
Suspect Screening and Semi-Quantification of Macrolide Antibiotics in Municipal Wastewater by High-Performance Liquid Chromatography—Precursor Ion Scan Tandem Mass Spectrometry

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Municipal Wastewater Sample Preparation
2.3. Liquid Chromatography–Tandem Mass Spectrometry
2.4. Liquid Chromatography–High-Resolution Mass Spectrometry
2.5. Quantification and Method Validation
3. Results and Discussion
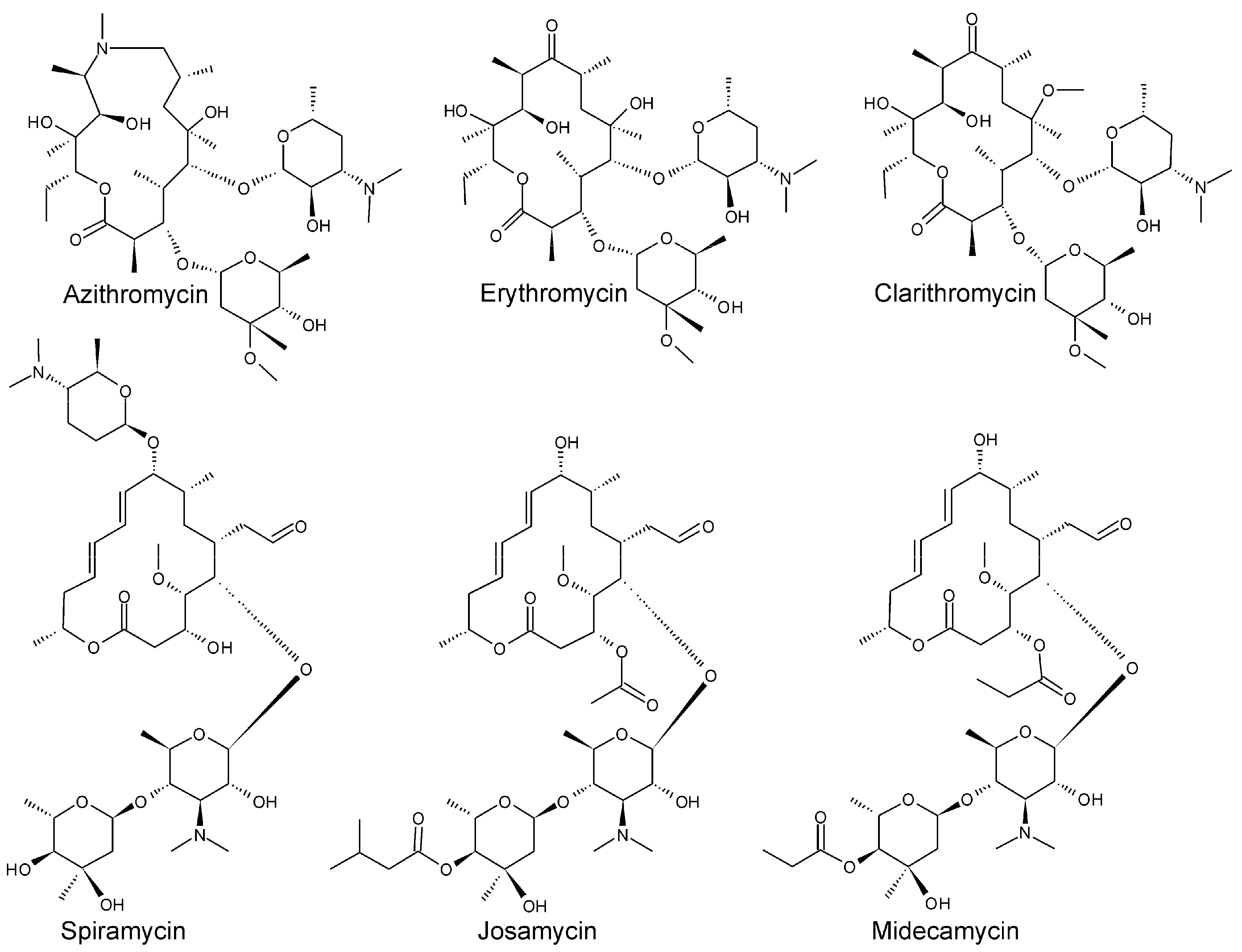
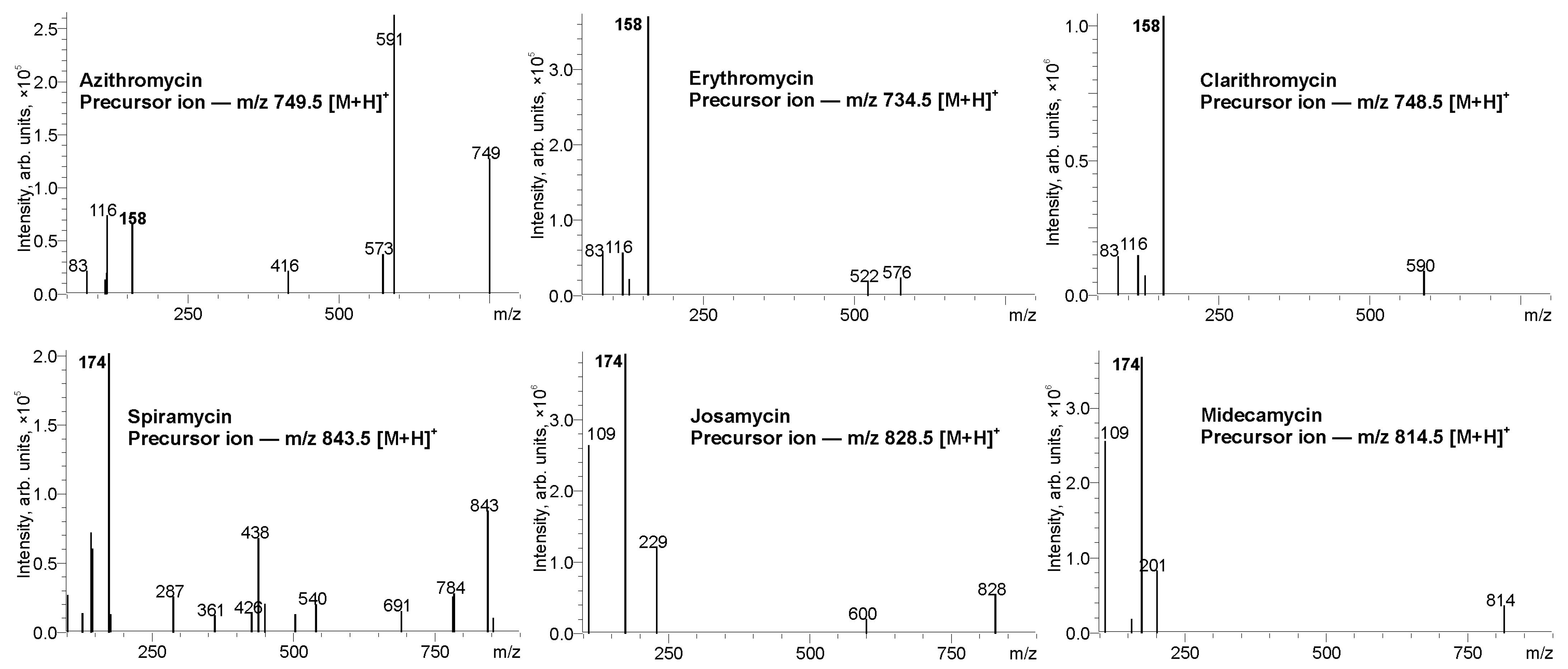
3.1. Tandem Mass Spectra of Macrolides and Selection of Diagnostic Product Ions
3.2. Method Development and Validation
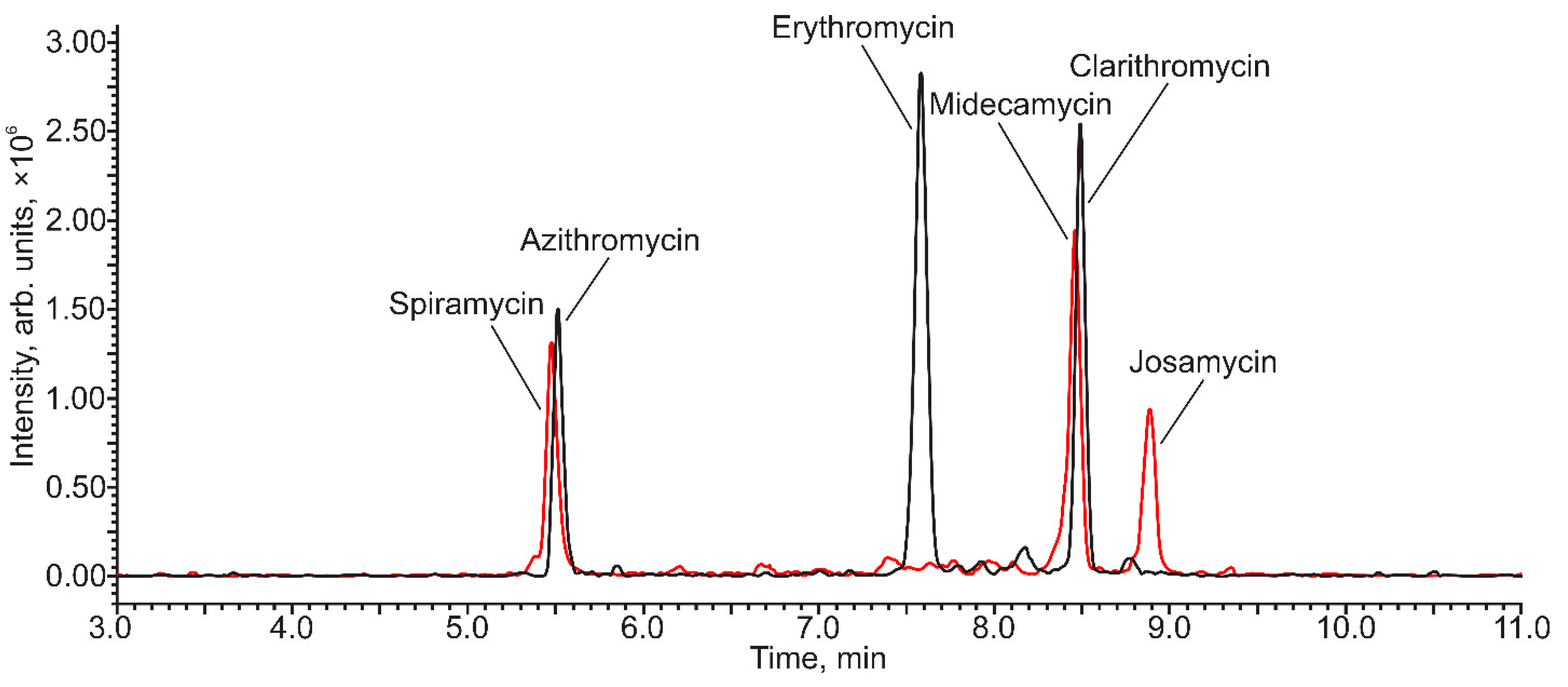
3.3. Targeted Analysis of Municipal Wastewater
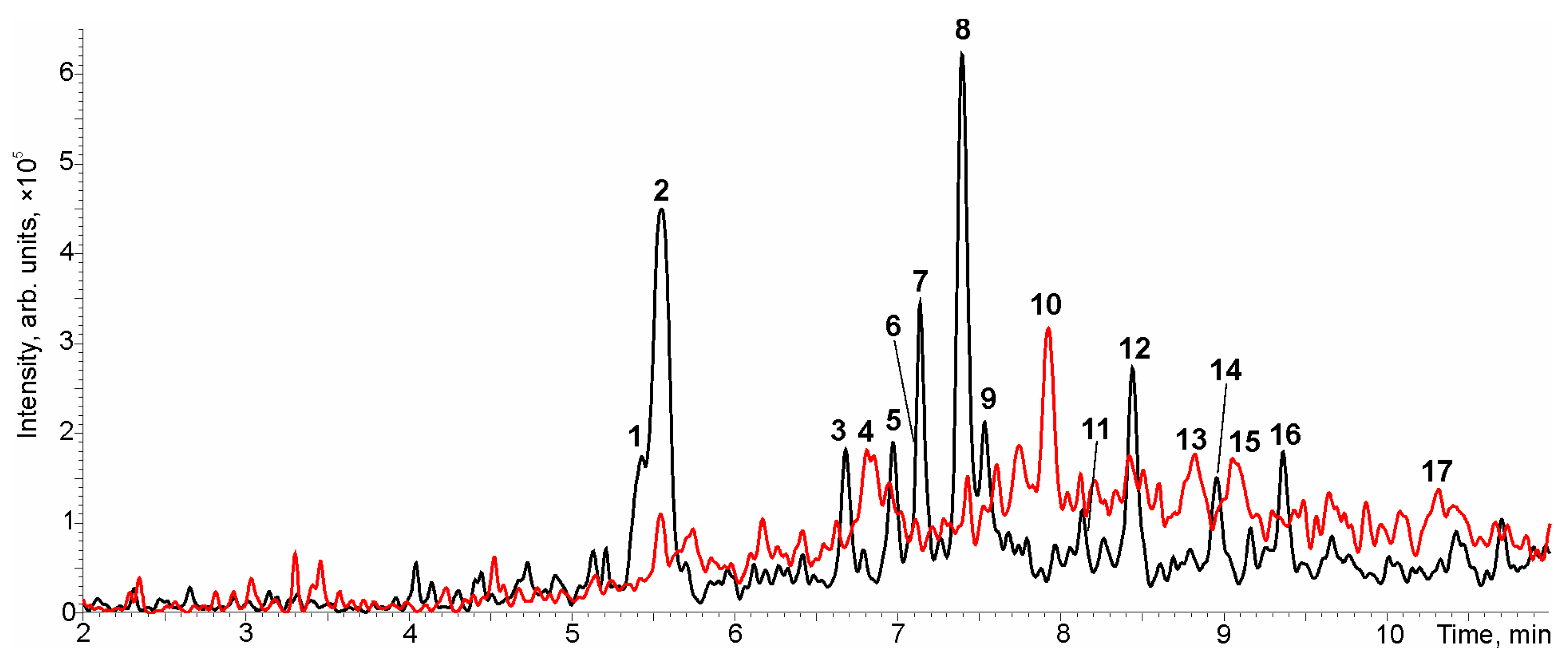
3.4. PrecIS Suspect Screening of Macrolides in Municipal Wastewater
3.5. Semi-Quantfication and Levels of Macrolides in Municipal Wastewater
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ye, X.; Sikirica, V.; Schein, J.R.; Grant, R.; Zarotsky, V.; Doshi, D.; Benson, C.J.; Riede, A.A. Treatment Failure Rates and Health Care Utilization and Costs among Patients with Community-Acquired Pneumonia Treated with Levofloxacin or Macrolides. in an Outpatient Setting: A Retrospective Claims Database Analysis. Clin. Ther. 2008, 30, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Culić, V.; Eraković, M.J. Parnham. Anti-inflammatory effects of macrolide antibiotics. Eur. J. Pharmcol. 2001, 429, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Van Boeckel, T.P.; Gandra, S.; Ashok, A.; Caudron, Q.; Grenfell, B.T.; Levin, S.A.; Laxminarayan, R. Global antibiotic consumption 2000 to 2010: An analysis of national pharmaceutical sales data. Lancet Infect. Dis. 2014, 14, 742–750. [Google Scholar] [CrossRef]
