
Predicting the Kinetic Coordination of Immune Response Dynamics in SARS-CoV-2 Infection: Implications for Disease Pathogenesis
 , ,
, ,  , and
, and
Abstract
:1. Introduction
- 1.
- to develop a calibrated mathematical model of antiviral innate and adaptive immune responses to SARS-CoV-2 during mild-to-moderate symptoms infection;
- 2.
- to infer the sensitivity of the peak viral load to the kinetics of innate and adaptive responses;
- 3.
- to quantify the infectiousness of the COVID-19 patients from the onset to the recovery phase of the infection;
- 4.
- to examine the effect of the accelerated or decelerated components of the immune response on the viral load and prolonged viral persistence;
- 5.
- to evaluate the person’s infectiousness and effectiveness of testing procedures.
2. Materials and Methods
2.1. Mathematical Model of Antiviral Immune Response
2.1.1. Virus Spreading in Sensitive Tissue
2.1.2. Innate Immune Defence Reaction
2.1.3. Antigen-Specific Immune Response
2.1.4. Effects of Inflammation and Tissue Damage
2.1.5. Initial Conditions
2.2. Reference Data on SARS-CoV-2 Infection
2.3. Calibration of the Model
- First stage (incubation period, 0–3 days): .
- Second stage (activation of immune response and peak of viral load, 4–7 days) and third stage (recovery period, 8–13 days): .
- Forth stage (post-symptomatic period, 14–19 days): .
