
Mitochondrial Dysfunction, Mitophagy and Their Correlation with Perinatal Complications: Preeclampsia and Low Birth Weight

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
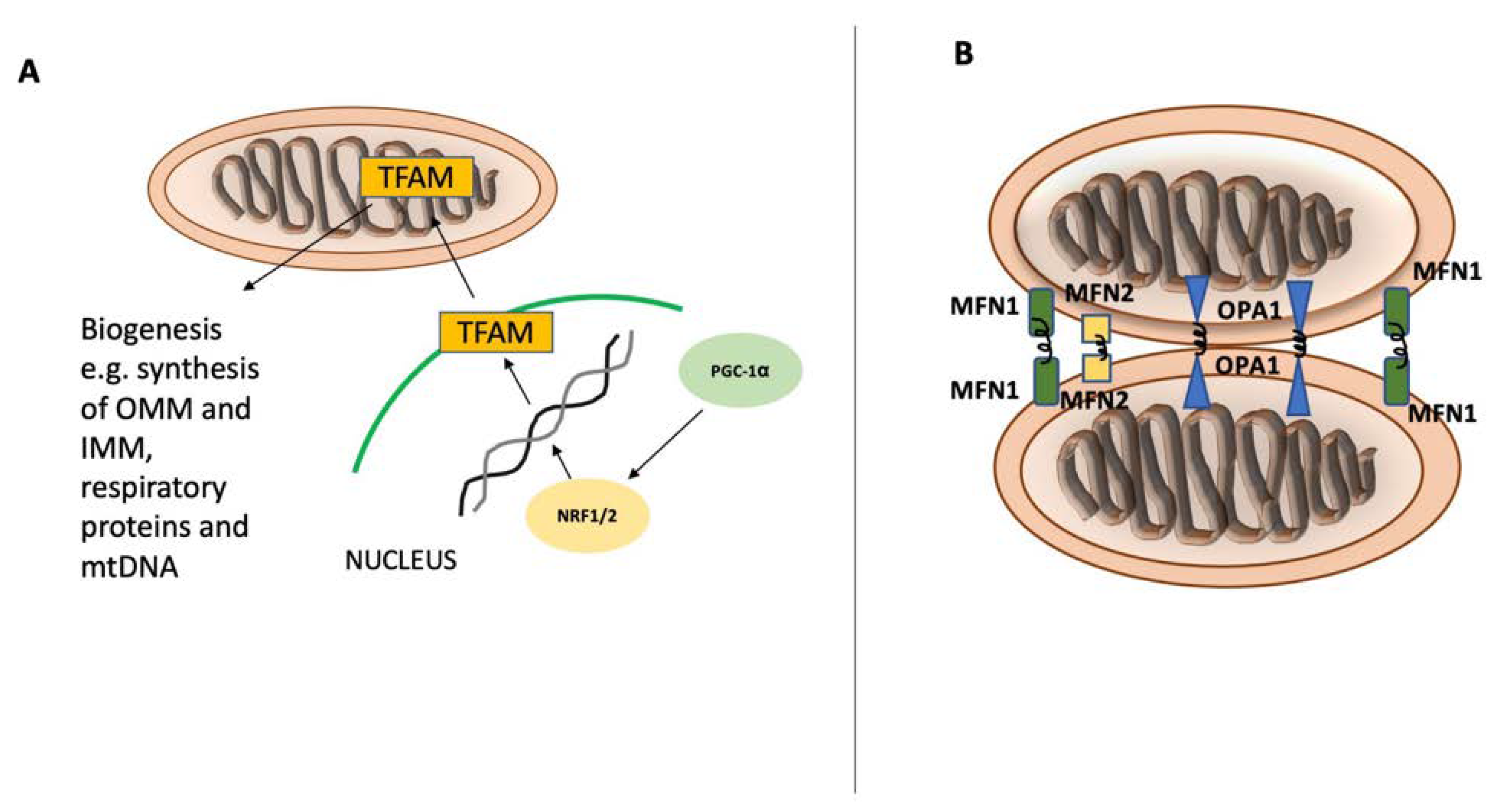
2. Mitochondrial Homeostasis
3. Insights into Preeclampsia
4. Preeclampsia and Mitochondria
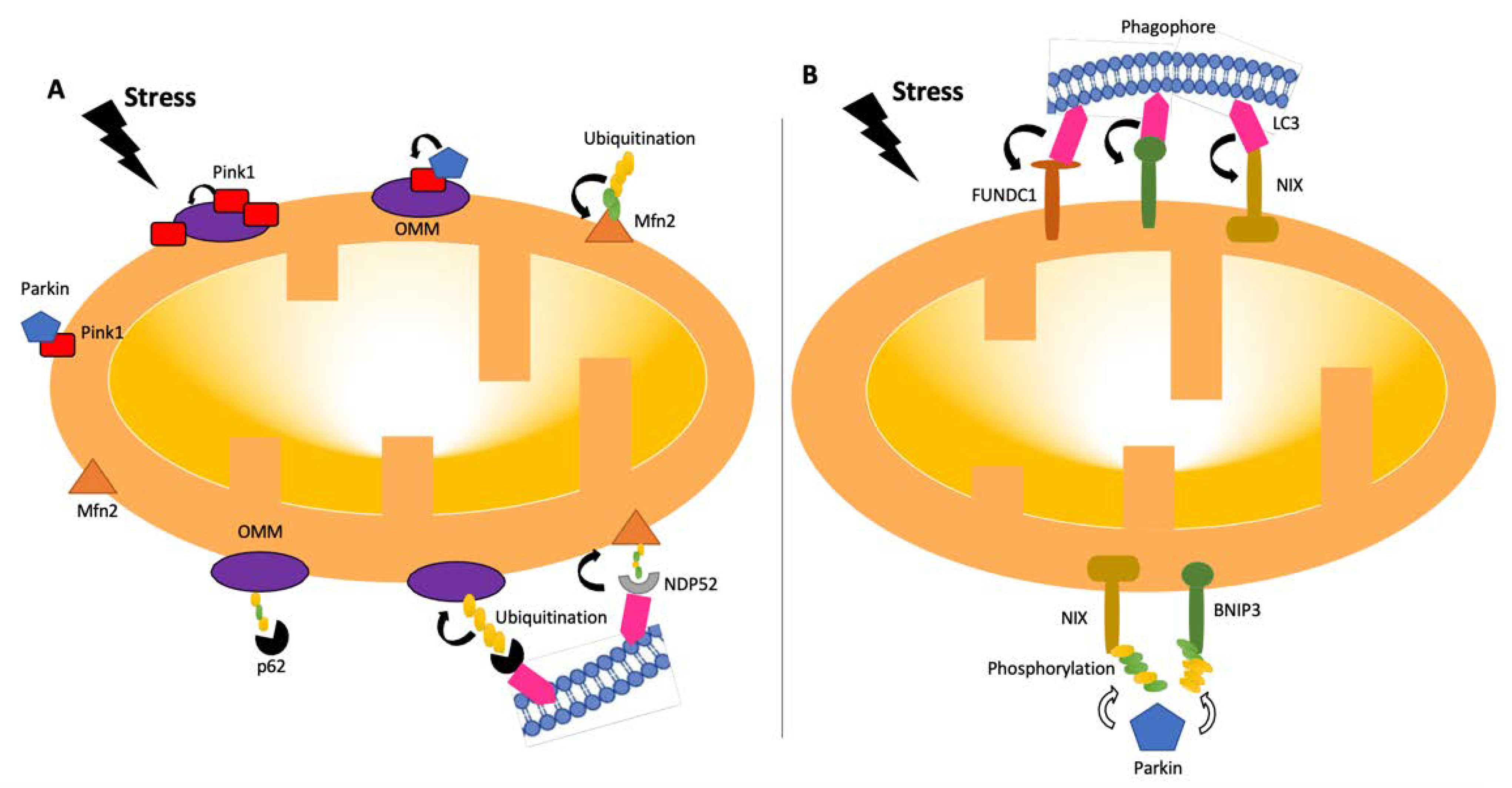
5. Markers of Mitophagy and Effects on Preeclampsia
5.1. BNIP3
5.2. FUNDC1
5.3. DRAM1
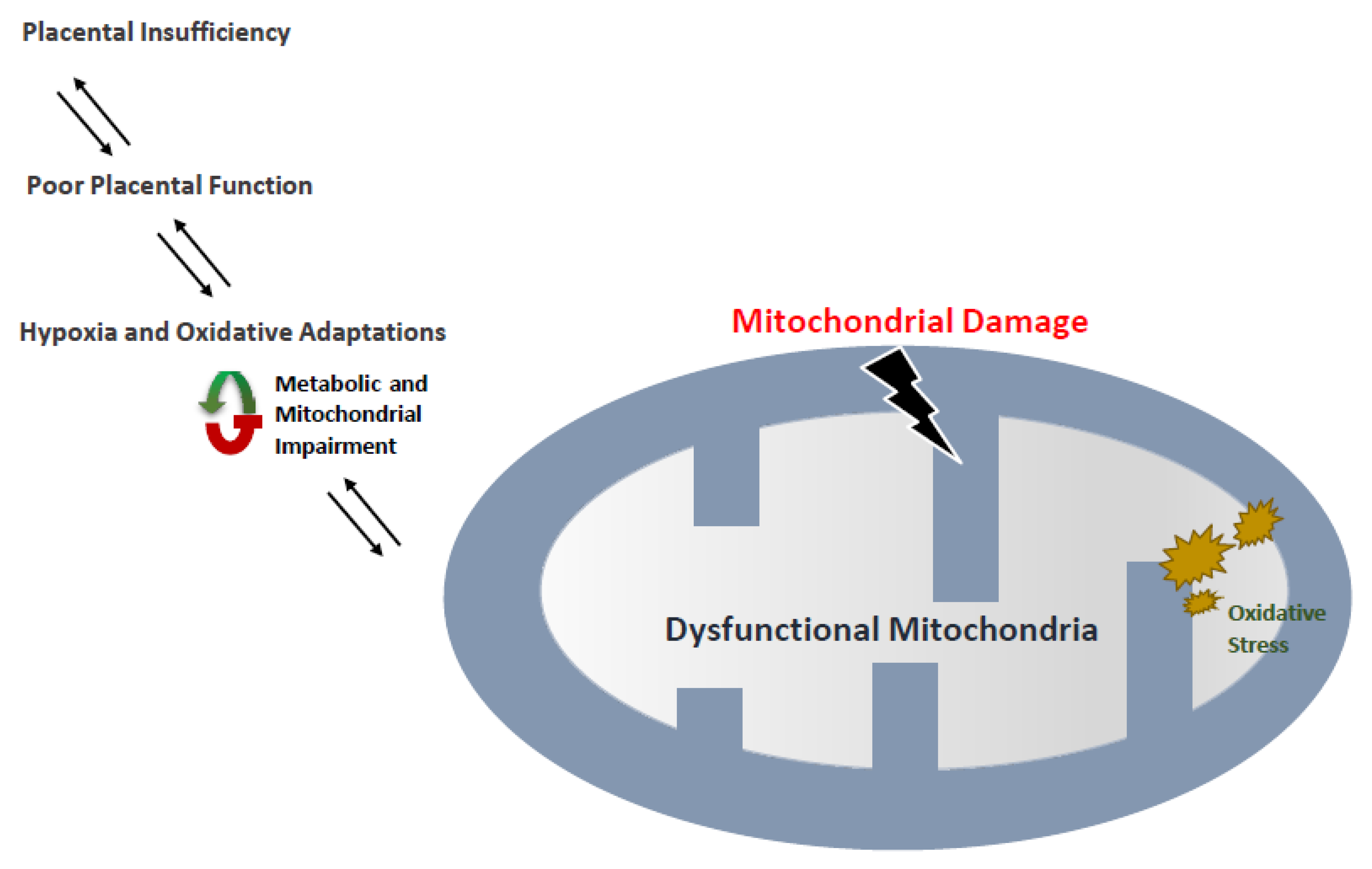
6. Intrauterine Growth Restriction (IUGR) and Low Birth Weight
7. Mitochondria and Its Association with Low-Birth-Weight Rate
8. Fetal Growth Restriction and Correlation with Mitophagy
9. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATF4 | Activating transcription factor 4 |
| ATP | Adenosine Triphosphate |
| BNIP3 | Bcl-2/adenovirus E1B 19-kDa interacting protein 3 |
| BP | Blood pressure |
| CERs | Ceramides |
| CYP11A1 | Cholesterol side-chain cleavage enzyme gene |
| COX IV | Cytochrome c oxidase IV |
| COX4 | Cytochrome C Oxidase Subunit 4 |
| DRAM1 | DNA damage-regulated autophagy modulator 1 |
| Drp1 | Dynamin-related protein 1 |
| ETC | Electron Transport Chain |
| ER | Endoplasmic Reticulum |
| ERR-α, ERR-β, ERR-γ | Estrogen-related Receptors -α,-β,-γ |
| FUNDC1 | FUN14 domain containing 1 |
| GCN2 | General control nonderepressible 2 |
| GSH | Glutathione |
| HSP6 | Histone Protein 60 |
| hUMSCs | Human Mesenchymal Stem Cells |
| IMM | Inner Mitochondrial Membrane |
| IUGR | Intrauterine growth restriction |
| LIR | LC3 interaction region |
| LC3 | Light Chain 3 |
| LBW | Low Birth Weight |
| mtDNA | Mitochondrial DNA |
| TFAM | Mitochondrial Transcription Factor A |
| MFN-1 | Mitofusin-1 |
| MFN-2 | Mitofusin-2 |
| NIX | NIP-3 like protein X |
| NDP52 | Nuclear dot protein 52 |
| NRF1 | Nuclear respiratory factor 1 |
| NRF2 | Nuclear respiratory factor 2 |
| OPA1 | Optic Atrophy 1 |
| OPTN | Optineurin |
| OMM | Outer Mitochondrial Membrane |
| OXPHOS | Oxidative phosphorylation system |
| PGC-1α | Peroxisome proliferator-activated receptor gamma co-activator 1-alpha |
| PE | Preeclampsia |
| PKA | Protein kinase A |
| PINK1 | PTEN-induced putative kinase 1 |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| TFB | Transcription Factor B |
References
