
A Critical Overview of Enzyme-Based Electrochemical Biosensors for L-Dopa Detection in Biological Samples

 ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
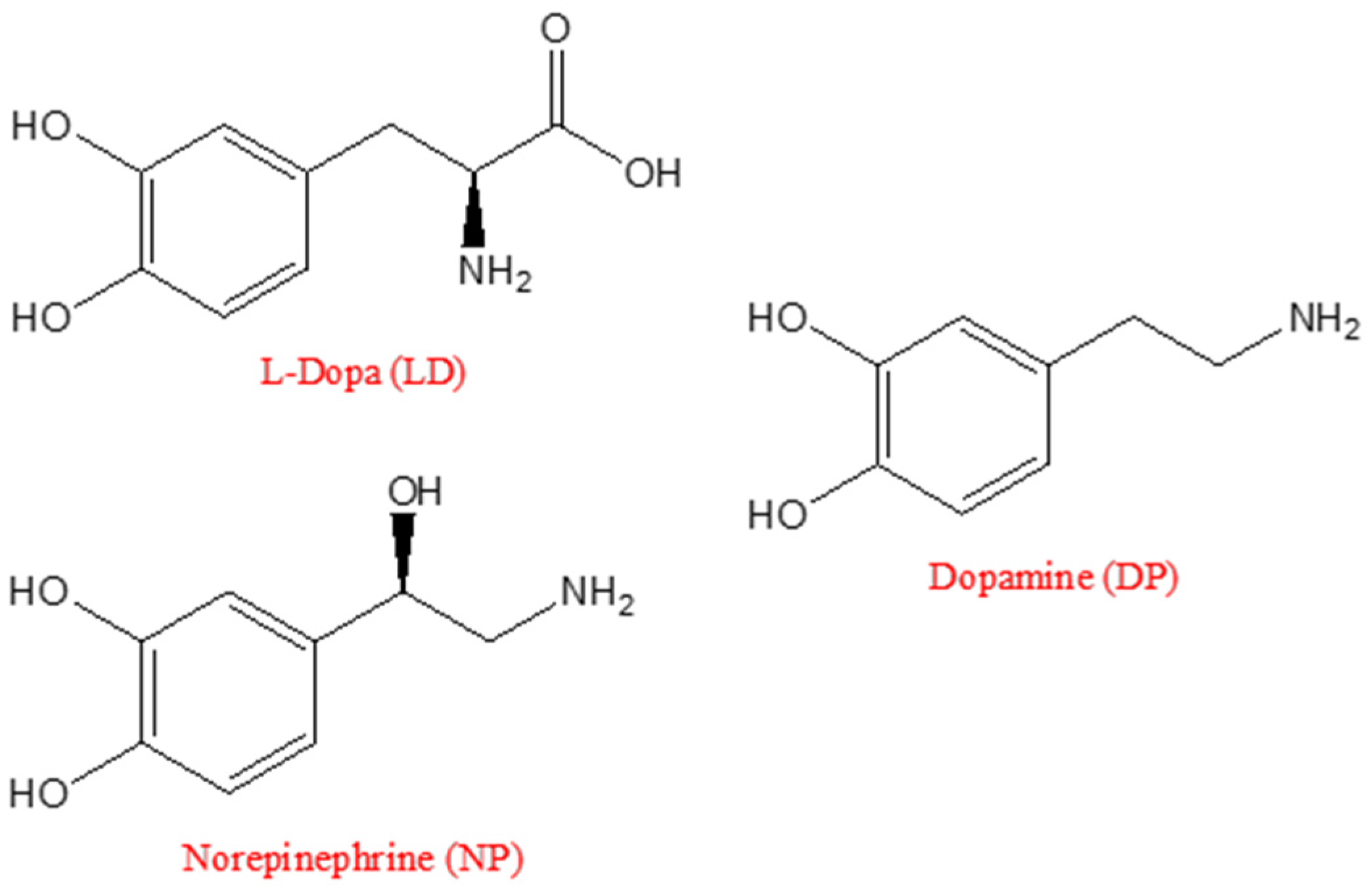
2. Enzymes for L-Dopa Biosensing
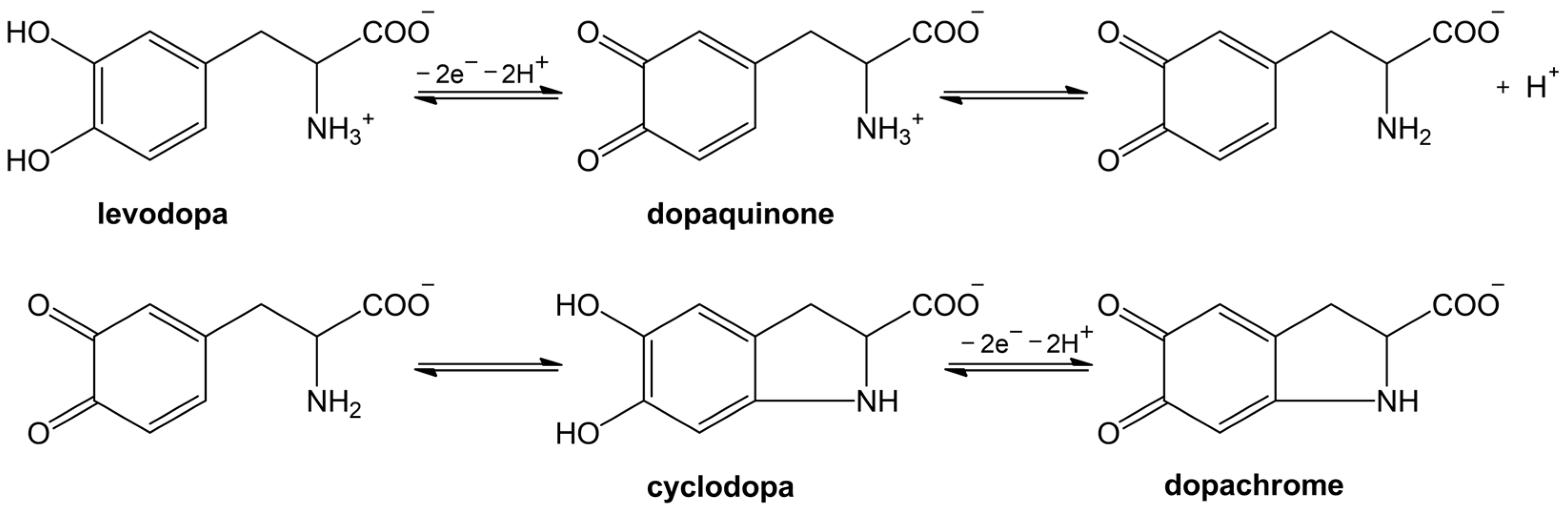
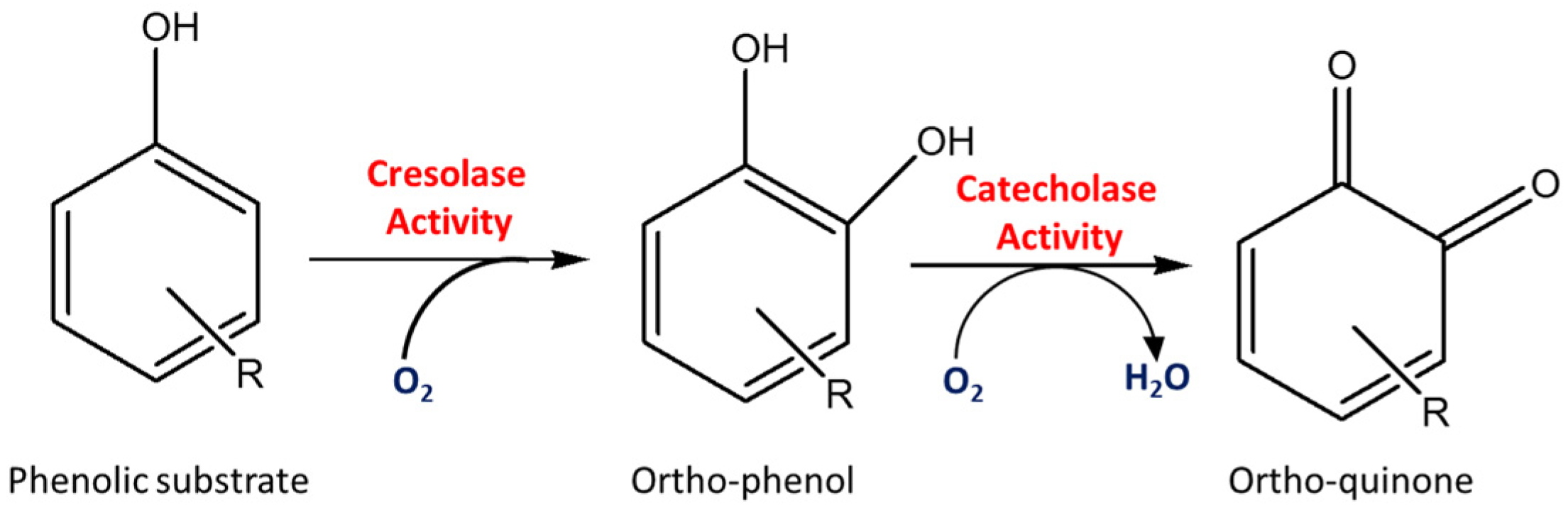
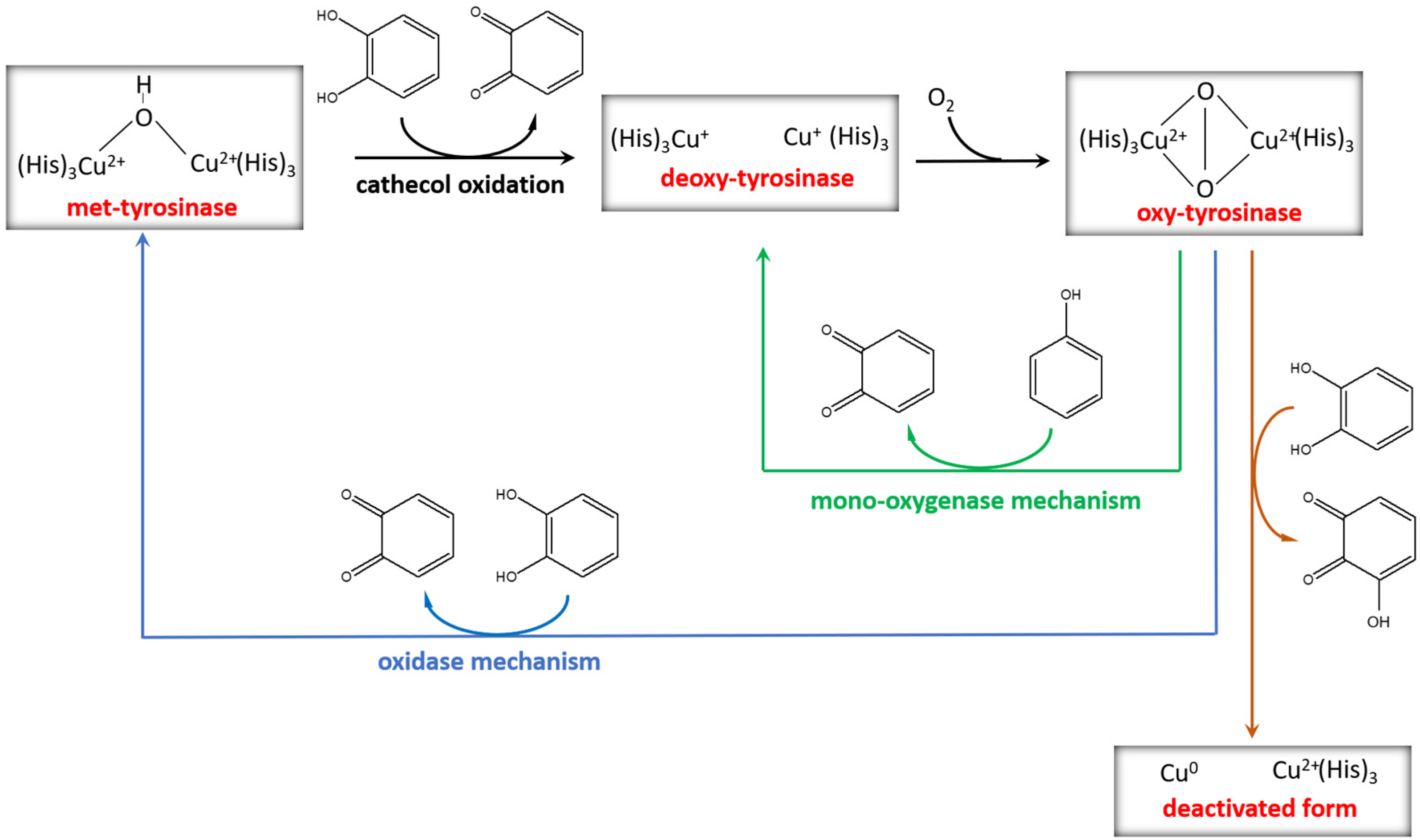
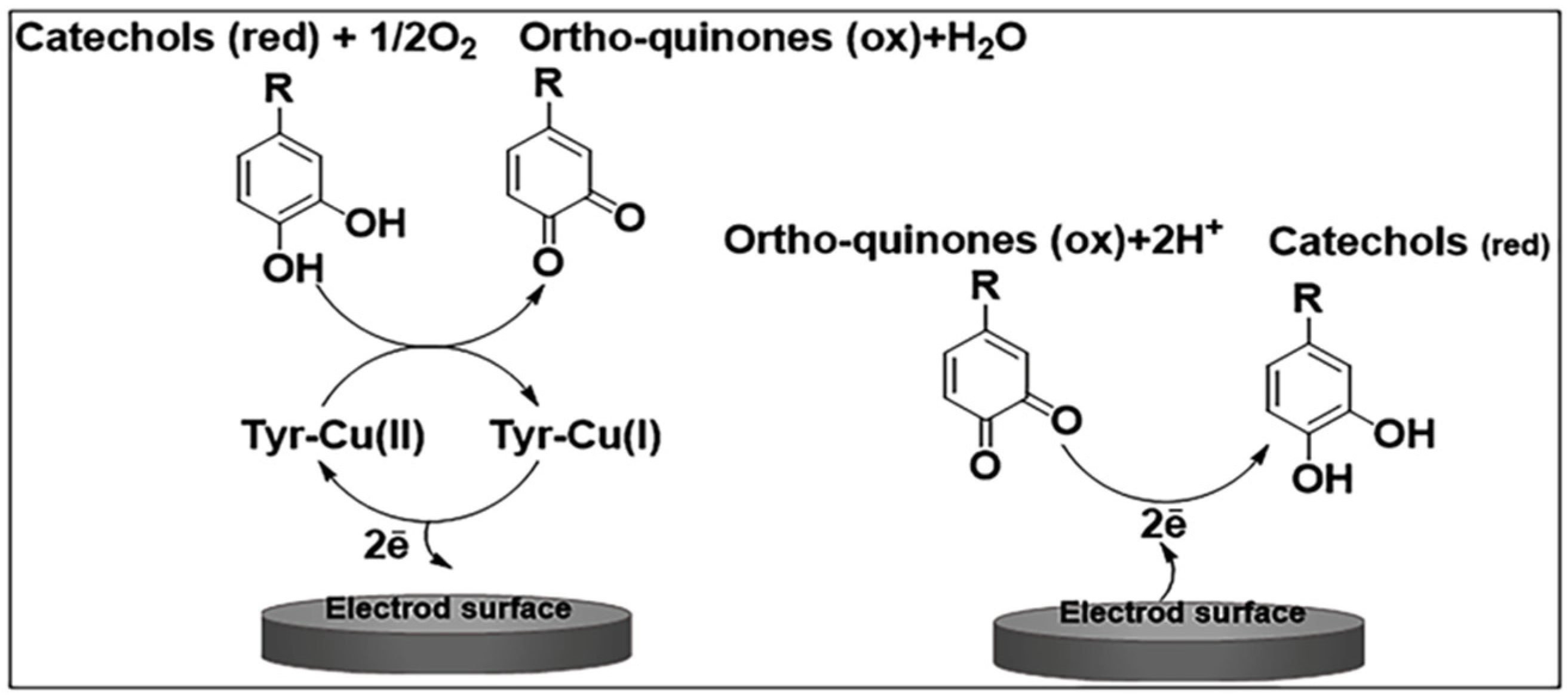
Tyrosinase
3. Tyrosinase-Based Biosensors
3.1. Tyrosinase Immobilization Method

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immobilization Method | Time for Enzyme Immobilization | Stability | Reference |
|---|---|---|---|
| Drop-casting of enzyme solution and immobilization by intermolecular interaction with the electrode nanocomposite | 24 h | After 17 days, 83.07% of enzyme activity was retained | [72] |
| Drop-casting of enzyme solution and immobilization by electrostatic interaction with the electrode nanocomposite | After 28 days, 64% of enzyme activity was retained | [71] | |
| Drop-casting of a mixture of tyrosinase, BSA, and GLU and immobilization by co-crosslinking; then electrode immersion in a GLU solution | -20′ for co-crosslinking -15′ in GLU solution -drying for 30′ | After about three weeks, sensitivity was around 80% of the initial value | [70] |
| Immersion of the electrode in a solution of tyrosinase and BSA for protein absorption and then co-crosslinking by GLU | -24 h for protein adsorption -30′ for GLU crosslinking | Enzyme activity maintained for at least two weeks | [67] |
| Dripping on the electrode of a mixed tyrosinase and ZIF-8/GO solution | Drying overnight | -Stable current response for 50 continuous cycles in artificial sweat at the scan rate of 50 mV/s -Good stability after storage for 7 days | [73] |
| Drop-casting of an enzyme solution and then crosslinking by GLU | Kept overnight after GLU addition | After one week, 97.3% of the initial response was retained | [65] |
| Enzyme-containing paste (graphite powder, tyrosinase, and mineral oil) | WE1: stability test carried out over a 110 min period by recording the SWV response for 100 μM L-Dopa in artificial ISF at 10 min time intervals (SD = 2.5, n = 11). WE2: stability test performed using a 100 μM L-Dopa in artificial ISF over 110 min period via repetitive measurements at 10 min intervals (SD 2%; n = 11) WE1 and WE2: stability test in the presence of common ISF proteins by repetitive measurements at 10 min intervals over a 2 h period (11 and 14% decrease of the response of the nonenzymatic and enzymatic sensors, respectively) | [75] | |
