Hemodynamic Effects of Alpha-Tropomyosin Mutations Associated with Inherited Cardiomyopathies: Multiscale Simulation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cardiac Muscle Mechanics and Regulation
2.2. Geometry of the Left Ventricle
2.3. Model of Circulation
2.4. Numerical Simulation and the Model Validation
2.5. Modeling Cell-Level Effects of Two Cardiomyopathy-Associated Mutations
2.6. Modeling LV remodeling for HCM and DCM-Associated Tpm Mutations
2.7. Modeling Changes in Passive Myocardial Stiffness Accompanying HCM and DCM
3. Results
3.1. Simulation of Hemodynamic Changes Caused by the Asp230Asn and Ile284Val Tpm Mutations Without the LV Remodeling
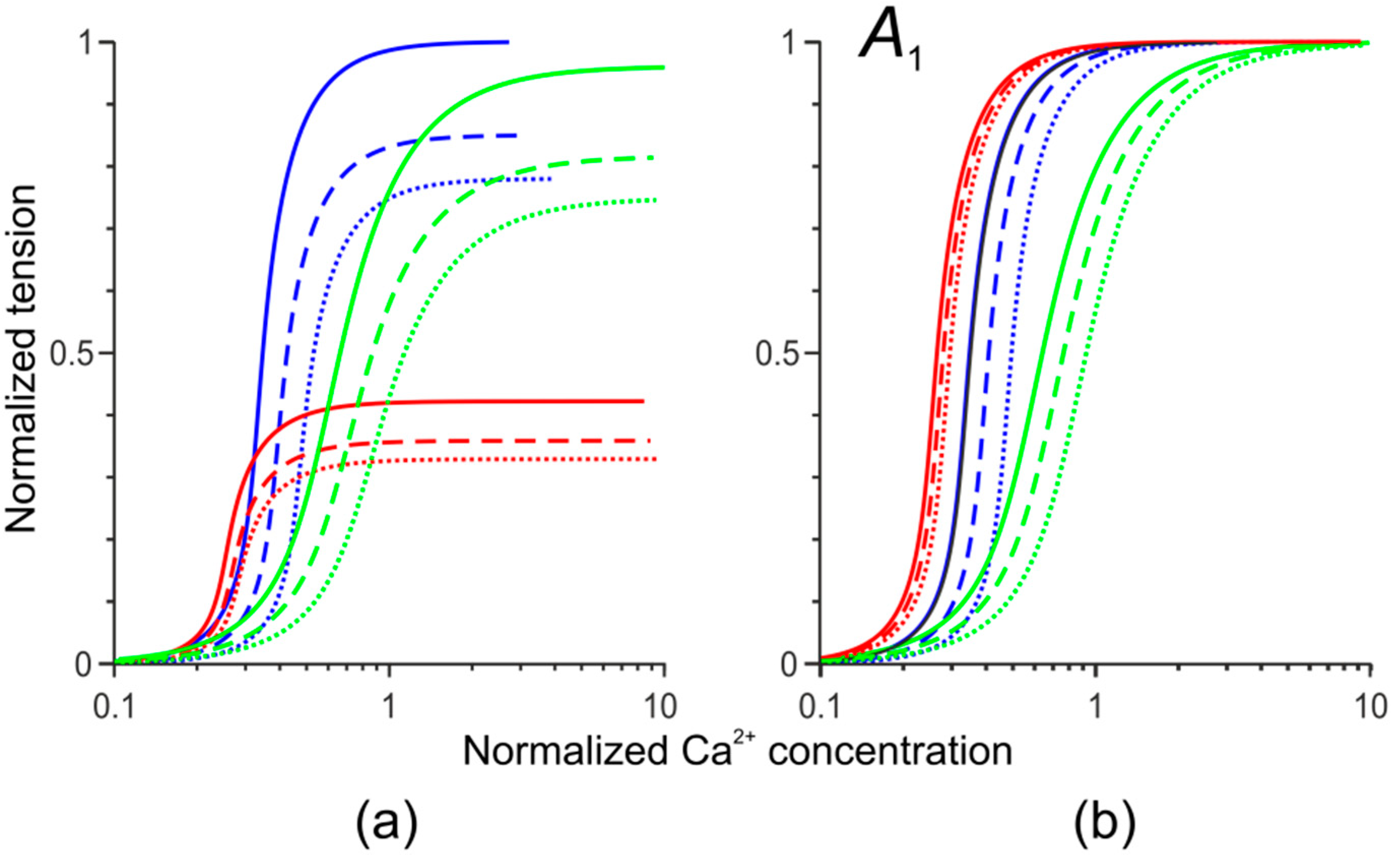
3.2. Changes in Ca2+ Transients Caused by the Tpm Mutations
3.3. Simulation of Hemodynamic Changes Caused by the Tpm Mutations and the LV Remodeling
3.4. Simulation of the Effects of the Tpm Mutations and the LV Remodeling on the Pressure-Volume Loops
4. Discussion
4.1. Cell-Level Changes in Ca2+ Regulation and Cardiac Function
4.2. Effects of LV Remodeling
4.3. PV Loops Depend on Not Only on Myocardial Contractility but Also LV Geometry
4.4. Relation to Previous Works
4.5. Limitations
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
A.1. Cell Model of Myocardium [18]
A.2. Left Ventricle Approximation
A.3. Hemodynamics Model [17]
A.4. Local Hydraulic Valve Resistances in the Cases of the Valve Pathologies
References
- Moraczewska, J. Thin filament dysfunctions caused by mutations in tropomyosin Tpm3.12 and Tpm1.1. J. Muscle Res. Cell Motil. 2020, 41, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Redwood, C.; Robinson, P. Alpha-tropomyosin mutations in inherited cardiomyopathies. J. Muscle Res. Cell Motil. 2013, 34, 285–294. [Google Scholar] [CrossRef]
- Watkins, H.; Ashraan, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef]
- Opie, L.H.; Commerford, P.J.; Gersh, B.J.; Pfeffer, M.A. Controversies in ventricular remodelling. Lancet 2006, 367, 356–367. [Google Scholar] [CrossRef]
- Nagueh, S.F.; Shah, G.; Wu, Y.; Torre-Amione, G.; King, N.M.; Lahmers, S.; Witt, C.C.; Becker, K.; Labeit, S.; Granzier, H.L. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004, 110, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Røe, Å.T.; Aronsen, J.M.; Skårdal, K.; Hamdani, N.; Linke, W.A.; Danielsen, H.E.; Sejersted, O.M.; Sjaastad, I.; Louch, W.E. Increased passive stiffness promotes diastolic dysfunction despite improved Ca2+ handling during left ventricular concentric hypertrophy. Cardiovasc. Res. 2017, 113, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Villemain, O.; Correia, M.; Mousseaux, E.; Baranger, J.; Zarka, S.; Podetti, I.; Soulat, G.; Damy, T.; Hagège, A.; Tanter, M.; et al. Myocardial stiffness evaluation using noninvasive shear wave imaging in healthy and hypertrophic cardiomyopathic adults. JACC Cardiovasc. Imaging 2019, 12, 1135–1145. [Google Scholar] [CrossRef]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef]