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominy, J.E.; Puigserver, P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a015008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodger, C.E.; McWilliams, T.G.; Ganley, I.G. Mammalian mitophagy—From in vitro molecules to in vivo models. FEBS J. 2018, 285, 1185–1202. [Google Scholar] [CrossRef] [Green Version]
- Berkane, N. [Gestational hypertensions: Definitions and consequences in outcome of pregnancy]. Ann. Fr. Anesth. Reanim. 2010, 29, e1–e6. [Google Scholar] [CrossRef]
- Savitz, D.A.; Danilack, V.A.; Engel, S.M.; Elston, B.; Lipkind, H.S. Descriptive epidemiology of chronic hypertension, gestational hypertension, and preeclampsia in New York State, 1995–2004. Matern. Child Health J. 2014, 18, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Shastri, S.; Sharma, P. Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clin. Med. Insights Pediatr. 2016, 10, 67–83. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Sferruzzi-Perri, A.N. Placental mitochondrial function in response to gestational exposures. Placenta 2021, 104, 124–137. [Google Scholar] [CrossRef]
- Zhou, X.; Zhao, X.; Zhou, W.; Qi, H.; Zhang, H.; Han, T.L.; Baker, P. Impaired placental mitophagy and oxidative stress are associated with dysregulated BNIP3 in preeclampsia. Sci. Rep. 2021, 11, 20469. [Google Scholar] [CrossRef]
- Cetin, I.; Alvino, G. Intrauterine growth restriction: Implications for placental metabolism and transport. A review. Placenta 2009, 30 (Suppl. A), S77–S82. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; Rogov, V.V. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol. Cell 2019, 76, 268–285. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Gubas, A.; Dikic, I. A guide to the regulation of selective autophagy receptors. FEBS J. 2022, 289, 75–89. [Google Scholar] [CrossRef]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 2016, 129, 2170–2181. [Google Scholar]