| Drop-casting of a mixture of tyrosinase and GLU and immobilization by crosslinking | 12 h | Stability tested continuously for about 1500 s | [66] |
| Drop-casting of a mixture of tyrosinase, BSA, and GLU and immobilization by co-crosslinking | 2 h | When not in use, the biosensors were stored for up to five days from the fabrication at 4 °C | [68] |
| (1) Chemisorption of cysteamine on the gold surface (2) Dipping of the aminated gold surface in GLU solution (3) Drop-casting of tyrosinase (4) Enzyme electrode is blocked with PBS containing BSA | (1) 18 h (2) 2 h (3) 24 h (4) 15′ | The electrodes were stable for more than a month when stored at 4 °C | [69] |
| Deposition of tyrosinase in a mixture with an immobilizing polymeric hydrogel | 24 h | [74] |
3.2. Electrode Modification and Detection Mode
3.3. Selectivity
3.4. Application to Real Sample Analysis
4. Biosensors Based on Laccase and PPO
5. Conclusions and Future Perspectives
- The dynamic nature of drug metabolism requires continuous and long-term measurements that can be performed if the biosensing device exhibits high operational stability. Unfortunately, in most of the works concerning biosensors for LD, stability has been tested on short time scales. The immobilization technique adopted strongly influences the possibility of preserving the activity and stability of the native enzyme. Crosslinking is an advantageous immobilization technique that has often been employed to immobilize tyrosinase. Multistep protocols have been devised, which are very time-consuming. These aspects should be considered and simplified in future studies. The key could be to properly adjust the concentration values of the protein and crosslinker, which strongly influence the mechanical properties of the resulting enzymatic layer, as recently demonstrated [70].