- Lehman, W. Switching muscles on and off in steps: The McKillop-Geeves three-state model of muscle regulation. Biophys. J. 2017, 112, 2459–2466. [Google Scholar] [CrossRef] [Green Version]
- Land, S.; Niederer, S.A. A spatially detailed model of isometric contraction based on competitive binding of troponin I explains cooperative interactions between tropomyosin and crossbridges. PLoS Comput. Biol. 2015, 1, e1004376. [Google Scholar] [CrossRef] [Green Version]
- Metalnikova, N.A.; Tsaturyan, A.K. A mechanistic model of Ca regulation of thin filaments in cardiac muscle. Biophys. J. 2013, 105, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.A.; Maytum, R.; Geeves, M.A. Cooperative regulation of myosin-actin interactions by a continuous flexible chain I: Actin-tropomyosin systems. Biophys. J. 2003, 84, 3155–3167. [Google Scholar] [CrossRef] [Green Version]
- Lakdawala, N.K.; Dellefave, L.; Redwood, C.R.; Sparks, E.; Cirino, A.L.; Depalma, S.; Colan, S.D.; Funke, B.; Zimmerman, R.S.; Robinson, P.; et al. Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: The distinctive natural history of sarcomeric dilated cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, M.L.; Tal Grinspan, L.; Holeman, T.A.; Jimenez, J.; Strom, J.; Tardiff, J.C. The structural basis of alpha-tropomyosin linked (Asp230Asn) familial dilated cardiomyopathy. J. Mol. Cell Cardiol. 2017, 108, 127–137. [Google Scholar] [CrossRef]
- Sequeira, V.; Wijnker, P.J.; Nijenkamp, L.L.; Kuster, D.W.; Najafi, A.; Witjas-Paalberends, E.R.; Regan, J.A.; Boontje, N.; Ten Cate, F.J.; Germans, T.; et al. Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ. Res. 2013, 112, 1491–1505. [Google Scholar] [CrossRef] [Green Version]
- Syomin, F.A.; Tsaturyan, A.K. Mechanical model of the left ventricle of the heart approximated by axisymmetric geometry. Russ. J. Numer. Anal. Math. Model. 2017, 32, 327–337. [Google Scholar] [CrossRef]
- Syomin, F.A.; Zberia, M.V.; Tsaturyan, A.K. Multiscale simulation of the effects of atrioventricular block and valve diseases on heart performance. Int. J. Numer. Method. Biomed. Eng. 2019, 35, e3216. [Google Scholar] [CrossRef]
- Syomin, F.A.; Tsaturyan, A.K. A simple model of cardiac muscle for multiscale simulation: Passive mechanics, crossbridge kinetics and calcium regulation. J. Theor. Biol. 2017, 420, 105–116. [Google Scholar] [CrossRef]