- Pryde, K.R.; Smith, H.L.; Chau, K.-Y.; Schapira, A.H. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016, 213, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Lin, C.; Wu, K.; Jiang, L.; Wang, X.; Li, W.; Zhuang, H.; Zhang, X.; Chen, H.; Li, S. FUNDC 1 regulates mitochondrial dynamics at the ER–mitochondrial contact site under hypoxic conditions. EMBO J. 2016, 35, 1368–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsch, L.E.; Moore, M.E.; Sarraf, S.A.; Pickrell, A.M. Ubiquitin and Receptor-Dependent Mitophagy Pathways and Their Implication in Neurodegeneration. J. Mol. Biol. 2020, 432, 2510–2524. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [Green Version]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S. PINK1 import regulation at a crossroad of mitochondrial fate: The molecular mechanisms of PINK1 import. J. Biochem. 2020, 167, 217–224. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, E.; Proics, E.; Rubio-Patino, C.; Obba, S.; Zunino, B.; Bossowski, J.P.; Rozier, R.M.; Chiche, J.; Mondragon, L.; Riley, J.S.; et al. Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells. Cell Rep. 2017, 20, 2846–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvedahl, A.; Sumpter, R., Jr.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Kubli, D.A.; Quinsay, M.N.; Huang, C.; Lee, Y.; Gustafsson, A.B. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2025–H2031. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.S.; Wojdyla, D.; Say, L.; Gulmezoglu, A.M.; Van Look, P.F. WHO analysis of causes of maternal death: A systematic review. Lancet 2006, 367, 1066–1074. [Google Scholar] [CrossRef]
- Kuklina, E.V.; Meikle, S.F.; Jamieson, D.J.; Whiteman, M.K.; Barfield, W.D.; Hillis, S.D.; Posner, S.F. Severe obstetric morbidity in the United States: 1998-2005. Obs. Gynecol. 2009, 113, 293–299. [Google Scholar] [CrossRef]
- Berkane, N.; Liere, P.; Oudinet, J.P.; Hertig, A.; Lefevre, G.; Pluchino, N.; Schumacher, M.; Chabbert-Buffet, N. From Pregnancy to Preeclampsia: A Key Role for Estrogens. Endocr. Rev. 2017, 38, 123–144. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.N.; Yang, Z.; Ma, R.Q. [Interaction of fatty acid oxidation with oxidative stress in preeclampsia-like mouse model at multiple stages of gestation]. Zhonghua Yi Xue Za Zhi 2011, 91, 2343–2347. [Google Scholar] [PubMed]