- World Health Organization. WHO Report on Surveillance of Antibiotic Consumption: 2016–2018 Early Implementation; WHO: Geneva, Switzerland, 2018; pp. 1–128. [Google Scholar]
- European Centre for Disease Prevention and Control. Antimicrobial Consumption. Annual Epidemiological Report for 2016; ECDC: Stockholm, Sweden, 2018; pp. 1–16. [Google Scholar]
- Grau, S.; Echeverria-Esnal, D.; Gómez-Zorrilla, S.; Navarrete-Rouco, M.E.; Masclans, J.R.; Espona, M.; Gracia-Arnillas, M.P.; Duran, X.; Comas, M.; Horcajada, J.P.; et al. Evolution of Antimicrobial Consumption during the First Wave of COVID-19. Pandemic. Antibiotics 2021, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Pani, A.; Lauriola, M.; Romandini, A.; Scaglione, F. Macrolides and viral infections: Focus on azithromycin in COVID-19 pathology. Int. J. Antimicrob. Agents 2020, 56, 106053. [Google Scholar] [CrossRef] [PubMed]
- Nippes, R.P.; Macruz, P.D.; da Silva, G.N.; Neves Olsen Scaliante, M.H. A critical review on environmental presence of pharmaceutical drugs tested for the COVID-19 treatment. Process Saf. Environ. Prot. 2021, 152, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.; Farooq, M.; Hanna, K. Environmental side effects of the injudicious use of antimicrobials in the era of COVID-19. Sci. Total Environ. 2020, 745, 141053. [Google Scholar] [CrossRef]
- Seiple, I.B.; Zhang, Z.; Jakubec, P.; Langlois-Mercier, A.; Wright, P.M.; Hog, D.T.; Yabu, K.; Allu, S.R.; Fukuzaki, T.; Caarlsen, P.M.; et al. A platform for the discovery of new macrolide antibiotics. Nature 2016, 533, 338–345. [Google Scholar] [CrossRef]
- Wang, W.; Wang, H.; Zhang, W.; Liang, H.; Gao, D. Occurrence, distribution, and risk assessment of antibiotics in the Songhua River in China. Environ. Sci. Pollut. Res. Int. 2017, 24, 19282–19292. [Google Scholar] [CrossRef]