- De Tombe, P.P.; Mateja, R.D.; Tachampa, K.; Ait Mou, Y.; Farman, G.P.; Irving, T.C. Myofilament length dependent activation. J. Mol. Cell Cardiol. 2010, 48, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Kopylova, G.; Berg, V.; Bershitsky, S.; Matyushenko, A.; Shchepkin, D. The effects of cardiomyopathy-associated mutations in the head-to-tail overlap junction of a-tropomyosin on the calcium regulation of the actin-myosin interaction. FEBS Open Bio 2019, 9, 111. [Google Scholar]
- Gupte, T.M.; Haque, F.; Gangadharan, B.; Sunitha, M.S.; Mukherjee, S.; Anandhan, S.; Rani, D.S.; Mukundan, N.; Jambekar, A.; Thangaraj, K.; et al. Mechanistic heterogeneity in contractile properties of α-Tropomyosin (TPM1) mutants associated with inherited cardiomyopathies. J. Biol. Chem. 2015, 290, 7003–7015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pravdin, S.F.; Berdyshev, V.I.; Panfilov, A.V.; Katsnelson, L.B.; Solovyova, O.; Markhasin, V.S. Mathematical model of the anatomy and fibre orientation field of the left ventricle of the heart. Biomed. Eng. Online 2013, 12, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Ven, F.; Bibeau, K.; De Larochelliere, E.; Tizon-Marcos, H.; Deneault-Bissonnette, S.; Pibarot, P.; Deschepper, C.F.; Larose, E. Cardiac morphology and function reference values derived from a large subset of healthy young Caucasian adults by magnetic resonance imaging. Eur. Heart J. Cardiovasc. Imaging 2016, 17, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Haland, T.F.; Hasselberg, N.E.; Almaas, V.M.; Dejgaard, L.A.; Saberniak, J.; Leren, I.S.; Berge, K.E.; Haugaa, K.H.; Edvardsen, T. The systolic paradox in hypertrophic cardiomyopathy. Open Heart 2017, 4, e000571. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yue, Y.; Wang, Y.; Deng, Y.; Liu, L.; Di, Y.; Sun, S.; Chen, D.; Fan, L.; Cao, J. Assessment of left ventricular systolic and diastolic abnormalities in patients with hypertrophic cardiomyopathy using real-time three-dimensional echocardiography and two-dimensional speckle tracking imaging. Cardiovasc. Ultrasound 2018, 16, 23. [Google Scholar] [CrossRef] [Green Version]
- Marko, J.F.; Siggia, E.D. Statistical mechanics of supercoiled DNA. Phys. Rev. E 1995, 52, 2912–2938. [Google Scholar] [CrossRef]
- Suga, H.; Sagawa, K. Instantaneous pressure-volume relationships and their ratio in the excised, supported canine left ventricle. Circ. Res. 1974, 35, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Sagawa, K. The end-systolic pressure-volume relation of the ventricle: Definition, modifications and clinical use. Circulation 1981, 63, 1223–1227. [Google Scholar] [CrossRef] [Green Version]