- Zarate, A.; Saucedo, R.; Valencia, J.; Manuel, L.; Hernandez, M. Early disturbed placental ischemia and hypoxia creates immune alteration and vascular disorder causing preeclampsia. Arch. Med. Res. 2014, 45, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Taysi, S.; Tascan, A.S.; Ugur, M.G.; Demir, M. Radicals, Oxidative/Nitrosative Stress and Preeclampsia. Mini. Rev. Med. Chem. 2019, 19, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Nakashima, A. A review of the mechanism for poor placentation in early-onset preeclampsia: The role of autophagy in trophoblast invasion and vascular remodeling. J. Reprod. Immunol. 2014, 101–102, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Fowden, A.L.; Thornburg, K.L. Placental Origins of Chronic Disease. Physiol. Rev. 2016, 96, 1509–1565. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef]
- Kumar, C.A.; Das, U.N. Lipid peroxides, anti-oxidants and nitric oxide in patients with pre-eclampsia and essential hypertension. Med. Sci. Monit. 2000, 6, 901–907. [Google Scholar]
- Zhou, X.; Han, T.L.; Chen, H.; Baker, P.N.; Qi, H.; Zhang, H. Impaired mitochondrial fusion, autophagy, biogenesis and dysregulated lipid metabolism is associated with preeclampsia. Exp. Cell Res. 2017, 359, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Udagawa, O.; Ishihara, T.; Maeda, M.; Matsunaga, Y.; Tsukamoto, S.; Kawano, N.; Miyado, K.; Shitara, H.; Yokota, S.; Nomura, M.; et al. Mitochondrial fission factor Drp1 maintains oocyte quality via dynamic rearrangement of multiple organelles. Curr. Biol. 2014, 24, 2451–2458. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Bener, M.B.; Jiang, Z.; Wang, T.; Esencan, E.; Scott Iii, R.; Horvath, T.; Seli, E. Mitofusin 1 is required for female fertility and to maintain ovarian follicular reserve. Cell Death Dis. 2019, 10, 560. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Guo, X.; Chen, R.; Feng, L. Downregulation of Mitofusin 2 in Placenta Is Related to Preeclampsia. BioMed Res. Int. 2016, 2016, 6323086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ausman, J.; Abbade, J.; Ermini, L.; Farrell, A.; Tagliaferro, A.; Post, M.; Caniggia, I. Ceramide-induced BOK promotes mitochondrial fission in preeclampsia. Cell Death Dis. 2018, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Vishnyakova, P.A.; Volodina, M.A.; Tarasova, N.V.; Marey, M.V.; Tsvirkun, D.V.; Vavina, O.V.; Khodzhaeva, Z.S.; Kan, N.E.; Menon, R.; Vysokikh, M.Y.; et al. Mitochondrial role in adaptive response to stress conditions in preeclampsia. Sci. Rep. 2016, 6, 32410. [Google Scholar] [CrossRef] [Green Version]