- Enzyme activity is very rarely determined after deposition, and its loading is also usually unknown and uncontrolled. Indeed, enzyme solution is usually deposited by drop-casting, which does not allow for precise control of the thickness and spatial distribution of the protein membrane onto the electrode surface. To this aim, as a future outlook, electrochemically assisted procedures could be devised, such as electrophoretic protein deposition, which is applicable to electrodes of any shape and size, resulting in particular suitability for the realization of miniaturized implantable devices.
- Similar considerations apply to the electrochemical transducer. Many new materials, such as carbon materials (carbon nanotubes, graphene), nanoparticles, nanodendrites, and conducting polymers, have been assembled as nanocomposites for sensor construction to improve their performance in terms of sensitivity and electrocatalytic properties. Although conducting polymers are deposited by electrochemical techniques (CV, chronoamperometry), nanomaterial deposition is mainly performed by drop-casting. Surface distribution and the load of the nanocomposites are, therefore, difficult to control. The development of modification protocols based on electrochemical techniques for both electrode modification and enzyme deposition would be desirable for realizing potentially wearable and/or implantable devices with high reproducibility.
- Another issue that requires further investigation is the assessment of the biocompatibility of microneedle sensors by on-body testing. For this purpose, it should be noted that the focus of emerging studies is devoted to the possibility of employing biological samples suitable for less invasive analysis. As an alternative to blood, sweat has been used in various studies to evaluate the LD content in humans. Promising results have already been obtained, even if a systematic study must still be carried out to validate the correlation between LD levels in sweat and ISF. Moreover, from a future perspective, clinical testing and validation in PD patients is required. Indeed, all the applications reported have been carried out on biological samples from healthy subjects. Unfortunately, only a few papers have determined the true concentration of LD in biological samples. At the same time, analyte spiking has often been used to validate the clinical utility of the developed biosensor. From a future perspective, the validation of the developed biosensors by a standard reference method would increase the validity of electrochemical devices for clinical application.