- Kanzaki, H.; Nakatani, S.; Yamada, N.; Urayama, S.; Miyatake, K.; Kitakaze, M. Impaired systolic torsion in dilated cardiomyopathy: Reversal of apical rotation at mid-systole characterized with magnetic resonance tagging method. Basic Res. Cardiol. 2006, 101, 465–470. [Google Scholar] [CrossRef]
- Omar, A.M.S.; Bansal, M.; Sengupta, P.P. Advances in echocardiographic imaging in heart failure with reduced and preserved ejection fraction. Circ. Res. 2016, 119, 357–374. [Google Scholar] [CrossRef]
- Rady, M.; Ulbrich, S.; Heidrich, F.; Jellinghaus, S.; Ibrahim, K.; Linke, A.; Sveric, K.M. Left ventricular torsion—a new echocardiographic prognosticator in patients with non-ischemic dilated cardiomyopathy. Circ. J. 2019, 83, 595–603. [Google Scholar] [CrossRef] [Green Version]
- van der Bijl, P.; Bootsma, M.; Hiemstra, Y.L.; Ajmone Marsan, N.; Bax, J.J.; Delgado, V. Left ventricular 2D speckle tracking echocardiography for detection of systolic dysfunction in genetic, dilated cardiomyopathies. Eur. Heart J. Cardiovasc. Imaging 2018, 20, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Urbano Moral, J.A.; Ariez Godinez, J.A.; Maron, M.S.; Malik, R.; Eagan, J.E.; Patel, A.R.; Pandian, N.G. Left ventricular twist mechanics in hypertrophic cardiomyopathy assessed by three-dimensional speckle tracking echocardiography. Am. J. Cardiol. 2011, 108, 1788–1795. [Google Scholar] [CrossRef]
- Soullier, C.; Obert, P.; Doucende, G.; Nottin, S.; Cade, S.; Perez-Martin, A.; Messner-Pellenc, P.; Schuster, I. Exercise response in hypertrophic cardiomyopathy. Circ. Cardiovasc. Imaging 2012, 5, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Abozguia, K.; Nallur-Shivu, G.; Phan, T.T.; Ahmed, I.; Kalra, R.; Weaver, R.A.; McKenna, W.J.; Sanderson, J.E.; Elliott, P.; Frenneaux, M.P. Left ventricular strain and untwist in hypertrophic cardiomyopathy: Relation to exercise capacity. Am. Heart J. 2010, 159, 825–832. [Google Scholar] [CrossRef]
- Carasso, S.; Yang, H.; Woo, A.; Vannan, M.A.; Jamorski, M.; Wigle, E.D.; Rakowski, H. Systolic myocardial mechanics in hypertrophic cardiomyopathy: Novel concepts and implications for clinical status. J. Am. Soc. Echocardiogr. 2008, 21, 675–683. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Hay, I.; Fetics, B.; Kass, D.A. Combined ventricular systolic and arterial stiffening in patients with heart failure and preserved ejection fraction. Circulation 2003, 107, 714–720. [Google Scholar] [CrossRef]
- Westermann, D.; Kasner, M.; Steendijk, P.; Spillmann, F.; Riad, A.; Weitmann, K.; Homann, W.; Poller, W.; Pauschinger, M.; Schultheiss, H.P.; et al. Role of left ventricular stiffness in heart failure with normal ejection fraction. Circulation 2008, 117, 2051–2060. [Google Scholar] [CrossRef]