- Kümmerer, K. Drugs in the environment: Emission of drugs, diagnostic aids and disinfectants into wastewater by hospitals in relation to other sources—A review. Chemosphere 2001, 45, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Munir, M.; Xagoraraki, I. Correlation of tetracycline and sulfonamide antibiotics with corresponding resistance genes and resistant bacteria in a conventional municipal wastewater treatment plant. Sci. Total Environ. 2012, 421, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hu, X.; Xu, T.; Zhang, H.; Sheng, D.; Yi, D. Prevalence of antibiotic resistance genes and their relationship with antibiotics in the Huangpu River and the drinking water sources, Shanghai, China. Sci. Total Environ. 2013, 458, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, I.T.; Santos, L. Antibiotics in the aquatic environments: A review of the European scenario. Environ. Int. 2016, 94, 736–757. [Google Scholar] [CrossRef] [PubMed]
- Seifrtová, M.; Nováková, L.; Lino, C.; Pena, A.; Solich, P. An overview of analytical methodologies for the determination of antibiotics in environmental waters. Anal. Chim. Acta 2009, 649, 158–179. [Google Scholar] [CrossRef] [PubMed]
- Sherazi, S.T.H.; Mahesar, S.A.; Sirajuddin, M.A.; Malah, M.A. Brief overview of frequently used macrolides and analytical techniques for their assessment. Cur. Anal. Chem. 2019, 15, 324–338. [Google Scholar] [CrossRef]
- Ozumchelouei, J.E.; Hamidian, A.H.; Zhang, Y.; Yang, M. Physico-Chemical Properties of Antibiotics: A review with an emphasis on detection in the aquatic environment. Water Environ. Res. 2019, 92, 177–188. [Google Scholar] [CrossRef]
- Senta, I.; Terzic, S.; Ahel, M. Simultaneous Determination of Sulfonamides, Fluoroquinolones, Macrolides and Trimethoprim in Wastewater and River Water by LC-Tandem-MS. Chromatographia 2008, 68, 747–758. [Google Scholar] [CrossRef]
