
Bio-Constrained Codes with Neural Network for Density-Based DNA Data Storage



Abstract
:1. Introduction
- To improve the net information density by storing a large amount of digital data in shorter DNA sequences.
- To construct the DNA codes that satisfy the combinatorial bio-constraints to overcome the reading errors.
- A novel computational model based on the LSTM neural network with a forward pass is proposed to generate the optimal DNA codes from the premiere DNA bases. To the best of our knowledge, such a model has not been studied in the prior studies.
- The combinatorial bio-constraints, including GC-content, RC constraint, and Hamming distance, are constructed for optimal DNA codes to avoid non-specific hybridization by overcoming sequencing errors and secondary structures.
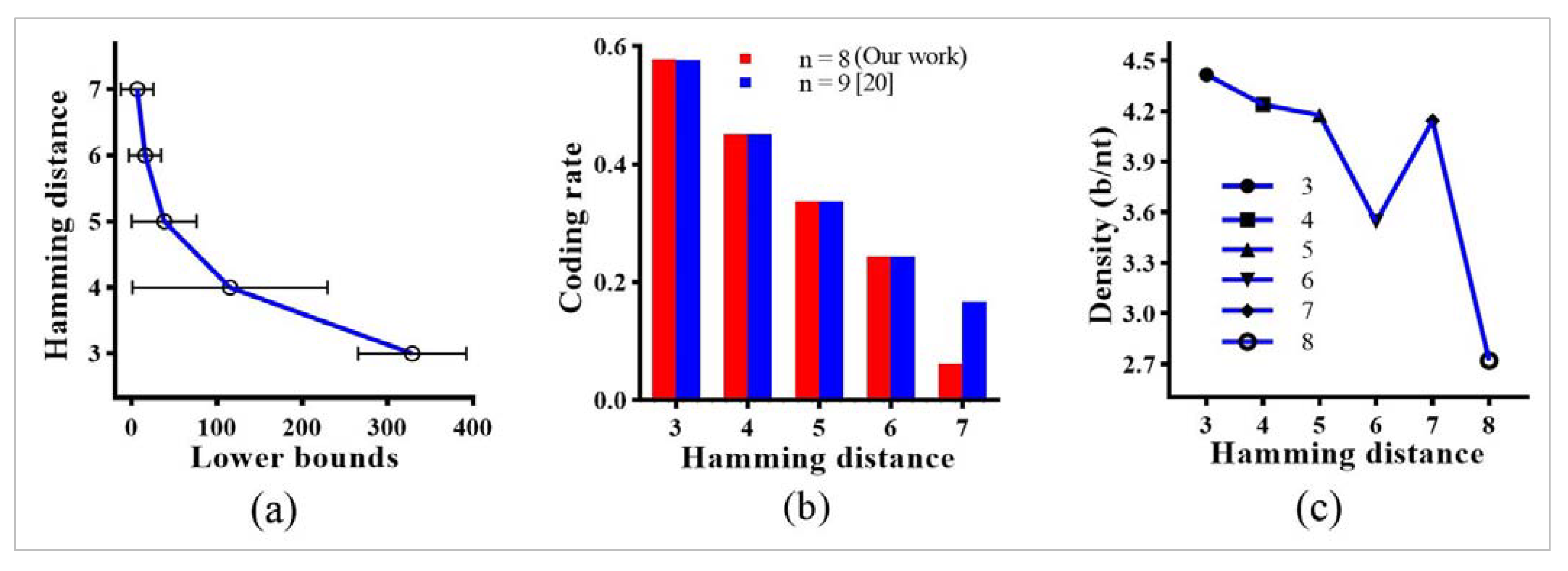
- The results receive many DNA coding sets satisfying the bio-constraints and significantly improving the DNA coding rates compared to the existing studies.