- Bozkurt, S. Mathematical modeling of cardiac function to evaluate clinical cases in adults and children. PLoS ONE 2019, 14, 1–20. [Google Scholar] [CrossRef]
- Kosta, S.; Negroni, J.; Lascano, E.; Dauby, P.C. Multiscale model of the human cardiovascular system: Description of heart failure and comparison of contractility indices. Math. Biosci. 2017, 284, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Lascano, E.C.; Felice, J.I.; Wray, S.; Kosta, S.; Dauby, P.C.; Cabrera-Fischer, E.I.; Negroni, J.A. Experimental assessment of a myocyte-based multiscale model of cardiac contractile dysfunction. J. Theor. Biol. 2018, 456, 16–28. [Google Scholar] [CrossRef]
- Bakir, A.; Al Abed, A.; Stevens, M.C.; Lovell, N.H.; Dokos, S. A multiphysics biventricular cardiac model: Simulations with a left-ventricular assist device. Front. Physiol. 2018, 9, 1259. [Google Scholar] [CrossRef] [PubMed]
- Genet, M.; Lee, L.C.; Baillargeon, B.; Guccione, J.M.; Kuhl, E. Modeling pathologies of diastolic and systolic heart failure. Ann. Biomed. Eng. 2016, 44, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Kassab, G.; Guccione, J. Mathematical modeling of cardiac growth and remodeling. Wires. Syst. Biol. Med. 2016, 8, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berberoglu, E.; Solmaz, H.O.; Goktepe, S. Computational modeling of coupled cardiac electromechanics incorporating cardiac dysfunctions. Eur. J. Mech. A Solid. 2014, 48, 60–73. [Google Scholar] [CrossRef]
- Sahli Costabal, F.; Choy, J.S.; Sack, K.L.; Guccione, J.M.; Kassab, G.S.; Kuhl, E. Multiscale characterization of heart failure. Acta Biomat. 2019, 86, 66–76. [Google Scholar] [CrossRef]
- Bianco, F.D.; Franzone, P.C.; Scacchi, S.; Fassina, L. Electromechanical effects of concentric hypertrophy on the left ventricle: A simulation study. Comput. Biol. Med. 2018, 99, 236–256. [Google Scholar] [CrossRef]
- Land, S.; Niederer, S.A.; Aronsen, J.M.; Espe, E.K.S.; Zhang, L.; Louch, W.E.; Sjaastad, I.; Sejersted, O.M.; Smith, N.P. An analysis of deformation-dependent electromechanical coupling in the mouse heart. J. Physiol. 2012, 590, 4553–4569. [Google Scholar] [CrossRef]
- Trayanova, N.A.; Constantino, J.; Gurev, V. Electromechanical models of the ventricles. Comput. Methods Biomech. Biomed. Engin. 2011, 301, H279–H286. [Google Scholar] [CrossRef]
- von Deuster, C.; Sammut, E.; Asner, L.; Nordsletten, D.; Lamata, P.; Stoeck, C.T.; Kozerke, S.; Razavi, R. Studying dynamic myofiber aggregate reorientation in dilated cardiomyopathy using in vivo magnetic resonance diffusion tensor imaging. Circ. Cardiovasc. Imaging 2016, 9, e005018. [Google Scholar] [CrossRef] [Green Version]