- Wang, Z.; Song, X.; Zhou, T.; Bian, K.; Zhang, F.; He, L. Simultaneous determination of ten macrolides drugs in feeds by high performance liquid chromatography with evaporation light scattering detection. RSC Adv. 2015, 5, 1491–1499. [Google Scholar] [CrossRef]
- Gros, M.; Rodríguez-Mozaz, S.; Barceló, D. Rapid analysis of multiclass antibiotic residues and some of their metabolites in hospital, urban wastewater and river water by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap. tandem mass spectrometry. J. Chromatog. A 2013, 1292, 173–188. [Google Scholar] [CrossRef] [Green Version]
- Benedetti, B.; Mauro, M.; Cavaliere, C.; Montone, C.M.; Fatone, F.; Frisonc, N.; Lagana, A.; Capriotti, A.L. Determination of multi-class emerging contaminants in sludge and recovery materials from waste water treatment plants: Development of a modified QuEChERS method coupled to LC–MS/MS. Microchem. J. 2020, 155, 104732. [Google Scholar] [CrossRef]
- Boix, C.; Ibáñez, M.; Sancho, J.V.; Rambla, J.; Aranda, J.L.; Ballester, S.; Hernandez, F. Fast determination of 40 drugs in water using large volume direct injection liquid chromatography–tandem mass spectrometry. Talanta 2015, 131, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Xu, Y.; Sang, J.; Zhu, B.; Wang, J. Characterization of a new component and impurities in josamycin by trap-free two-dimensional liquid chromatography coupled to ion trap time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2019, 33, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Zweigenbaum, J.A.; Thurman, E.M. Analysis of 70 Environmental Protection Agency priority pharmaceuticals in water by EPA Method 1694. J. Chromatog. A 2010, 1217, 5674–5686. [Google Scholar] [CrossRef] [PubMed]
- Walczak-Skierska, J.; Szultka-Młyńska, M.; Pauter, K.; Buszewski, B. Study of chromatographic behavior of antibiotic drugs and their metabolites based on quantitative structure-retention relationships with the use of HPLC-DAD. J. Pharm. Biomed. Anal. 2020, 184, 113187. [Google Scholar] [CrossRef]