- Vishnyakova, P.A.; Volodina, M.A.; Tarasova, N.V.; Marey, M.V.; Kan, N.E.; Khodzhaeva, Z.S.; Vysokikh, M.Y.; Sukhikh, G.T. Alterations in antioxidant system, mitochondrial biogenesis and autophagy in preeclamptic myometrium. BBA Clin. 2017, 8, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Cizeau, J.; Vande Velde, C.; Park, J.H.; Bozek, G.; Bolton, J.; Shi, L.; Dubik, D.; Greenberg, A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J. Biol. Chem. 1999, 274, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Zhao, W.; Lv, H.; Li, W.P.; Chen, Z.J.; Zhang, C. Transcriptomic Profiling in Human Decidua of Severe Preeclampsia Detected by RNA Sequencing. J. Cell Biochem. 2018, 119, 607–615. [Google Scholar] [CrossRef]
- Ma, J.; Yang, J.; Lv, S.; Gao, M.; Sun, Y.; Chen, Z.J.; Zhang, C. Dysfunction of B-cell lymphoma 2/adenovirus E1B 19KD interacting protein 3 in decidua is involved in the pathogenesis of preeclampsia. J. Hypertens. 2019, 37, 2048–2060. [Google Scholar] [CrossRef]
- Vangrieken, P.; Al-Nasiry, S.; Bast, A.; Leermakers, P.A.; Tulen, C.B.M.; Schiffers, P.M.H.; van Schooten, F.J.; Remels, A.H.V. Placental Mitochondrial Abnormalities in Preeclampsia. Reprod. Sci. 2021, 28, 2186–2199. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Chen, Z.; Siraj, S.; Liu, L.; Chen, Q. MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy. Autophagy 2017, 13, 1244–1245. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Chen, L.; Huang, Y.; Zhu, X.; Yu, Y. Increased FUN14 domain containing 1 (FUNDC1) ubiquitination level inhibits mitophagy and alleviates the injury in hypoxia-induced trophoblast cells. Bioengineered 2022, 13, 3620–3633. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Qi, L.; Wu, J.C.; Qin, Z.H. DRAM1 regulates autophagy flux through lysosomes. PLoS ONE 2013, 8, e63245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Lin, Y.; Chen, L.; Zeng, F.; Zhang, L.; Huang, Y.; Huang, P.; Liao, L.; Yu, Y. Role of DRAM1 in mitophagy contributes to preeclampsia regulation in mice. Mol. Med. Rep. 2020, 22, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Bener, M.B.; Jiang, Z.; Wang, T.; Esencan, E.; Scott, R.; Horvath, T.; Seli, E. Mitofusin 2 plays a role in oocyte and follicle