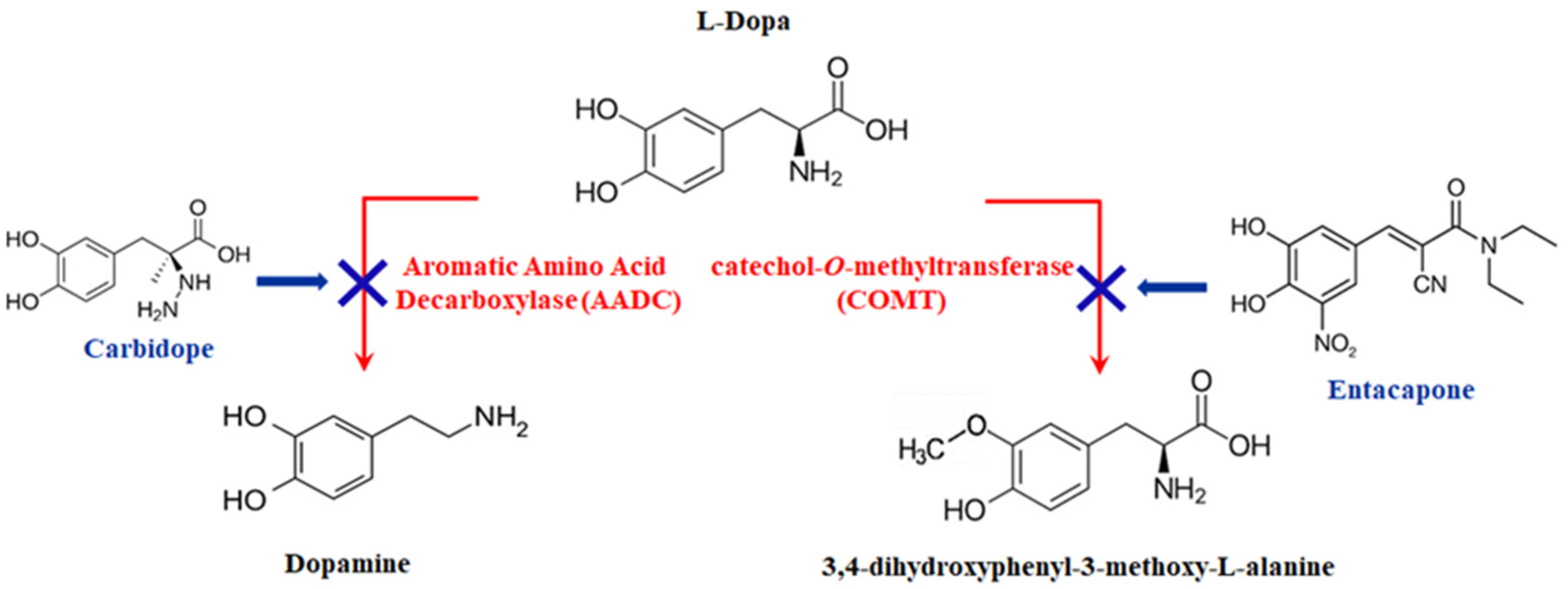
- Selectivity is a further critical issue arising from the examination of tyrosinase biosensors. As previously discussed, tyrosinase has broad substrate specificity, catalyzing naturally occurring compounds in biological samples such as dopamine, serotonin, adrenaline, and compounds like carbidopa found as a result of drug administration, as well as LD. Among the strategies adopted to solve interference problems, dual-sensing platforms have been proposed based on parallel, simultaneous, and independent enzymatic and nonenzymatic electrochemical detection [67,75]. Nafion has often been used as a protecting membrane towards negatively charged compounds such as ascorbic acid and uric acid. A drawback of this material is its susceptibility to membrane fouling, which limits the operational stability of biosensors. Alternative protective layers should be explored. More or less effective strategies have been developed to solve interference problems. However, this issue has not been properly solved and addressed. Biosensor selectivity has often been evaluated by testing a few potential interferent compounds, as well as at physiological concentration levels typical of healthy individuals, which can differ from those found in plasma from PD patients. Tyrosine and serotonin plasma levels, for instance, are lower than they would be under normal settings [90,91], whereas homocysteine plasma levels rise in levodopa-treated individuals [92]. This aspect should be taken into consideration in future interference studies.
- The employment of other enzymes as biocatalysts would not help to improve LD biorecognition. PPOs, for example, despite being easily available from fruit sources, show a wide substrate response. In work concerning PPO-based biosensors, these enzymes were employed to detect various catecholamines, among which were LD. Moreover, any application to real sample analysis is not reported, or concerns samples that are different from biological ones.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xing, B.; Li, Y.C.; Gao, W.J. Norepinephrine versus dopamine and their interaction in modulating synaptic function in the prefrontal cortex. Brain Res. 2016, 1641, 217–233. [Google Scholar] [CrossRef]
- Hornykiewicz, O. L-DOPA: From a biologically inactive amino acid to a successful therapeutic agent: Historical review article. Amino Acids 2002, 23, 65–70. [Google Scholar] [CrossRef]
- Khan, S.T.; Ahmed, S.; Gul, S.; Khan, A.; Al-Harrasi, A. Search for safer and potent natural inhibitors of Parkinson’s disease. Neurochem. Int. 2021, 149, 105135. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s disease: From pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 29, 1783–1793. [Google Scholar] [CrossRef]
- Min, K.; Park, K.; Park, D.H.; Yoo, Y.J. Overview on the biotechnological production of l-DOPA. Appl. Microbiol. Biotechnol. 2015, 99, 575–584. [Google Scholar] [CrossRef]
- Fasano, A.; Canning, C.G.; Hausdorff, J.M.; Lord, S.; Rochester, L. Falls in Parkinson’s disease: A complex and evolving picture. Mov. Disord. 2017, 32, 1524–1536. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Chaudhuri, K.R. Unmet needs in Parkinson disease: Motor and non-motor. Park. Relat. Disord. 2020, 80, S7–S12. [Google Scholar] [CrossRef]
- Dirnberger, G.; Jahanshahi, M. Executive dysfunction in Parkinson’s disease: A review. J. Neuropsychol. 2013, 7, 193–224. [Google Scholar] [CrossRef]
- Beitz, J.M. Parkinson’s disease: A review. Front. Biosci. 2014, 6, 65–74. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef]
- Salat, D.; Tolosa, E. Levodopa in the treatment of Parkinson’s disease: Current status and new developments. J. Parkinsons. Dis. 2013, 3, 255–269. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef]
- Poewe, W.; Antonini, A.; Zijlmans, J.C.; Burkhard, P.R.; Vingerhoets, F. Levodopa in the treatment of Parkinson’s disease: An old drug still going strong. Clin. Interv. Aging 2010, 5, 229–238. [Google Scholar] [CrossRef]
- Thanvi, B.R.; Lo, T.C.N. Long term motor complications of levodopa: Clinical features, mechanisms, and management strategies. Postgr. Med. J. 2004, 80, 452–458. [Google Scholar] [CrossRef]
- Jankovic, J.; Stacy, M. Medical management of levodopa-associated motor complications in patients with Parkinson’s disease. CNS Drugs 2007, 21, 677–692. [Google Scholar] [CrossRef]
- Rezak, M. Current Pharmacotherapeutic Treatment Options in Parkinson’s Disease. Dis.-a-Month 2007, 53, 214–222. [Google Scholar] [CrossRef]
- Nutt, J.G. Pharmacokinetics and pharmacodynamics of levodopa. Mov. Disord. 2008, 23, 580–584. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B.; Aschner, M. Novel Pharmacotherapies in Parkinson’s Disease. Neurotox. Res. 2021, 39, 1381–1390. [Google Scholar] [CrossRef]
- Valdés, R.H.; Puzer, L.; Gomes, M.; Marques, C.E.S.J.; Aranda, D.A.G.; Bastos, M.L.; Gemal, A.L.; Antunes, O.A.C. Production of L-DOPA under heterogeneous asymmetric catalysis. Catal. Commun. 2004, 5, 631–634. [Google Scholar] [CrossRef]
- Patil, S.A.; Apine, O.A.; Surwase, S.N.; Jadhav, J.P. Biological sources of L-DOPA: An alternative approach. Adv. Park. Dis. 2013, 2, 81–87. [Google Scholar] [CrossRef]
- Lampariello, L.; Cortelazzo, A.; Guerranti, R.; Sticozzi, C.; Valacchi, G. The magic velvet bean of mucuna pruriens. J. Tradit. Complement. Med. 2012, 2, 331–339. [Google Scholar] [CrossRef]