- Bichon, E.; Béasse, A.; Prevost, S.; Christien, S.; Courant, F.; Monteau, F.; Le Bizec, B. Improvement of estradiol esters monitoring in bovine hair by dansylation and liquid chromatography/tandem mass spectrometry analysis in multiple reaction. monitoring and precursor ion scan modes. Rapid Commun. Mass Spectrom. 2012, 26, 819–827. [Google Scholar] [CrossRef]
- Falev, D.I.; Ul’yanovskii, N.V.; Ovchinnikov, D.V.; Faleva, A.V.; Kosyakov, D.S. Screening and semi-quantitative determination of pentacyclic triterpenoids in plants by liquid chromatography–tandem mass spectrometry in precursor ion scan mode. Phytochem. Anal. 2020, 32, 252–261. [Google Scholar] [CrossRef]
- Zheng, F.; Xiao, H.M.; Zhu, Q.F.; Yu, Q.W.; Feng, Y.Q. Profiling of benzimidazoles and related metabolites in pig serum based on SiO2@NiO solid-phase extraction combined precursor ion scan with high resolution orbitrap mass spectrometry. Food Chem. 2019, 284, 279–286. [Google Scholar] [CrossRef]
- U.S. Environmental Protection Agency. Method 1694: Pharmaceuticals and Personal Care Products in Water, Soil, Sediment, and Biosolids by HPLC/MS/MS; U.S. EPA: Washington, DC, USA, 2007; pp. 1–77. [Google Scholar]
- Mokh, S.; El Khatib, M.; Koubar, M.; Daher, Z.; Al Iskandarani, M. Innovative SPE-LC-MS/MS technique for the assessment of 63 pharmaceuticals and the detection of antibiotic-resistant-bacteria: A case study natural water sources in Lebanon. Sci. Total Environ. 2017, 609, 830–840. [Google Scholar] [CrossRef]
- Fohner, A.E.; Sparreboom, A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Macrolide antibiotic pathway, pharmacokinetics/pharmacodynamics. Pharmacogen. Genomics 2017, 27, 164–167. [Google Scholar] [CrossRef] [Green Version]