development, and is required to maintain ovarian follicular reserve during reproductive aging. Aging (Albany NY) 2019, 11, 3919–3938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qu, B.; Yu, W.; Zhu, Y.; Yan, X.; Shen, H.; Zhao, J. Role of surface ectoderm-specific mitofusin 2 in the corneal morphologic development of mice. Am. J. Transl. Res. 2019, 11, 3620–3628. [Google Scholar] [PubMed]
- Sharma, D.; Farahbakhsh, N.; Shastri, S.; Sharma, P. Intrauterine growth restriction—Part 2. J. Matern. Fetal. Neonatal. Med. 2016, 29, 4037–4048. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Shastri, S.; Farahbakhsh, N.; Sharma, P. Intrauterine growth restriction—Part 1. J. Matern. Fetal. Neonatal. Med. 2016, 29, 3977–3987. [Google Scholar] [CrossRef]
- Hendrix, N.; Berghella, V. Non-placental causes of intrauterine growth restriction. Semin. Perinatol. 2008, 32, 161–165. [Google Scholar] [CrossRef]
- Laskowska, M.; Laskowska, K.; Leszczyńska-Gorzelak, B.; Oleszczuk, J. Asymmetric dimethylarginine in normotensive pregnant women with isolated fetal intrauterine growth restriction: A comparison with preeclamptic women with and without intrauterine growth restriction. J. Matern. Fetal. Neonatal. Med. 2011, 24, 936–942. [Google Scholar] [CrossRef]
- Blencowe, H.; Cousens, S.; Oestergaard, M.Z.; Chou, D.; Moller, A.B.; Narwal, R.; Adler, A.; Vera Garcia, C.; Rohde, S.; Say, L.; et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: A systematic analysis and implications. Lancet 2012, 379, 2162–2172. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.C.; Katz, J.; Blencowe, H.; Cousens, S.; Kozuki, N.; Vogel, J.P.; Adair, L.; Baqui, A.H.; Bhutta, Z.A.; Caulfield, L.E.; et al. National and regional estimates of term and preterm babies born small for gestational age in 138 low-income and middle-income countries in 2010. Lancet Glob. Health 2013, 1, e26–e36. [Google Scholar] [CrossRef]
- Organization W.H. P07 Disorders Related to Short Gestation and Low Birth Weight, Not Elsewhere Classified. XVI Certain Conditions Originating in Perinatal Period. International Statistical Classification of Diseases and Related Health Problems 10th Revision (ICD-10) Version for 2010. Available online: https://icd.who.int/browse10/2010/en#/P07 (accessed on 1 August 2022).