2. Literature Review
2.1. Deep Neural Networks for DNA Codes
2.2. DNA Coding with Combinatorial Bio-Constraints
3. Preliminaries and Notations
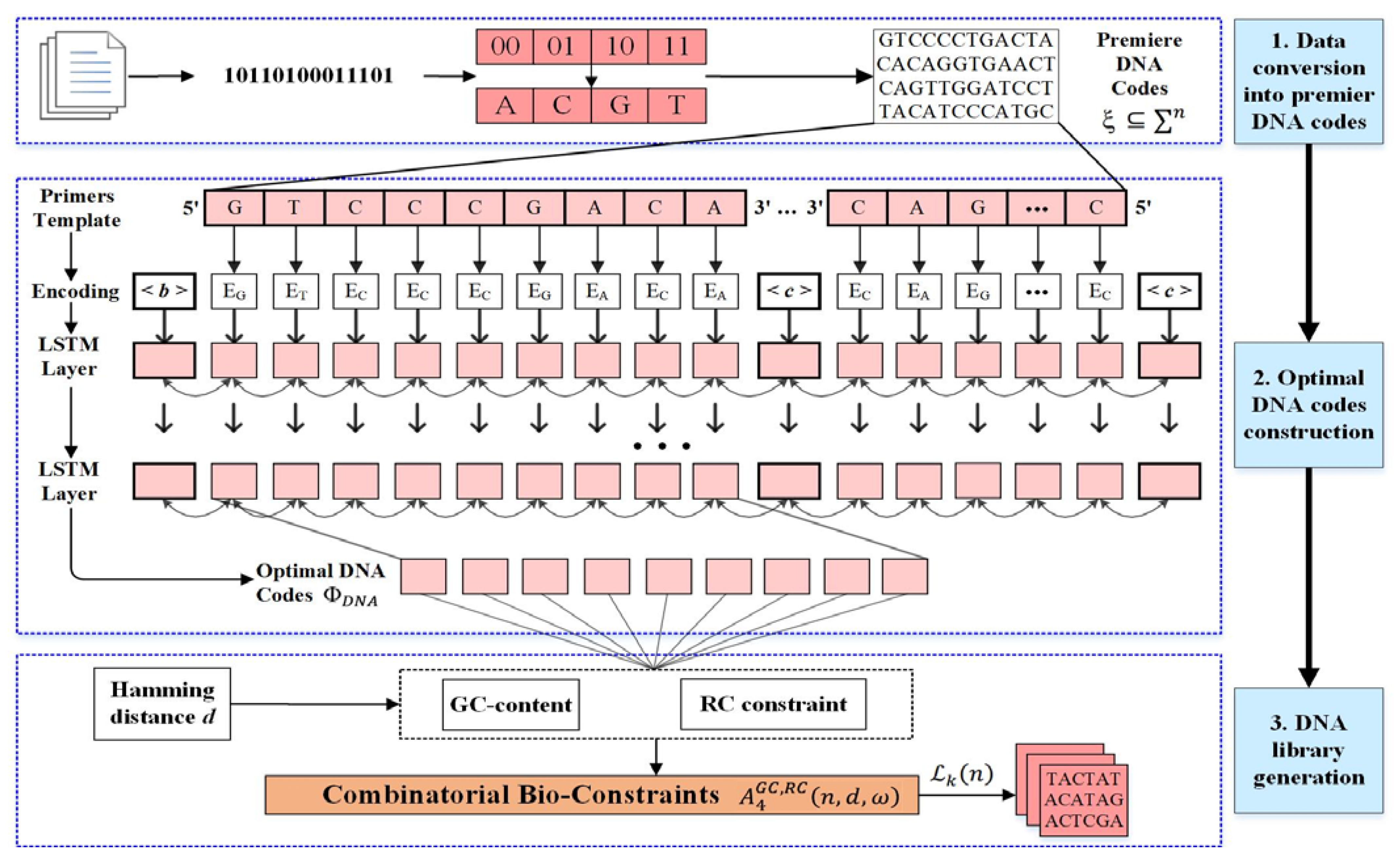
4. Proposed Model
- Transform the digital data into the sequence of bases (A, C, G, and T).
- Encode the DNA bases into optimal DNA codes.
- Create the bio-constraint codes for the DNA library construction.