- Kibwage, I.O.; Janssen, G.; Busson, R.; Hoogmartens, J.; Vanderhaeghe, H.; Verbist, L. Identification of novel erythromycin derivatives in mother liquor concentrates of Streptomyces erythraeus. J. Antibiot. 1987, 40, 1–6. [Google Scholar] [CrossRef]
- Rodvold, K.A. Clinical Pharmacokinetics of Clarithromycin. Clin. Pharmacokinet. 1999, 37, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Weiss, K.; Maletzki, D.; Schüssler, W.; Schudoma, D.; Kopf, W.; Kühnen, U. Aquatic toxicity of the macrolide antibiotic clarithromycin and its metabolites. Chemosphere 2015, 120, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Gago-Ferrero, P.; Schymanski, E.L.; Bletsou, A.A.; Aalizadeh, R.; Hollender, J.; Thomaidis, N.S. Extended Suspect and Non-Target Strategies to Characterize Emerging Polar Organic Contaminants in Raw Wastewater with LC-HRMS/MS. Environ. Sci. Technol. 2015, 49, 12333–12341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, T.; Dougherty, T.J.; Magee, T.V. Macrolide Antibiotics. Compr. Med. Chem. 2007, 7, 519–566. [Google Scholar] [CrossRef]
- Van den Bossche, L.; Daidone, F.; Van Schepdael, A.; Hoogmartens, J.; Adams, E. Characterization of impurities in josamycin using dual liquid chromatography combined with mass spectrometry. J. Pharm. Biomed. Anal. 2013, 73, 66–76. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Larsson, D.G.J. Concentrations of antibiotics predicted to select for resistant bacteria: Proposed limits for environmental regulation. Environ. Int. 2016, 86, 140–149. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Weight, Da | Precursor Ion, m/z | Product Ion, m/z | Q1 Bias, V | Collision Energy, eV | Q2 Bias, V |
|---|---|---|---|---|---|---|
| Azithromycin | 748.5 | 749.5 | 158 | −33.9 | 41.3 | −27.4 |
| 83 * | −37.1 | 53.0 | −33.9 | |||
| Spiramycin | 842.5 | 843.5 | 174 | −14.5 | 34.5 | −14.5 |
| 142 * | −14.5 | 38.5 | −17.8 | |||
| Erythromycin | 733.5 | 734.5 | 158 | −33.9 | 32.7 | −27.4 |
| 83 * | −33.9 | 49.8 | −33.9 | |||
