitated by focused ultrasound and recombinant adeno-associated virus. Gene Ther. 2015, 22, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mead, B.P.; Kim, N.; Miller, G.W.; Hodges, D.; Mastorakos, P.; Klibanov, A.L.; Mandell, J.W.; Hirsh, J.; Suk, J.S.; Hanes, J.; et al. Novel Focused Ultrasound Gene Therapy Approach Noninvasively Restores Dopaminergic Neuron Function in a Rat Parkinson’s Disease Model. Nano Lett. 2017, 17, 3533–3542. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, L.; Cai, X.; Guo, R.; Wang, P.; Wu, L.; Yin, T.; Liao, S.; Lu, Z. Treatment of Parkinson’s disease in rats by Nrf2 transfection using MRI-guided focused ultrasound delivery of nanomicrobubbles. Biochem. Biophys. Res. Commun. 2017, 482, 75–80. [Google Scholar] [CrossRef]
- Iodice, V.; Low, D.A.; Vichayanrat, E.; Mathias, C.J. Cardiovascular autonomic dysfunction in MSA and Parkinson’s disease: Similarities and differences. J. Neurol. Sci. 2011, 310, 133–138. [Google Scholar] [CrossRef]
- Tada, M.; Onodera, O.; Tada, M.; Ozawa, T.; Piao, Y.S.; Kakita, A.; Takahashi, H.; Nishizawa, M. Early Development of Autonomic Dysfunction May Predict Poor Prognosis in Patients with Multiple System Atrophy. Arch. Neurol. 2007, 64, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Squair, J.W.; Berney, M.; Castro Jimenez, M.; Hankov, N.; Demesmaeker, R.; Amir, S.; Paley, A.; Hernandez-Charpak, S.; Dumont, G.; Asboth, L.; et al. Implanted System for Orthostatic Hypotension in Multiple-System Atrophy. N. Engl. J. Med. 2022, 386, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Stagg, C.J.; Nitsche, M.A. Physiological Basis of Transcranial Direct Current Stimulation. Neuroscientist 2011, 17, 37–53. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sriram, S.; Root, K.; Chacko, K.; Patel, A.; Lucke-Wold, B. Surgical Management of Synucleinopathies. Biomedicines 2022, 10, 2657. https://doi.org/10.3390/biomedicines10102657
Sriram S, Root K, Chacko K, Patel A, Lucke-Wold B. Surgical Management of Synucleinopathies. Biomedicines. 2022; 10(10):2657. https://doi.org/10.3390/biomedicines10102657
Chicago/Turabian StyleSriram, Sai, Kevin Root, Kevin Chacko, Aashay Patel, and Brandon Lucke-Wold. 2022. "Surgical Management of Synucleinopathies" Biomedicines 10, no. 10: 2657. https://doi.org/10.3390/biomedicines10102657