- Denne, T. Analysis of Levodopa Content in Commercial Formulations of Mucuna pruriens Seeds Used in Integrative Treatment of Parkinson’s Disease. Mov. Disord. 2019, 34, S37–S38. [Google Scholar]
- Tesoro, C.; Ciriello, R.; Lelario, F.; Di Capua, A.; Pascale, R.; Bianco, G.; Dell’Agli, M.; Piazza, S.; Guerrieri, A.; Scrano, L.; et al. Development and Validation of a Reversed-Phase HPLC Method with UV Detection for the Determination of L-Dopa in Vicia faba L. Broad Beans. Molecules 2022, 27, 7468. [Google Scholar] [CrossRef]
- Tolokán, A.; Klebovich, I.; Balogh-Nemes, K.; Horvai, G. Automated determination of levodopa and carbidopa in plasma by high-performance liquid chromatography-electrochemical detection using an on-line flow injection analysis sample pretreatment unit. J. Chromatogr. B Biomed. Appl. 1997, 698, 201–207. [Google Scholar] [CrossRef]
- Rondelli, I.; Acerbi, D.; Mariotti, F.; Ventura, P. Simultaneous determination of levodopa methyl ester, levodopa, 3-O-methyldopa and dopamine in plasma by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. B Biomed. Sci. Appl. 1994, 653, 17–23. [Google Scholar] [CrossRef]
- Tesoro, C.; Lelario, F.; Ciriello, R.; Bianco, G.; Di Capua, A.; Acquavia, M.A. An Overview of Methods for L-Dopa Extraction and Analytical Determination in Plant Matrices. Separations 2022, 9, 224. [Google Scholar] [CrossRef]
- César, I.d.C.; Byrro, R.M.D.; de Santana e Silva Cardoso, F.F.; Mundim, I.M.; de Souza Teixeira, L.; Pontes da Silva, E.; Gomes, S.A.; Bonfim, R.R.; Pianetti, G.A. Simultaneous quantitation of levodopa and 3-O-methyldopa in human plasma by HPLC-ESI-MS/MS: Application for a pharmacokinetic study with a levodopa/benserazide formulation. J. Pharm. Biomed. Anal. 2011, 56, 1094–1100. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, G.; Hu, Q.; Fang, Y. Separation and determination of levodopa and carbidopa in composite tablets by capillary zone electrophoresis with amperometric detection. Anal. Chim. Acta 2001, 431, 287–292. [Google Scholar] [CrossRef]
- Coello, J.; Maspoch, S.; Villegas, N. Simultaneous kinetic-spectrophotometric determination of levodopa and benserazide by bi- and three-way partial least squares calibration. Talanta 2000, 53, 627–637. [Google Scholar] [CrossRef]
- Madrakian, T.; Afkhami, A.; Mohammadnejad, M. Simultaneous spectrofluorimetric determination of levodopa and propranolol in urine using feed-forward neural networks assisted by principal component analysis. Talanta 2009, 78, 1051–1055. [Google Scholar] [CrossRef]
- He, W.W.; Zhou, X.W.; Lu, J.Q. Capillary electrophoresis-chemiluminecence detection of levodopa and benserazide in Medopar tablet. Chin. Chem. Lett. 2007, 18, 91–93. [Google Scholar] [CrossRef]
- Zhao, S.; Bai, W.; Wang, B.; He, M. Determination of levodopa by capillary electrophoresis with chemiluminescence detection. Talanta 2007, 73, 142–146. [Google Scholar] [CrossRef]
- State, R.G.; van Staden, J. (KOOS) F. Electrochemical sensors used in the determination of L-Dopa. Electrochem. Sci. Adv. 2022, 2, e2100040. [Google Scholar] [CrossRef]
- Beitollahi, H.; Safaei, M.; Tajik, S. Electrochemical deduction of levodopa by utilizing modified electrodes: A review. Microchem. J. 2020, 152, 104287. [Google Scholar] [CrossRef]
- Crapnell, R.D.; Banks, C.E. Electroanalytical Overview: The Determination of Levodopa (L-DOPA). ACS Meas. Sci. Au 2023, 3, 84–97. [Google Scholar] [CrossRef]
- Eslami, M.; Namazian, M.; Zare, H.R. Electrochemical behavior of 3,4-dihydroxyphenylalanine in aqueous solution. Electrochim. Acta 2013, 88, 543–551. [Google Scholar] [CrossRef]
- Thévenot, D.R.; Toth, K.; Durst, R.A.; Wilson, G.S. Electrochemical biosensors: Recommended definitions and classification. Anal. Lett. 2001, 34, 635–659. [Google Scholar] [CrossRef]
- Munteanu, F.D.; Lindgren, A.; Emnéus, J.; Gorton, L.; Ruzgas, T.; Csöregi, E.; Ciucu, A.; Van Huystee, R.B.; Gazaryan, I.G.; Lagrimini, L.M. Bioelectrochemical monitoring of phenols and aromatic amines in flow injection using novel plant peroxidases. Anal. Chem. 1998, 70, 2596–2600. [Google Scholar] [CrossRef]
- Ziyan, E.; Pekyardimci, Ş. Characterization of polyphenol oxidase from Jerusalem artichoke (Helianthus tuberosus). Turk. J. Chem. 2003, 27, 217–225. [Google Scholar]
- Burton, S.G. Biocatalysis with polyphenol oxidase: A review. Catal. Today 1994, 22, 459–487. [Google Scholar] [CrossRef]
- Solem, E.; Tuczek, F.; Decker, H. Tyrosinase versus Catechol Oxidase: One Asparagine Makes the Difference. Angew. Chemie-Int. Ed. 2016, 55, 2884–2888. [Google Scholar] [CrossRef]
- Gul, I.; Ahmad, M.S.; Naqvi, S.M.S.; Hussain, A.; Wali, R.; Farooqi, A.A.; Ahmed, I. Polyphenol oxidase (PPO) based biosensors for detection of phenolic compounds: A Review. J. Appl. Biol. Biotechnol. 2017, 5, 72–85. [Google Scholar] [CrossRef]
- Claus, H. Laccases: Structure, reactions, distribution. Micron 2004, 35, 93–96. [Google Scholar] [CrossRef]
- Klabunde, T.; Eicken, C.; Sacchettini, J.C.; Krebs, B.; Henson, M.J.; Solomon, E.I. Crystal structure of a plant catechol oxidase containing a dicopper center. Chemtracts 2000, 13, 97–102. [Google Scholar] [CrossRef]
- Raymundo-Pereira, P.A.; Silva, T.A.; Caetano, F.R.; Ribovski, L.; Zapp, E.; Brondani, D.; Bergamini, M.F.; Marcolino, L.H.; Banks, C.E.; Oliveira, O.N.; et al. Polyphenol oxidase-based electrochemical biosensors: A review. Anal. Chim. Acta 2020, 1139, 198–221. [Google Scholar] [CrossRef]