| Midecamycin | 813.5 | 814.5 | 109 | −21.0 | 45.2 | −43.5 |
| 174 * | −21.0 | 33.9 | −17.8 | |||
| Clarithromycin | 747.5 | 748.5 | 158 | −33.9 | 32.0 | −14.5 |
| 83 * | −33.9 | 50.2 | −33.9 | |||
| Josamycin | 827.5 | 828.5 | 109 | −21.0 | 43.6 | −17.8 |
| 174 * | −21.0 | 35.9 | −17.8 |
| Analyte | Linear Range, µg L−1 | s | a | R2 | LOD | LOQ | ||
|---|---|---|---|---|---|---|---|---|
| Instrumental µg L−1 | with SPE ng L−1 | Instrumental µg L−1 | with SPE ng L−1 | |||||
| Azithromycin | 210–10,000 | 325.5 | −2563 | 0.999 | 60 | 120 | 210 | 400 |
| Spiramycin | 290–10,000 | 323.4 | −1226 | 0.999 | 90 | 150 | 290 | 495 |
| Erythromycin | 13–1000 | 10,220 | 1209 | 0.999 | 4 | 5 | 13 | 18 |
| Midecamycin | 17–1000 | 16,630 | 648.3 | 0.999 | 5 | 6 | 17 | 21 |
| Clarythromycin | 12–1000 | 21,060 | 667.0 | 0.999 | 4 | 4 | 12 | 13 |
| Josamycin | 14–1000 | 17,760 | 3212 | 0.999 | 4 | 5 | 14 | 16 |
| Analyte | Concentration in Municipal Wastewater, ng L−1 | ||
|---|---|---|---|
| PrecIS | MRM | Δ, % | |
| Azithromycin | 1220 ± 160 | 1010 ± 140 | 20 |
| Spiramycin | – * | – | |
| Erythromycin | – | 0.47 ± 0.1 | |
| Midecamycin | – | 1.24 ± 0.4 | |
| Clarythromycin | 21.3 ± 3.3 | 21.5 ± 0.2 | 0.9 |
| Josamycin | 13.8 ± 7.3 | 13.5 ± 0.7 | 2.4 |
| No * | tR, min | [M + H]+ m/z | Elemental Composition | Tentative Identification | Estimated Level, ng L−1 |
|---|---|---|---|---|---|
| 1 | 5.40 | 606.3848 | C30H55O11N | 3-O-Decladinosyl-14-hydroxycalrithromycin | 16.2 ± 2.4 |
| 2 | 5.50 | 749.5151 | C38H72O12N2 | Azithromycin | 1220 ± 160 |
| 3 | 6.68 | 590.3899 | C30H55O10N | 3-O-Decladinosyl-calrithromycin | 13.1 ± 0.7 |
| 4 | 6.81 | 860.4645 | C42H69O17N | Dihydroxyjosamycin | 21.5 ± 6.0 |
| 5 | 6.97 | 782.4896 | C38H71O15N | Clarithromycin derivative (PubChem CID 139596847) | 11.3 ± 3.3 |
| 6 | 7.13 | 748.4842 | C38H69O13N | Methylerythromycin | 13.5 ± 2.7 |