4.1. NN-Based DNA Codes
- The DNA primer length is generally 15–30 nt [6]. The best length for PCR amplification primers is usually 20 nt; we also train our model at this limit.
- The length of repeated bases in a primer is generally ≤4 nt [5]. The consecutive appearance of any particular base makes the unstable DNA structure. We set consecutive base lengths ≤ 3 nt.
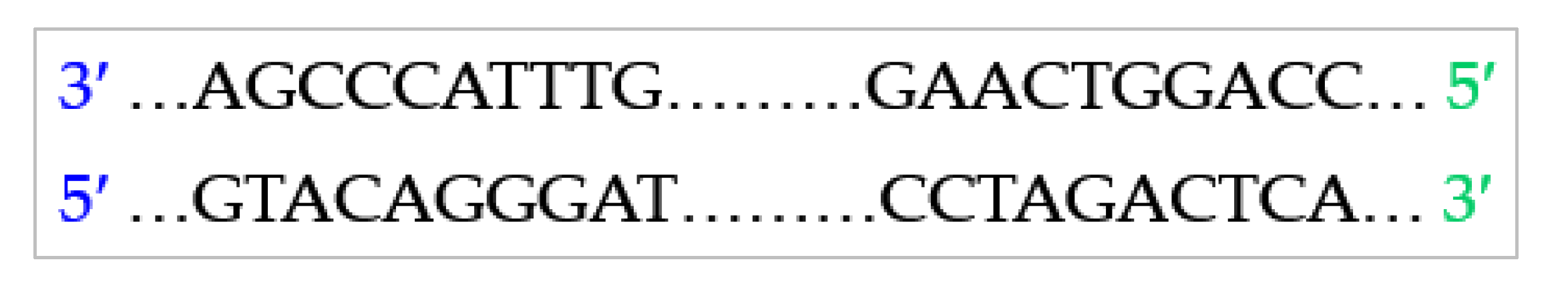
4.2. Combinatorial Constraints
| Algorithm 1. Proposed algorithm to construct DNA library |
| Input: Premiere DNA codes ξ , optimal DNA codes , GC-content , code length , Hamming distance , and reverse constraint; |
| Output: DNA library
|
| return: DNA library for DNA data storage. |
5. Result Evaluations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, M.; Wu, J.; Dai, J.; Jiang, Q.; Qu, Q.; Huang, X.; Wang, Y. A self-contained and self-explanatory DNA storage system. Sci. Rep. 2021, 11, 18063. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, S.M.H.T.; Gabrys, R.; Milenkovic, O. Portable and Error-Free DNA-Based Data Storage. Sci. Rep. 2017, 7, 5011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlich, Y.; Zielinski, D. DNA Fountain enables a robust and efficient storage architecture. Science 2017, 355, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blawat, M.; Gaedke, K.; Hütter, I.; Chen, X.-M.; Turczyk, B.; Inverso, S.; Pruitt, B.W.; Church, G.M. Forward Error Correction for DNA Data Storage. Procedia Comput. Sci. 2016, 80, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Grass, R.N.; Heckel, R.; Puddu, M.; Paunescu, D.; Stark, W.J. Robust Chemical Preservation of Digital Information on DNA in Silica with Error-Correcting Codes. Angew. Chem. Int. Ed. 2015, 54, 2552–2555. [Google Scholar] [CrossRef]
- Goldman, N.; Bertone, P.; Chen, S.; Dessimoz, C.; LeProust, E.M.; Sipos, B.; Birney, E. Towards practical, high-capacity, low-maintenance information storage in synthesized DNA. Nature 2013, 494, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Church, G.M.; Gao, Y.; Kosuri, S. Next-Generation Digital Information Storage in DNA. Science 2012, 337, 1628. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Wong, K.-C. Future DNA computing device and accompanied tool stack: Towards high-throughput computation. Future Gener. Comput. Syst. 2021, 117, 111–124. [Google Scholar] [CrossRef]
- Wang, Y.; Noor-A-Rahim, M.; Gunawan, E.; Guan, Y.L.; Poh, C.L. Construction of Bio-Constrained Code for DNA Data Storage. IEEE Commun. Lett. 2019, 23, 963–966. [Google Scholar] [CrossRef]
- Limbachiya, D.; Gupta, M.K.; Aggarwal, V. Family of Constrained Codes for Archival DNA Data Storage. IEEE Commun. Lett. 2018, 22, 1972–1975. [Google Scholar] [CrossRef]
- Benerjee, K.G.; Banerjee, A. On DNA Codes With Multiple Constraints. IEEE Commun. Lett. 2021, 25, 365–368. [Google Scholar] [CrossRef]