- Tarasov, A.; Stozhko, N.; Bukharinova, M.; Khamzina, E. Biosensors Based on Phenol Oxidases (Laccase, Tyrosinase, and Their Mixture) for Estimating the Total Phenolic Index in Food-Related Samples. Life 2023, 13, 291. [Google Scholar] [CrossRef]
- Bourquelot, E.; Bertrand, A. Le bleuissement et le noircissement des champignons. Comp. Rend. Soc. Biol. 1985, 47, 582–584. [Google Scholar]
- Sánchez-Ferrer, Á.; Neptuno Rodríguez-López, J.; García-Cánovas, F.; García-Carmona, F. Tyrosinase: A comprehensive review of its mechanism. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. 1995, 1247, 1–11. [Google Scholar] [CrossRef]
- Selinheimo, E.; NiEidhin, D.; Steffensen, C.; Nielsen, J.; Lomascolo, A.; Halaouli, S.; Record, E.; O’Beirne, D.; Buchert, J.; Kruus, K. Comparison of the characteristics of fungal and plant tyrosinases. J. Biotechnol. 2007, 130, 471–480. [Google Scholar] [CrossRef]
- Zaidi, K.U.; Ali, A.S.; Ali, S.A.; Naaz, I. Microbial tyrosinases: Promising enzymes for pharmaceutical, food bioprocessing, and environmental industry. Biochem. Res. Int. 2014, 2014, 854687. [Google Scholar] [CrossRef]
- Greggio, E.; Bergantino, E.; Carter, D.; Ahmad, R.; Costin, G.E.; Hearing, V.J.; Clarimon, J.; Singleton, A.; Eerola, J.; Hellström, O.; et al. Tyrosinase exacerbates dopamine toxicity but is not genetically associated with Parkinson’s disease. J. Neurochem. 2005, 93, 246–256. [Google Scholar] [CrossRef]
- Mayer, A.M. Polyphenol oxidases in plants and fungi: Going places? A review. Phytochemistry 2006, 67, 2318–2331. [Google Scholar] [CrossRef]
- Nawaz, A.; Shafi, T.; Khaliq, A.; Mukhtar, H.; ul Haq, I.; Thanvi, B.R.; Lo, T.C.N. Tyrosinase: Sources, Structure and Applications. Int. J. Biotechnol. Bioeng. 2017, 3, 135–141. [Google Scholar] [CrossRef]
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef]
- Zolghadri, S.; Bahrami, A.; Hassan Khan, M.T.; Munoz-Munoz, J.; Garcia-Molina, F.; Garcia-Canovas, F.; Saboury, A.A. A comprehensive review on tyrosinase inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef]
- Espín, J.C.; Varón, R.; Fenoll, L.G.; Gilabert, M.A.; García-Ruíz, P.A.; Tudela, J.; García-Cánovas, F. Kinetic characterization of the substrate specificity and mechanism of mushroom tyrosinase. Eur. J. Biochem. 2000, 267, 1270–1279. [Google Scholar] [CrossRef]
- Sassolas, A.; Blum, L.J.; Leca-Bouvier, B.D. Immobilization strategies to develop enzymatic biosensors. Biotechnol. Adv. 2012, 30, 489–511. [Google Scholar] [CrossRef]
- Wiseman, A. Handbook of Enzyme Biotechnology; JohnWiley & Sons, Halsted Press, Inc.: New York, NY, USA, 1985; pp. 147–207. [Google Scholar]
- Guerrieri, A.; Ciriello, R.; Crispo, F.; Bianco, G. Detection of choline in biological fluids from patients on haemodialysis by an amperometric biosensor based on a novel anti-interference bilayer. Bioelectrochemistry 2019, 129, 135–143. [Google Scholar] [CrossRef]
- Ciriello, R.; De Gennaro, F.; Frascaro, S.; Guerrieri, A. A novel approach for the selective analysis of L-lysine in untreated human serum by a co-crosslinked L-lysine–α-oxidase/overoxidized polypyrrole bilayer based amperometric biosensor. Bioelectrochemistry 2018, 124, 47–56. [Google Scholar] [CrossRef]
- Ciriello, R.; Guerrieri, A. A Crosstalk- and Interferent-Free Dual Electrode Amperometric Biosensor for the Simultaneous Determination of Choline and Phosphocholine. Sensors 2021, 21, 3545. [Google Scholar] [CrossRef]
- Moon, J.M.; Teymourian, H.; De la Paz, E.; Sempionatto, J.R.; Mahato, K.; Sonsa-ard, T.; Huang, N.; Longardner, K.; Litvan, I.; Wang, J. Non-Invasive Sweat-Based Tracking of L-Dopa Pharmacokinetic Profiles Following an Oral Tablet Administration. Angew. Chemie-Int. Ed. 2021, 60, 19074–19078. [Google Scholar] [CrossRef]
- Tai, L.C.; Liaw, T.S.; Lin, Y.; Nyein, H.Y.Y.; Bariya, M.; Ji, W.; Hettick, M.; Zhao, C.; Zhao, J.; Hou, L.; et al. Wearable Sweat Band for Noninvasive Levodopa Monitoring. Nano Lett. 2019, 19, 6346–6351. [Google Scholar] [CrossRef]
- Fang, L.; Ren, H.; Mao, X.; Zhang, S.; Cai, Y.; Xu, S.; Zhang, Y.; Li, L.; Ye, X.; Liang, B. Differential Amperometric Microneedle Biosensor for Wearable Levodopa Monitoring of Parkinson’s Disease. Biosensors 2022, 12, 102. [Google Scholar] [CrossRef]
- Brunetti, B.; Valdés-Ramírez, G.; Litvan, I.; Wang, J. A disposable electrochemical biosensor for l-DOPA determination in undiluted human serum. Electrochem. Commun. 2014, 48, 28–31. [Google Scholar] [CrossRef]
- Pinho, A.; Viswanathan, S.; Ribeiro, S.; Oliveira, M.B.P.P.; Delerue-Matos, C. Electroanalysis of urinary l-dopa using tyrosinase immobilized on gold nanoelectrode ensembles. J. Appl. Electrochem. 2012, 42, 131–137. [Google Scholar] [CrossRef]
- Cembalo, G.; Ciriello, R.; Tesoro, C.; Guerrieri, A.; Bianco, G.; Lelario, F.; Acquavia, M.A.; Di Capua, A. An Amperometric Biosensor Based on a Bilayer of Electrodeposited Graphene Oxide and Co-Crosslinked Tyrosinase for L-Dopa Detection in Untreated Human Plasma. Molecules 2023, 28, 5239. [Google Scholar] [CrossRef]
- Mollamohammadi, F.; Faridnouri, H.; Zare, E.N. Electrochemical Biosensing of L-DOPA Using Tyrosinase Immobilized on Carboxymethyl Starch-Graft-Polyaniline@MWCNTs Nanocomposite. Biosensors 2023, 13, 562. [Google Scholar] [CrossRef]