| 7 | 7.14 | 766.4948 | C38H71O14N | 10-Hydroxy-4′-(2-hydroxyethylmethylamino)-erythromycin | 11.0 ± 4.6 |
| 8 | 7.40 | 764.4793 | C38H69O14N | 14-Hydroxyclarithromycin | 47.0 ± 0.9 |
| 9 | 7.52 | 764.4793 | C38H69O14N | Hydroxyclarithromycin | 9.8 ± 2.4 |
| 10 | 7.92 | 844.4693 | C42H69O16N | Hydroxyjosamycin | 19.2 ± 9.1 |
| 11 | 8.13 | 716.4580 | C37H65O12N | Anhydroerythromycin | 7.5 ± 1.0 |
| 12 | 8.43 | 748.4842 | C38H69O13N | Clarithromycin | 21.3 ± 3.3 |
| 13 | 8.82 | 828.4740 | C42H69O15N | Josamycin | 13.8 ± 7.3 |
| 14 | 8.94 | 734.4689 | C37H67O13N | Pseudo-erythromycin A-6,9-hemiketal | 8.8 ± 0.6 |
| 15 | 9.06 | 892.4538 | C42H69O19N | Tetrahydroxyjosamycin | 3.6 ± 2.1 |
| 16 | 9.36 | 672.4319 | C35H61O11N | 12-Allyloxy-3-O-decladinosyl-calrithromycin | 12.4 ± 0.1 |
| 17 | 10.30 | 948.4801 | C45H73O20N | Josamycin derivative | 6.5 ± 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voronov, I.S.; Falev, D.I.; Ul’yanovskii, N.V.; Kosyakov, D.S. Suspect Screening and Semi-Quantification of Macrolide Antibiotics in Municipal Wastewater by High-Performance Liquid Chromatography—Precursor Ion Scan Tandem Mass Spectrometry. Chemosensors 2023, 11, 44. https://doi.org/10.3390/chemosensors11010044
Voronov IS, Falev DI, Ul’yanovskii NV, Kosyakov DS. Suspect Screening and Semi-Quantification of Macrolide Antibiotics in Municipal Wastewater by High-Performance Liquid Chromatography—Precursor Ion Scan Tandem Mass Spectrometry. Chemosensors. 2023; 11(1):44. https://doi.org/10.3390/chemosensors11010044
Chicago/Turabian StyleVoronov, Ilya S., Danil I. Falev, Nikolay V. Ul’yanovskii, and Dmitry S. Kosyakov. 2023. "Suspect Screening and Semi-Quantification of Macrolide Antibiotics in Municipal Wastewater by High-Performance Liquid Chromatography—Precursor Ion Scan Tandem Mass Spectrometry" Chemosensors 11, no. 1: 44. https://doi.org/10.3390/chemosensors11010044