- Rasool, A.; Qu, Q.; Jiang, Q.; Wang, Y. A Strategy-Based Optimization Algorithm to Design Codes for DNA Data Storage System. In Algorithms and Architectures for Parallel Processing; Springer International Publishing: Xiamen, China, 2022; pp. 284–299. [Google Scholar]
- Chee, Y.M.; Ling, S. Improved lower bounds for constant GC-content DNA codes. IEEE Trans. Inf. Theory 2008, 54, 391–394. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.X.; Yordanov, B.; Gaunt, A.; Wang, M.X.; Dai, P.; Chen, Y.J.; Zhang, K.; Fang, J.Z.; Dalchau, N.; Li, J.M.; et al. A deep learning model for predicting next-generation sequencing depth from DNA sequence. Nat. Commun. 2021, 12, 4387. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, L.; Yu, G.; Wang, D.; Xiao, C.-L.; Wang, K. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 2019, 10, 2449. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wu, J.; Huang, B.; Liu, Y. High-density information storage and random access scheme using synthetic DNA. 3 Biotech 2021, 11, 328. [Google Scholar] [CrossRef]
- Cao, B.; Li, X.; Zhang, X.; Wang, B.; Zhang, Q.; Wei, X. Designing Uncorrelated Address Constrain for DNA Storage by DMVO Algorithm. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 1. [Google Scholar] [CrossRef]
- King, O.D. Bounds for DNA codes with constant GC-content. Electron. J. Comb. 2003, 10, R33. [Google Scholar] [CrossRef]
- Milenkovic, O.; Kashyap, N. On the design of codes for DNA computing. In Coding and Cryptography; Ytrehus, O., Ed.; Springer: Berlin, Heidelberg, Germany, 2006; Volume 3969, pp. 100–119. [Google Scholar]
- Aboluion, N.; Smith, D.H.; Perkins, S. Linear and nonlinear constructions of DNA codes with Hamming distance d, constant GC-content and a reverse-complement constraint. Discret. Math. 2012, 312, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Koumakis, L. Deep learning models in genomics; are we there yet? Comput. Struct. Biotechnol. J. 2020, 18, 1466–1473. [Google Scholar] [CrossRef]
- Montana, D.J.; Davis, L. Training Feedforward Neural Networks Using Genetic Algorithms. In Proceedings of the Eleventh International Joint Conference on Artificial Intelligence, Detroit, MI, USA, 20–25 August 1989. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Muzammal, M.; Nasrulin, B. Renovating blockchain with distributed databases: An open source system. Future Gener. Comput. Syst. 2019, 90, 105–117. [Google Scholar] [CrossRef]
- Jin, X.; Nie, R.; Zhou, D.; Yao, S.; Chen, Y.; Yu, J.; Wang, Q. A novel DNA sequence similarity calculation based on simplified pulse-coupled neural network and Huffman coding. Phys. A Stat. Mech. Its Appl. 2016, 461, 325–338. [Google Scholar] [CrossRef]
- Deng, L.; Wu, H.; Liu, X.; Liu, H. DeepD2V: A Novel Deep Learning-Based Framework for Predicting Transcription Factor Binding Sites from Combined DNA Sequence. Int. J. Mol. Sci. 2021, 22, 5521. [Google Scholar] [CrossRef]