- Aliya, M.; Zare, E.N.; Faridnouri, H.; Ghomi, M.; Makvandi, P. Sulfonated Starch-Graft-Polyaniline@Graphene Electrically Conductive Nanocomposite: Application for Tyrosinase Immobilization. Biosensors 2022, 12, 939. [Google Scholar] [CrossRef]
- Xiao, J.; Fan, C.; Xu, T.; Su, L.; Zhang, X. An electrochemical wearable sensor for levodopa quantification in sweat based on a metal–Organic framework/graphene oxide composite with integrated enzymes. Sens. Actuators B Chem. 2022, 359, 131586. [Google Scholar] [CrossRef]
- Jubete, E.; Ochoteco, E.; Loinaz, I.; Pomposo, J.A.; Grande, H.; Linazasoro, G. Electrochemical biosensor development for detection of l-dopa levels in plasma during parkinson illness. In Proceedings of the SENSORS, 2008 IEEE, Lecce, Italy, 26–29 October 2008; pp. 239–241. [Google Scholar] [CrossRef]
- Goud, K.Y.; Moonla, C.; Mishra, R.K.; Yu, C.; Narayan, R.; Litvan, I.; Wang, J. Wearable Electrochemical Microneedle Sensor for Continuous Monitoring of Levodopa: Toward Parkinson Management. ACS Sens. 2019, 4, 2196–2204. [Google Scholar] [CrossRef]
- Aydemir, N.; Malmström, J.; Travas-Sejdic, J. Conducting polymer based electrochemical biosensors. Phys. Chem. Chem. Phys. 2016, 18, 8264–8277. [Google Scholar] [CrossRef]
- Malhotra, B.; Dhand, C.; Lakshminarayanan, R.; Dwivedi, N.; Mishra, S.; Solanki, P.; Venkatesh, M.; Beuerman, R.W.; Ramakrishna, S. Polyaniline-based biosensors. Nanobiosens. Dis. Diagnosis 2015, 4, 25–46. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, F.; Yu, X.; Liu, H.; Fu, Y.; Wang, Z.; Jiang, L.; Li, X. Polyelectrolyte Multilayer as Matrix for Electrochemical Deposition of Gold Clusters: Toward Super-Hydrophobic Surface. J. Am. Chem. Soc. 2004, 126, 3064–3065. [Google Scholar] [CrossRef]
- Kulkarni, T.; Slaughter, G. Application of semipermeable membranes in glucose biosensing. Membranes 2016, 6, 55. [Google Scholar] [CrossRef]
- Carbone, M.E.; Ciriello, R.; Guerrieri, A.; Salvi, A.M. Poly(o-aminophenol) electrosynthesized onto platinum at acidic and neutral ph: Comparative investigation on the polymers characteristics and on their inner and outer interfaces. Int. J. Electrochem. Sci. 2014, 9, 2047–2066. [Google Scholar] [CrossRef]
- Carbone, M.E.; Ciriello, R.; Guerrieri, A.; Salvi, A.M. XPS investigation on the chemical structure of a very thin, insulating, film synthesized on platinum by electropolymerization of o-aminophenol (oAP) in aqueous solution at neutral pH. Surf. Interface Anal. 2014, 46, 1081–1085. [Google Scholar] [CrossRef]
- Rathod, B.G.; Patel, N.M. Development of validated RP-HPLC method for the estimation of L-Dopa from Mucuna pruriens, its extracts and in Aphrodisiac formulation. Int. J. Pharma Sci. Res. 2014, 5, 508–513. [Google Scholar]
- Timur, S.; Pazarloǧlu, N.; Pilloton, R.; Telefoncu, A. Thick film sensors based on laccases from different sources immobilized in polyaniline matrix. Sens. Actuators B Chem. 2004, 97, 132–136. [Google Scholar] [CrossRef]
- Leite, O.D.; Lupetti, K.O.; Fatibello-Filho, O.; Vieira, I.C.; de M Barbosa, A. Synergic effect studies of the bi-enzymatic system laccaseperoxidase in a voltammetric biosensor for catecholamines. Talanta 2003, 59, 889–896. [Google Scholar] [CrossRef]
- Josypčuk, O.; Barek, J.; Josypčuk, B. Amperometric Determination of Catecholamines by Enzymatic Biosensors in Flow Systems. Electroanalysis 2018, 30, 1163–1171. [Google Scholar] [CrossRef]
- Chawla, S.; Narang, J.; Pundir, C.S. An amperometric polyphenol biosensor based on polyvinyl chloride membrane. Anal. Methods 2010, 2, 1106–1111. [Google Scholar] [CrossRef]
- Narang, J.; Chawla, S.; Chauhan, N.; Dahiya, M.; Pundir, C.S. Construction of an amperometric polyphenol biosensor based on PVA membrane. J. Food Meas. Charact. 2013, 7, 22–28. [Google Scholar] [CrossRef]
- Sandeep, S.; Santhosh, A.S.; Swamy, N.K.; Suresh, G.S.; Melo, J.S.; Nithin, K.S. Electrochemical detection of L-dopa using crude Polyphenol oxidase enzyme immobilized on electrochemically reduced RGO-Ag nanocomposite modified graphite electrode. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2018, 232–235, 15–21. [Google Scholar] [CrossRef]
- Shoja, Y.; Rafati, A.A.; Ghodsi, J. Glassy carbon electrode modified with horse radish peroxidase/organic nucleophilic-functionalized carbon nanotube composite for enhanced electrocatalytic oxidation and efficient voltammetric sensing of levodopa. Mater. Sci. Eng. C 2016, 58, 835–845. [Google Scholar] [CrossRef]
- Gątarek, P.; Sekulska-Nalewajko, J.; Bobrowska-Korczaka, B.; Pawełczyk, M.; Jastrzębski, K.; Głąbiński, A.; Kałużna-Czaplińska, J. Plasma Metabolic Disturbances in Parkinson’s Disease Patients. Biomedicines 2022, 10, 3005. [Google Scholar] [CrossRef]
- Tong, Q.; Zhang, L.; Yuan, Y.; Jiang, S.; Zhang, R.; Xu, Q.; Ding, J.; Li, D.; Zhou, X.; Zhang, K. Reduced plasma serotonin and 5-hydroxyindoleacetic acid levels in Parkinson’s disease are associated with nonmotor symptoms. Park. Relat. Disord. 2015, 21, 882–887. [Google Scholar] [CrossRef]
- Paul, R.; Borah, A. L-DOPA-induced hyperhomocysteinemia in Parkinson’s disease: Elephant in the room. Biochim. Biophys. Acta-Gen. Subj. 2016, 1860, 1989–1997. [Google Scholar] [CrossRef]








| Modified Electrode | Detection Mode | Linear Range | Sensitivity | Limit of Detection | Reference |
|---|---|---|---|---|---|