- Wardlaw, T.M. Low Birthweight: Country, Regional and Global Estimates; UNICEF: New York, NY, USA, 2004; Available online: https://www.who.int/publications/i/item/9280638327 (accessed on 1 August 2022).
- Mathewson, K.J.; Chow, C.H.; Dobson, K.G.; Pope, E.I.; Schmidt, L.A.; Van Lieshout, R.J. Mental health of extremely low birth weight survivors: A systematic review and meta-analysis. Psychol. Bull. 2017, 143, 347–383. [Google Scholar] [CrossRef] [PubMed]
- Remington, J.S.; Klein, J.O.; Wilson, C.B.; Baker, C.J. (Eds.) Infectious Diseases of the Fetus and Newborn Infant, 6th ed; W.B. Saunders: Philadelphia, PA, USA, 2006; pp. 1269–1313. [Google Scholar] [CrossRef] [Green Version]
- Bujold, E.; Roberge, S.; Lacasse, Y.; Bureau, M.; Audibert, F.; Marcoux, S.; Forest, J.-C.; Giguère, Y. Prevention of Preeclampsia and Intrauterine Growth Restriction With Aspirin Started in Early Pregnancy: A Meta-Analysis. Obstet. Gynecol. 2010, 116, 402–414. [Google Scholar] [CrossRef]
- Surico, D.; Bordino, V.; Cantaluppi, V.; Mary, D.; Gentilli, S.; Oldani, A.; Farruggio, S.; Melluzza, C.; Raina, G.; Grossini, E. Preeclampsia and intrauterine growth restriction: Role of human umbilical cord mesenchymal stem cells-trophoblast cross-talk. PLoS ONE 2019, 14, e0218437. [Google Scholar] [CrossRef] [Green Version]
- Priliani, L.; Febinia, C.A.; Kamal, B.; Shankar, A.H.; Malik, S.G. Increased mitochondrial DNA copy number in maternal peripheral blood is associated with low birth weight in Lombok, Indonesia. Placenta 2018, 70, 1–3. [Google Scholar] [CrossRef]
- van Gisbergen, M.W.; Voets, A.M.; Starmans, M.H.W.; de Coo, I.F.M.; Yadak, R.; Hoffmann, R.F.; Boutros, P.C.; Smeets, H.J.M.; Dubois, L.; Lambin, P. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat. Res./Rev. Mutat. Res. 2015, 764, 16–30. [Google Scholar] [CrossRef]
- Lattuada, D.; Colleoni, F.; Martinelli, A.; Garretto, A.; Magni, R.; Radaelli, T.; Cetin, I. Higher mitochondrial DNA content in human IUGR placenta. Placenta 2008, 29, 1029–1033. [Google Scholar] [CrossRef]
- Trotta, A.P.; Gelles, J.D.; Serasinghe, M.N.; Loi, P.; Arbiser, J.L.; Chipuk, J.E. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J. Biol. Chem. 2017, 292, 11727–11739. [Google Scholar] [CrossRef] [Green Version]
- Youn, D.H.; Kim, Y.; Kim, B.J.; Jeong, M.S.; Lee, J.; Rhim, J.K.; Kim, H.C.; Jeon, J.P. Mitochondrial dysfunction associated with autophagy and mitophagy in cerebrospinal fluid cells of patients with delayed cerebral ischemia following subarachnoid hemorrhage. Sci. Rep. 2021, 11, 16512. [Google Scholar] [CrossRef]
- Zhu, H.L.; Shi, X.T.; Xu, X.F.; Zhou, G.X.; Xiong, Y.W.; Yi, S.J.; Liu, W.B.; Dai, L.M.; Cao, X.L.; Xu, D.X.; et al. Melatonin protects against environmental stress-induced fetal growth restriction via suppressing ROS-mediated GCN2/ATF4/BNIP3-dependent mitophagy in placental trophoblasts. Redox Biol. 2021, 40, 101854. [Google Scholar] [CrossRef]