- Tseng, W.Y.; Dou, J.; Reese, T.G.; Wedeen, V.J. Imaging myocardial fiber disarray and intramural strain hypokinesis in hypertrophic cardiomyopathy with MRI. J. Magn. Reson. Imaging 2006, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kessler, E.L.; Boulaksil, M.; van Rijen, H.V.M.; Vos, M.A.; Veen, T.A.B. Passive ventricular remodeling in cardiac disease: Focus on heterogeneity. Front. Physiol. 2014, 5, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guccione, J.M.; McCulloch, A.D.; Waldman, L.K. Passive material properties of intact ventricular myocardium determined from a cylindrical model. J. Biomech. Eng. 1991, 113, 42–55. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LV Hemodynamic Characteristics | HCM + Normal Passive Stiffness | HCM + Stiff Titin Component | HCM + Stiff Isotropic Component | DCM + Normal Passive Stiffness | DCM + Soft Titin Component |
|---|---|---|---|---|---|
| Low Preload | |||||
| End-diastolic pressure, mm Hg | 6.4 | 4.3 | |||
| Peak pressure, mm Hg | 115.2 | 112.6 | 92.5 | 105.4 | 108.5 |
| End-diastolic volume, mL | 92.2 | 88.3 | 73.2 | 157.5 | 165.1 |
| Stroke volume, mL | 67.7 | 64.1 | 48.3 | 64 | 66.1 |
| Ejection fraction, % | 73 | 73 | 66 | 41 | 40 |
| Average Preload | |||||
| End-diastolic pressure, mm Hg | 8.7 | 5.1 | |||
| Peak pressure, mm Hg | 128.4 | 123.1 | 99.3 | 110.7 | 113.2 |
| End-diastolic volume, mL | 104.9 | 99.2 | 80 | 169.8 | 176.6 |
| Stroke volume, mL | 79 | 74 | 54.8 | 67.3 | 68.9 |
| Ejection fraction, % | 75 | 75 | 69 | 40 | 40 |
| High Preload | |||||
| End-diastolic pressure, mm Hg | 11.5 | 5.8 | |||
| Peak pressure, mm Hg | 141.1 | 133 | 108.6 | 115.2 | 118.3 |
| End-diastolic volume, mL | 117.9 | 109.5 | 88.8 | 181.5 | 191 |
| Stroke volume, mL | 89.9 | 83.1 | 63 | 70.2 | 72.1 |
| Ejection fraction, % | 76 | 76 | 71 | 39 | 38 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syomin, F.; Khabibullina, A.; Osepyan, A.; Tsaturyan, A. Hemodynamic Effects of Alpha-Tropomyosin Mutations Associated with Inherited Cardiomyopathies: Multiscale Simulation. Mathematics 2020, 8, 1169. https://doi.org/10.3390/math8071169
Syomin F, Khabibullina A, Osepyan A, Tsaturyan A. Hemodynamic Effects of Alpha-Tropomyosin Mutations Associated with Inherited Cardiomyopathies: Multiscale Simulation. Mathematics. 2020; 8(7):1169. https://doi.org/10.3390/math8071169
Chicago/Turabian StyleSyomin, Fyodor, Albina Khabibullina, Anna Osepyan, and Andrey Tsaturyan. 2020. "Hemodynamic Effects of Alpha-Tropomyosin Mutations Associated with Inherited Cardiomyopathies: Multiscale Simulation" Mathematics 8, no. 7: 1169. https://doi.org/10.3390/math8071169