- Song, W.; Cai, K.; Zhang, M.; Yuen, C. Codes with Run-Length and GC-Content Constraints for DNA-Based Data Storage. IEEE Commun. Lett. 2018, 22, 2004–2007. [Google Scholar] [CrossRef]
- Siegel, P. Codes for Mass Data Storage Systems (Second Edition) (K. H. Schouhamer Immink; 2004) [Book review]. IEEE Trans. Inf. Theory 2006, 52, 5614–5616. [Google Scholar] [CrossRef]
- Félix, B. On the embedding capacity of DNA strands under substitution, insertion, and deletion mutations. In Proceedings of the International Society for Optics and Photonics, San Jose, CA, USA, 17–21 January 2010. [Google Scholar]
- Heckel, R.; Shomorony, I.; Ramchandran, K.; David, N. Fundamental limits of DNA storage systems. In Proceedings of the 2017 IEEE International Symposium on Information Theory (ISIT), Aachen, Germany, 25–30 June 2017; pp. 3130–3134. [Google Scholar]
- Tulpan, D.; Smith, D.H.; Montemanni, R. Thermodynamic Post-Processing versus GC-Content Pre-Processing for DNA Codes Satisfying the Hamming Distance and Reverse-Complement Constraints. IEEE-ACM Trans. Comput. Biol. Bioinform. 2014, 11, 441–452. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Jacobson, A.B. Fast algorithm for predicting the secondary structure of single-stranded rna. Proc. Natl. Acad. Sci. USA 1980, 77, 6309–6313. [Google Scholar] [CrossRef] [Green Version]
- Peter Clote, R.B. Computational Molecular Biology: An Introduction; Wiley Series in Mathematical and Computational Biology; Wiley: Hoboken, NJ, USA, 2000. [Google Scholar]
- Wu, Y.T.; Yuan, M.; Dong, S.P.; Lin, L.; Liu, Y.Q. Remaining useful life estimation of engineered systems using vanilla LSTM neural networks. Neurocomputing 2018, 275, 167–179. [Google Scholar] [CrossRef]
- Rasool, A.; Jiang, Q.; Qu, Q.; Ji, C. WRS: A Novel Word-embedding Method for Real-time Sentiment with Integrated LSTM-CNN Model. In Proceedings of the 2021 IEEE International Conference on Real-time Computing and Robotics (RCAR), Xining, China, 15–19 July 2021; pp. 590–595. [Google Scholar]
- Harding, S.E.; Channell, G.; Phillips-Jones, M.K. The discovery of hydrogen bonds in DNA and a re-evaluation of the 1948 Creeth two-chain model for its structure. Biochem. Soc. Trans. 2018, 46, 1171–1182. [Google Scholar] [CrossRef] [Green Version]
- Marathe, A.; Condon, A.; Corn, R.M. On Combinatorial DNA Word Design. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2001, 83, 201–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charalambides, C.A. Enumerative Combinatorics, CRC Press Series on Discrete Mathematics and Its Applications; Chapman & Hall/CRC: Boca Raton, FL, USA, 2002. [Google Scholar]
- Wei, H.; Schwartz, M. Improved Coding over Sets for DNA-Based Data Storage. IEEE Trans. Inf. Theory 2021, 68, 118–129. [Google Scholar] [CrossRef]
- Cannon, J.; Bosma, W.; Fieker, C.; Steel, A.K. Handbook of Magma Functions. 2011. Available online: https://www.math.uzh.ch/sepp/magma-2.20.4-cr/HandbookVolume09 (accessed on 16 July 2021).