- Zhu, H.L.; Shi, X.T.; Xu, X.F.; Xiong, Y.W.; Yi, S.J.; Zhou, G.X.; Liu, W.B.; Huang, M.M.; Gao, L.; Zhang, C.; et al. Environmental cadmium exposure induces fetal growth restriction via triggering PERK-regulated mitophagy in placental trophoblasts. Environ. Int. 2021, 147, 106319. [Google Scholar] [CrossRef] [PubMed]
- Madeleneau, D.; Buffat, C.; Mondon, F.; Grimault, H.; Rigourd, V.; Tsatsaris, V.; Letourneur, F.; Vaiman, D.; Barbaux, S.; Gascoin, G. Transcriptomic analysis of human placenta in intrauterine growth restriction. Pediatr. Res. 2015, 77, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Ruis-González, M.D.; Cañete, M.D.; Gómez-Chaparro, J.L.; Abril, N.; Cañete, R.; López-Barea, J. Alterations of protein expression in serum of infants with intrauterine growth restriction and different gestational ages. J. Proteom. 2015, 119, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Guitart-Mampel, M.; Juarez-Flores, D.L.; Youssef, L.; Moren, C.; Garcia-Otero, L.; Roca-Agujetas, V.; Catalan-Garcia, M.; Gonzalez-Casacuberta, I.; Tobias, E.; Milisenda, J.C.; et al. Mitochondrial implications in human pregnancies with intrauterine growth restriction and associated cardiac remodelling. J. Cell Mol. Med. 2019, 23, 3962–3973. [Google Scholar] [CrossRef]
- Nicolson, G.L. Mitochondrial Dysfunction and Chronic Disease: Treatment With Natural Supplements. Integr. Med. (Encinitas) 2014, 13, 35–43. [Google Scholar] [PubMed]
- Carter, A.M. Placental oxygen consumption. Part I: In vivo studies—A review. Placenta 2000, 21 (Suppl. A), S31–S37. [Google Scholar] [CrossRef]
- Sferruzzi-Perri, A.N.; Higgins, J.S.; Vaughan, O.R.; Murray, A.J.; Fowden, A.L. Placental mitochondria adapt developmentally and in response to hypoxia to support fetal growth. Proc. Natl. Acad. Sci. USA 2019, 116, 1621–1626. [Google Scholar] [CrossRef] [Green Version]
- Doblado, L.; Lueck, C.; Rey, C.; Samhan-Arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in Human Diseases. Int. J. Mol. Sci. 2021, 22, 3903. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yildirim, R.M.; Ergun, Y.; Basar, M. Mitochondrial Dysfunction, Mitophagy and Their Correlation with Perinatal Complications: Preeclampsia and Low Birth Weight. Biomedicines 2022, 10, 2539. https://doi.org/10.3390/biomedicines10102539
Yildirim RM, Ergun Y, Basar M. Mitochondrial Dysfunction, Mitophagy and Their Correlation with Perinatal Complications: Preeclampsia and Low Birth Weight. Biomedicines. 2022; 10(10):2539. https://doi.org/10.3390/biomedicines10102539
Chicago/Turabian StyleYildirim, Raziye Melike, Yagmur Ergun, and Murat Basar. 2022. "Mitochondrial Dysfunction, Mitophagy and Their Correlation with Perinatal Complications: Preeclampsia and Low Birth Weight" Biomedicines 10, no. 10: 2539. https://doi.org/10.3390/biomedicines10102539