| GC/carboxymethyl starch-graft-PANI /MWCNT nanocomposite | DPV (oxidation) | 10–300 µM | 0.035 µA/µM | 32 µM | [71] |
| GC/sulfonated starch-graft-PANI@graphene nanocomposite | DPV (reduction) | 0.5–109 µM | 0.0002 µA/µM | 15.0 µM | [72] |
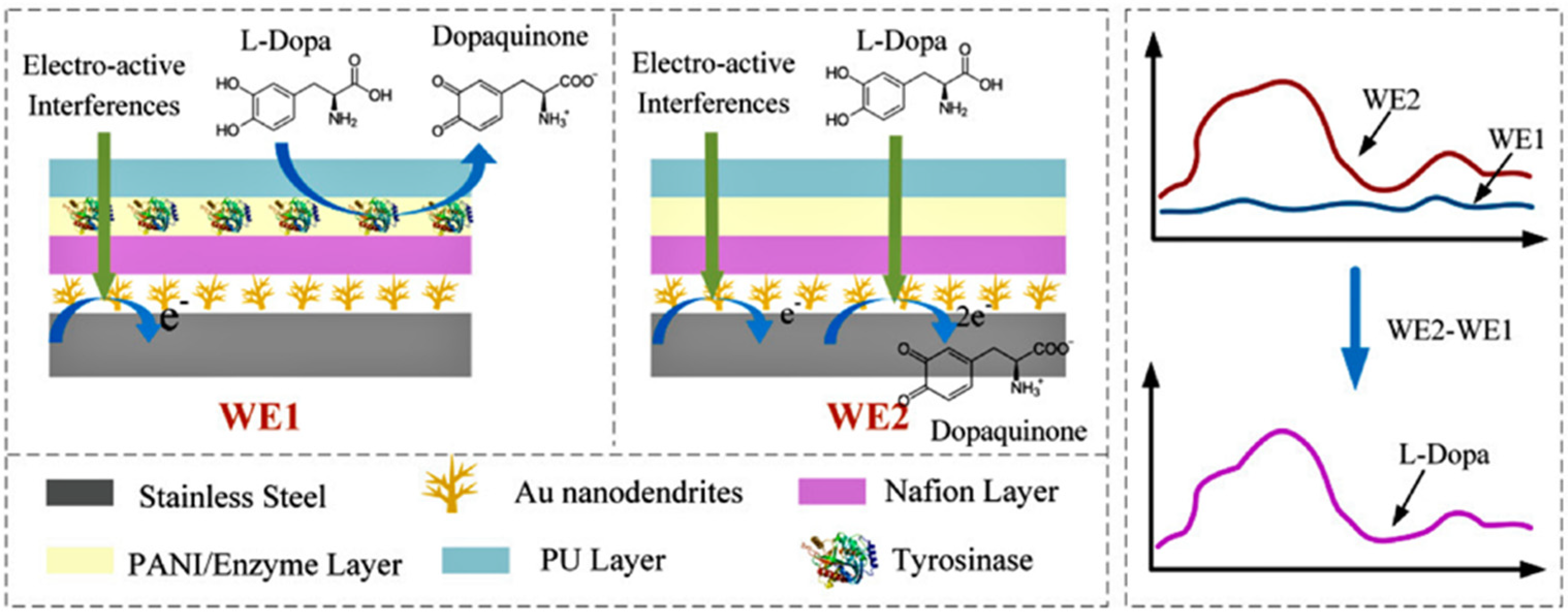
| Differential stainless-steel microneedles WE1: Aunanodentrites/Nafion/PANI/Tyrosinase/PU WE2: Aunanodentrites/Nafion/PANI/BSA/PU | Chronoamperometry (Differential current response, oxidation at 0.3 V) | 0–20 µM | 0.469 nA/µM | 0.18 µM | [67] |
| Flexible printed gold electrode/Zeolitic imidazolate framework/graphene oxide | Chronoamperometry (Oxidation at 0.3 V) | 1–95 µM | 0.047 µA/µM | 0.45 µM | [73] |
| Screen-printed carbon electrode/porous hydrogel | Chronoamperometry (reduction at −0.3 V) | 5–30 µM | not reported | 0.3 µM | [65] |
| GC/electrochemically deposited GO | Chronoamperometry (reduction at −0.1 V) | 1–210 µM | 3.21 µA/mM | 0.84 µM | [70] |
| Carbon paste microneedle electrodes WE1: unmodified carbon past, 65 wt. % graphite powder and 35 wt. % mineral oil WE2: modified carbon past, 55 wt. % graphite powder, 10 wt. % tyrosinase mushroom enzyme, and 35 wt. % mineral oil | WE1:SWV (oxidation) WE2: chronoamperometry (applied potential of 0.1 V in PB solution or 0.3 V in artificial ISF) | WE1: In PB 5–100 μM and 100–300 μM In ISF 20–160 µM WE2: In PB and ISF 20–300 µM | WE1: 0.037 μA/μM in ISF WE2: 0.048 nA/μM in ISF | WE1: in ISF 0.5 µM WE2: in ISF 0.25 µM | [75] |
| Au/Cr conductive layer/gold nanodendrites/polythionin | Amperometric (Oxidation at 0.34 V) | 0–20 µM | 15 nA/μM in PBs 1.7 nA/μM in sweat | 1.25 μM in sweat | [66] |
| Carbon electrode/CNT/polythionine | Chronoamperometry (reduction at −0.31 V) | 0.8–22.3 μM | 0.0619 A/M | 2.5 µM | [68] |
| Gold nanoelectrode ensemble | FIA, amperometric (reduction at −0.2 V) | 10−3–10−8 M | not reported | 1 × 10−9 M | [69] |
| MWCNT | Chronoamperometry (Reduction at −0.3 V) | not reported | not reported | not reported | [74] |
| Anti-Interference Strategy | Interferents and LD Tested | Real Samples | Wearable/System Integration | Reference |
|---|---|---|---|---|
| uric acid and ascorbic acid 30 µM, LD 30 µM | [71] | |||
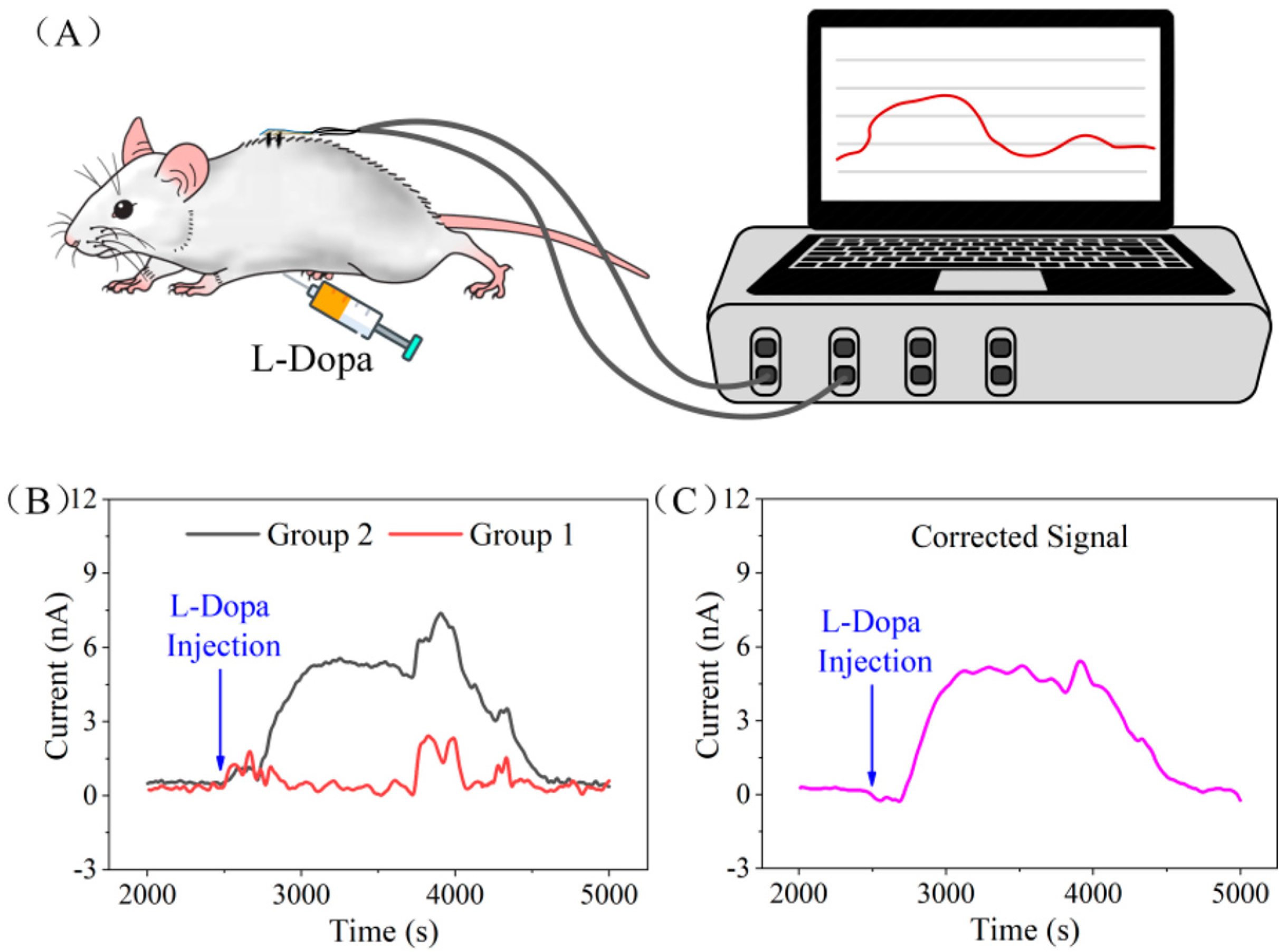
| Differential structure: -WE1 detects the electrochemical signal of the interferents -WE2 detects the mixed signal of the interferents and L-Dopa | uric acid and ascorbic acid 50 µM, glucose 200 µM, LD 30 µM | In vitro: bovine serum with a skin model (rat skin) In vivo: rat’s abdominal cavity | Minimally invasive subcutaneous biosensor | [67] |
| uric acid 20 μM, glucose 100 μM, lactate 20 μM, ascorbic acid 20 μM, LD 10 µM | Sweat levodopa concentrations after consuming broad beans (Recovery from 98.85 to 99.79%) | Flexible, wearable electrochemical sensor for the noninvasive in situ detection of LD in sweat. Integration with a wireless electronic circuit | [73] | |
| Nafion membrane as a protective layer | ascorbic acid 100 μM, dopamine 0.32 µM, carbidopa 0.2 μM, serotonin 0.15 μM, homocysteine 20 µM, tyrosine 2.5 μM, LD 2.5 μM | Human plasma (Recovery from 90.8 to 102.4%) | [70] | |
| Nafion membrane as an anti-interference barrier | L-Dopa/C-Dopa in 4:1 and 1:1 concentration ratio Caffeine 50 μM, ascorbic acid 100 μM, uric acid 400 μM, dopamine 10 nM, acetaminophen 100 μM, tyrosine and resorcinol 10 μM, LD 10 µM | Fingertip sweat | Noninvasive semi-continuous tracking of sweat LD levels upon administration of standard pill formulations based on a single fingertip touch | [65] |
| Orthogonally measured electrochemical signals, redox, and biocatalytic Nafion coating as permsaelective protective layer | ascorbic acid, uric acid, tyrosine, and theophylline 150 μM, LD 50 μM using artificial ISF medium | In vitro artificial ISF (pH 7.4) | Wearable, can penetrate through skin mimicking phantom gel and mice skin | [75] |
| Nafion coating as an antifouling layer | Uric acid 20 μM, glucose 166 μM, ascorbic acid 16μM, LD 10 µM | Sweat generated via iontophoresis and physical activities after Vicia faba consumption | Wearable sweatband for prolonged, continuous, and noninvasive drug monitoring in human subjects after fava bean intake | [66] |
| Nafion coating as a protective layer | Human serum | [68] | ||
| Glucose, ascorbic acid and urea 10 mM, LD 10−6 M | Human urine (Recovery 96%) | [69] | ||
| dopamine and epinephrine | [74] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesoro, C.; Cembalo, G.; Guerrieri, A.; Bianco, G.; Acquavia, M.A.; Di Capua, A.; Lelario, F.; Ciriello, R. A Critical Overview of Enzyme-Based Electrochemical Biosensors for L-Dopa Detection in Biological Samples. Chemosensors 2023, 11, 523. https://doi.org/10.3390/chemosensors11100523
Tesoro C, Cembalo G, Guerrieri A, Bianco G, Acquavia MA, Di Capua A, Lelario F, Ciriello R. A Critical Overview of Enzyme-Based Electrochemical Biosensors for L-Dopa Detection in Biological Samples. Chemosensors. 2023; 11(10):523. https://doi.org/10.3390/chemosensors11100523
Chicago/Turabian StyleTesoro, Carmen, Giuseppa Cembalo, Antonio Guerrieri, Giuliana Bianco, Maria Assunta Acquavia, Angela Di Capua, Filomena Lelario, and Rosanna Ciriello. 2023. "A Critical Overview of Enzyme-Based Electrochemical Biosensors for L-Dopa Detection in Biological Samples" Chemosensors 11, no. 10: 523. https://doi.org/10.3390/chemosensors11100523