- Paluncic, F.; Abdel-Ghaffar, K.A.S.; Ferreira, H.C.; Clarke, W.A. A Multiple Insertion/Deletion Correcting Code for Run-Length Limited Sequences. IEEE Trans. Inf. Theory 2012, 58, 1809–1824. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| n/d | d = 3 | d = 4 | d = 5 | d = 6 | d = 7 | d = 8 | d = 9 | d = 10 |
|---|---|---|---|---|---|---|---|---|
| 4 | 11 12 i | |||||||
| 5 | 17 21 | 7 7 | ||||||
| 6 | 44 59 | 16 19 | 6 7 | |||||
| 7 | 110 143 i | 36 52 i | 11 19 i | 4 4 | ||||
| 8 | 289 303 | 86 115 i | 29 36 | 9 10 | 4 4 | |||
| 9 | 662 864 i | 199 291 i | 59 61 | 15 31i | 8 7 d | 4 5 | ||
| 10 | 1810 1973 | 525 604 i | 141 171 i | 43 51 i | 7 21 i | 5 6 | 4 4 | |
| 11 | 4320 5764 i | 1235 1716 i | 284 401 i | 82 125 i | 29 41 i | 9 17 i | 4 5 | 4 4 |
| 12 | 12,068 11,618 d | 3326 4986 i | 662 617 d | 190 711 i | 58 72 | 22 29 i | 8 11 i | 4 4 |
| 13 | 41,867 57,322 | 7578 8113 | 1432 2564 i | 1201 1391 | 123 368 | 39 71 i | 13 21 | 6 8 i |
| n/d | d = 3 | d = 4 | d = 5 | d = 6 | d = 7 | d = 8 | d = 9 |
|---|---|---|---|---|---|---|---|
| 4 | 12 12 | ||||||
| 5 | 20 29 i | 8 14 i | |||||
| 6 | 58 63 i | 24 27 i | 8 12 i | ||||
| 7 | 125 118 d | 44 51 | 17 22 | 7 10 i | |||
| 8 | 324 334 i | 106 124 i | 35 41 i | 14 17 i | 5 8 i | ||
| 9 | 713 921 i | 223 237 | 64 94 i | 24 23 d | 10 14 i | 5 7 i | |
| 10 | 1906 2010 | 555 913 i | 159 163 | 51 48 d | 20 21 | 10 12 i | 4 4 |
| n/d | d = 3 | d = 4 | d = 5 | d = 6 | d = 7 | d = 8 | d = 9 | d = 10 |
|---|---|---|---|---|---|---|---|---|
| 4 | 6 6 | |||||||
| 5 | 15 27 | 3 4 | ||||||
| 6 | 44 67 | 16 21 | 4 4 | |||||
| 7 | 135 243 | 36 69 | 11 19 | 2 2 | ||||
| 8 | 528 617 | 128 148 | 28 42 | 12 15 | 2 2 | |||
| 9 | 1354 1827 | 275 430 | 67 121 | 21 36 | 8 11 | 2 2 | ||
| 10 | 4542 5914 | 860 1181 | 210 271 | 54 77 | 17 27 | 8 8 | 2 2 | |
| 11 | 14,405 23,713 | 2457 6429 | 477 961 | 117 557 | 37 59 | 14 23 | 5 8 | 2 2 |
| 12 | 59,136 67,761 | 14,784 19,132 | 1848 2062 | 924 1092 | 87 131 | 29 41 | 12 18 | 4 6 |
| GAGTCTAGAC | CTGTATGCAT | TACTAGACAG |
| GTCTGACATA | CACTACTGAC | ACTGTAGCAT |
| ATGACTCACT | GATACGACAT | CTACGTAGCA |
| TACTGTCACG | ACATCTGTCA | TGCACATGAC |
| AGCATACTCA | TACATCTGCT | GACATGACAG |
| CGATGTACTG | AGACGATGTC | TGTAGCTACA |
| CAGTAGATCA | TACGATCGAG | AGATCGACTG |
| GACTCATGAC | CACGTCTGAT | GCATAGTATC |
| ACTGACTACT | ACGCAGATAC | TGCGATACTA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasool, A.; Qu, Q.; Wang, Y.; Jiang, Q. Bio-Constrained Codes with Neural Network for Density-Based DNA Data Storage. Mathematics 2022, 10, 845. https://doi.org/10.3390/math10050845
Rasool A, Qu Q, Wang Y, Jiang Q. Bio-Constrained Codes with Neural Network for Density-Based DNA Data Storage. Mathematics. 2022; 10(5):845. https://doi.org/10.3390/math10050845
Chicago/Turabian StyleRasool, Abdur, Qiang Qu, Yang Wang, and Qingshan Jiang. 2022. "Bio-Constrained Codes with Neural Network for Density-Based DNA Data Storage" Mathematics 10, no. 5: 845. https://doi.org/10.3390/math10050845