2.4. Sensitivity Analysis
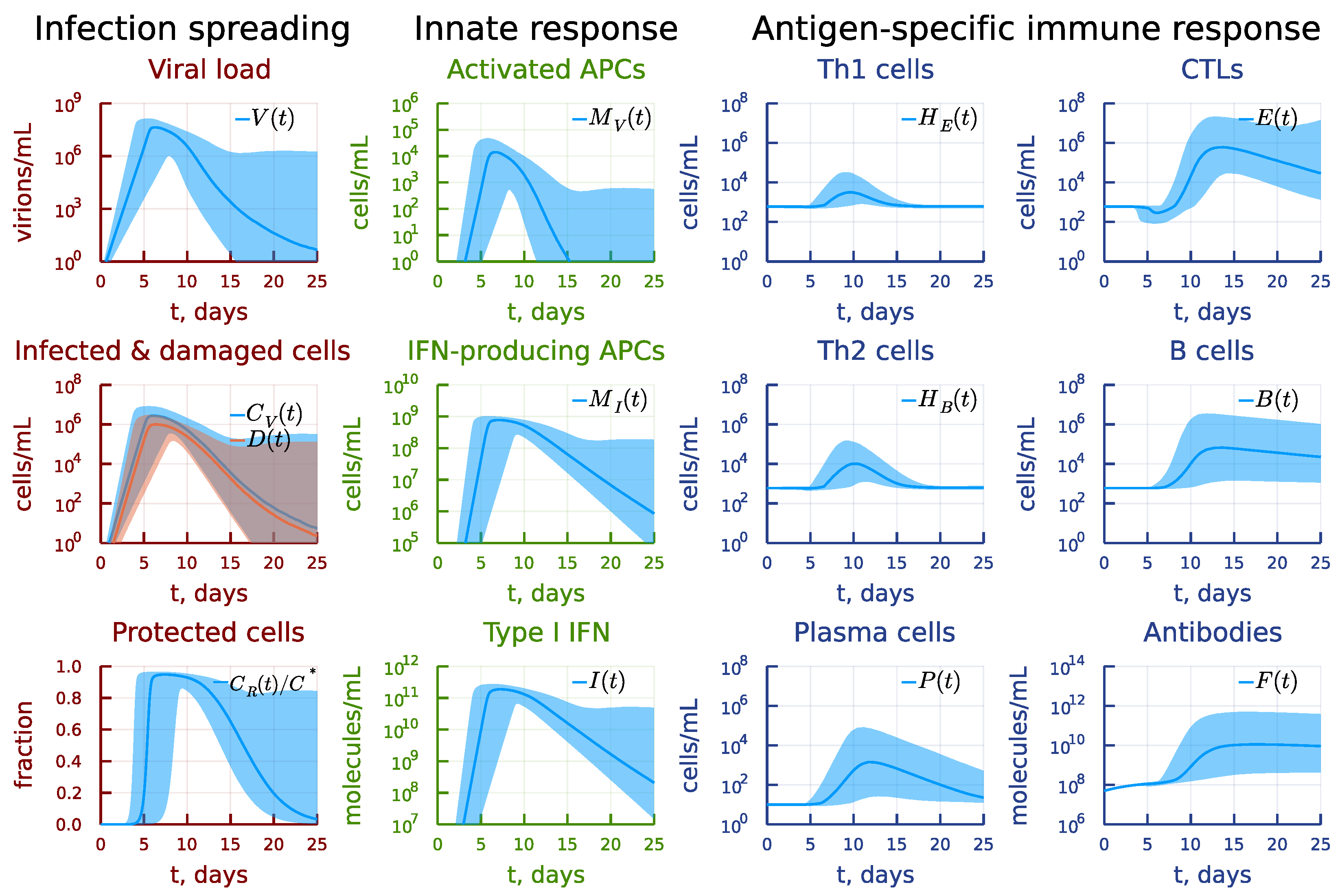
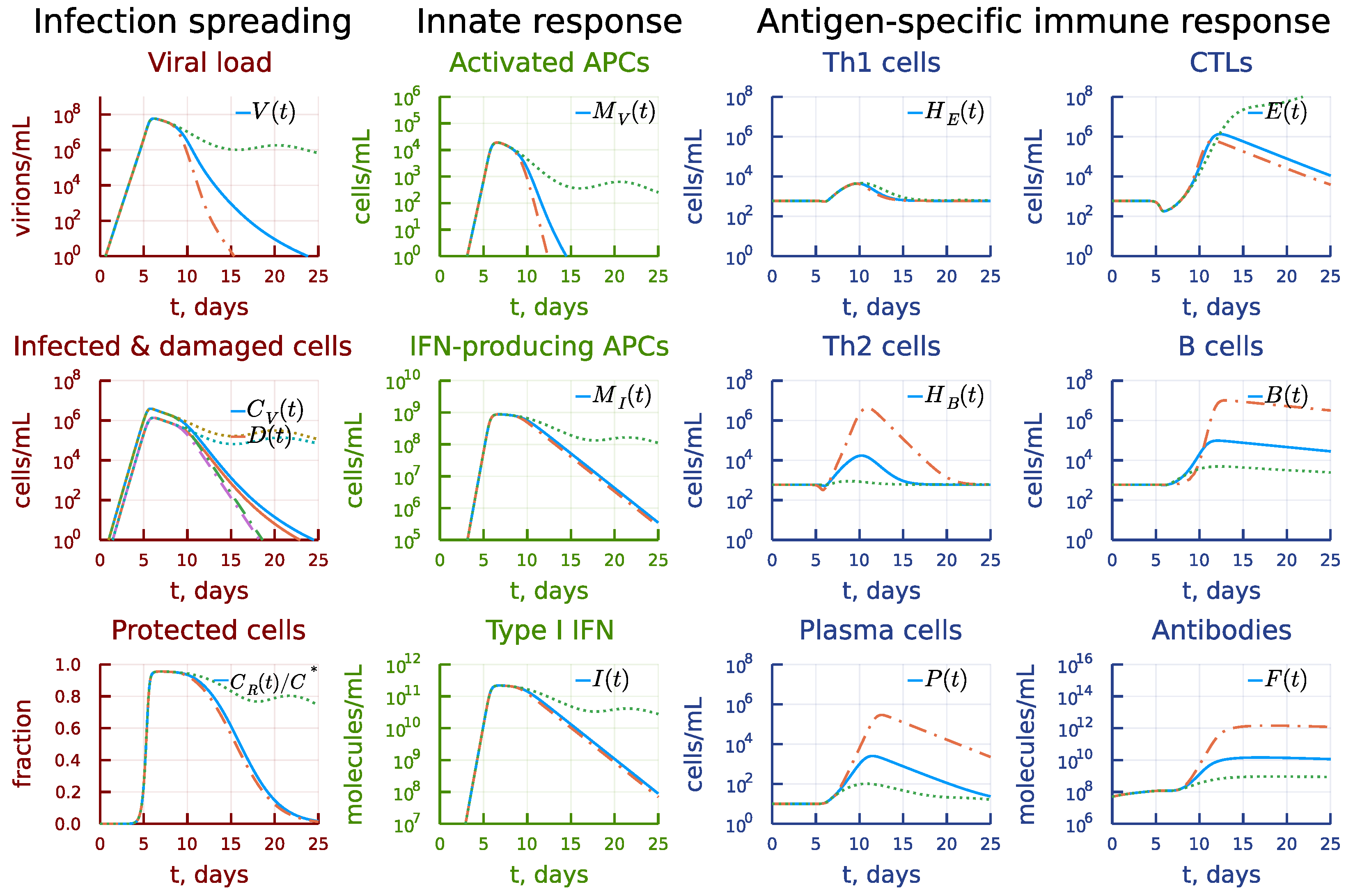
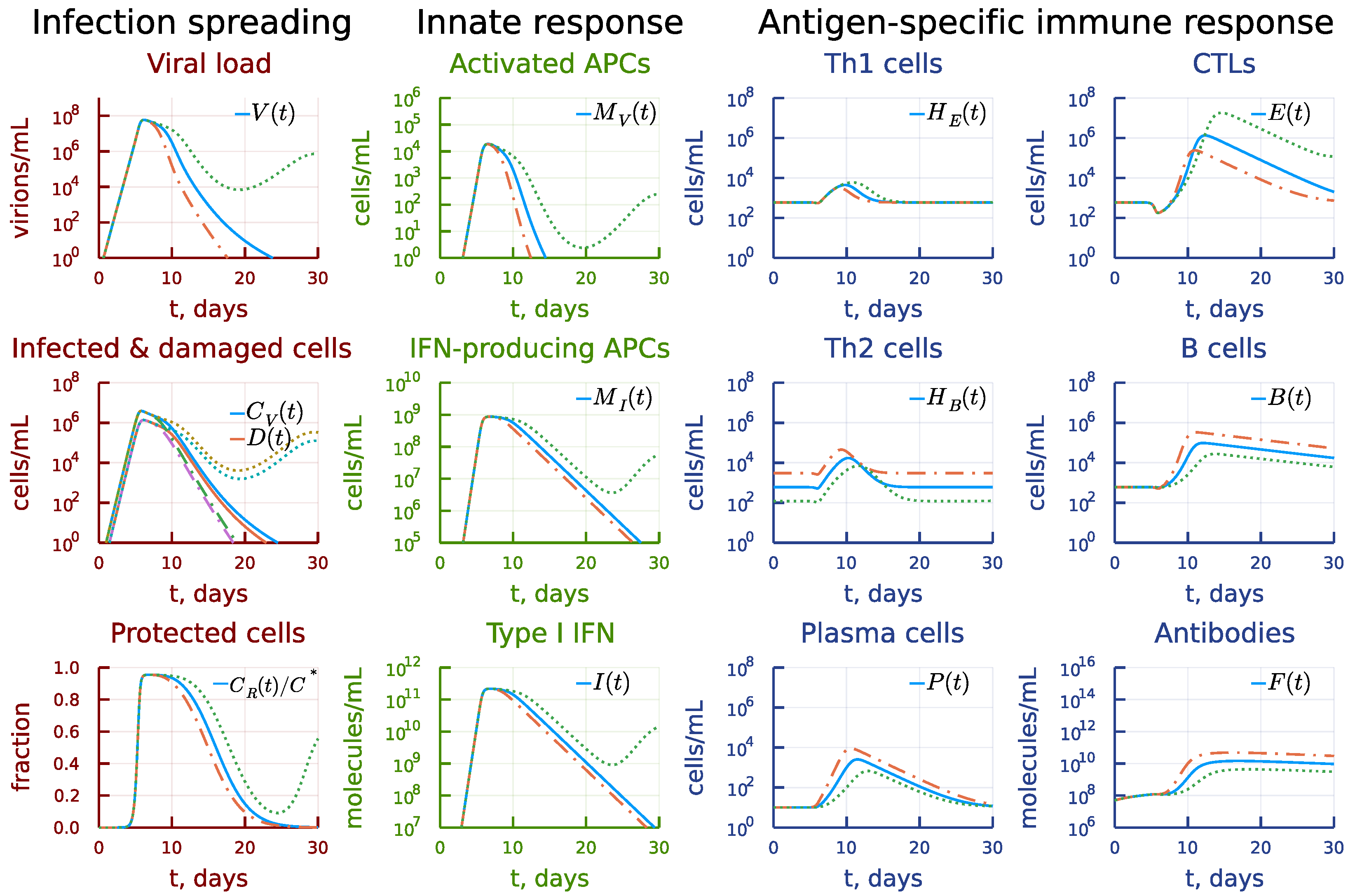
3. Results
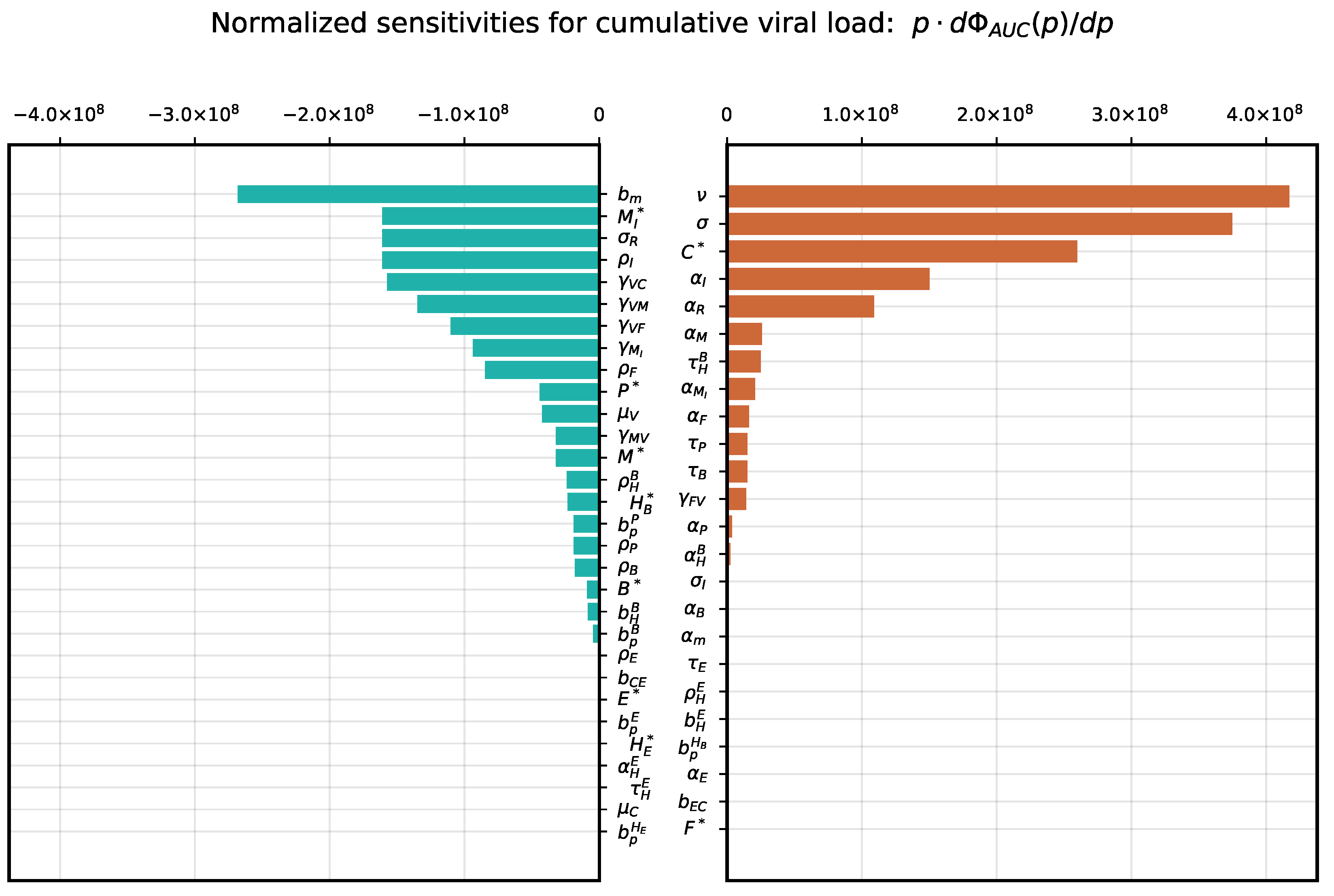
3.1. Local Sensitivity Analysis
3.2. Global Sensitivity Analysis
3.3. Induction of Antigen-Presenting Cells
3.4. Induction of Type I IFN Response
3.5. Disregulation of CTL and B-Cell Responses
3.6. Asymmetry of Th1 versus Th2 Responses
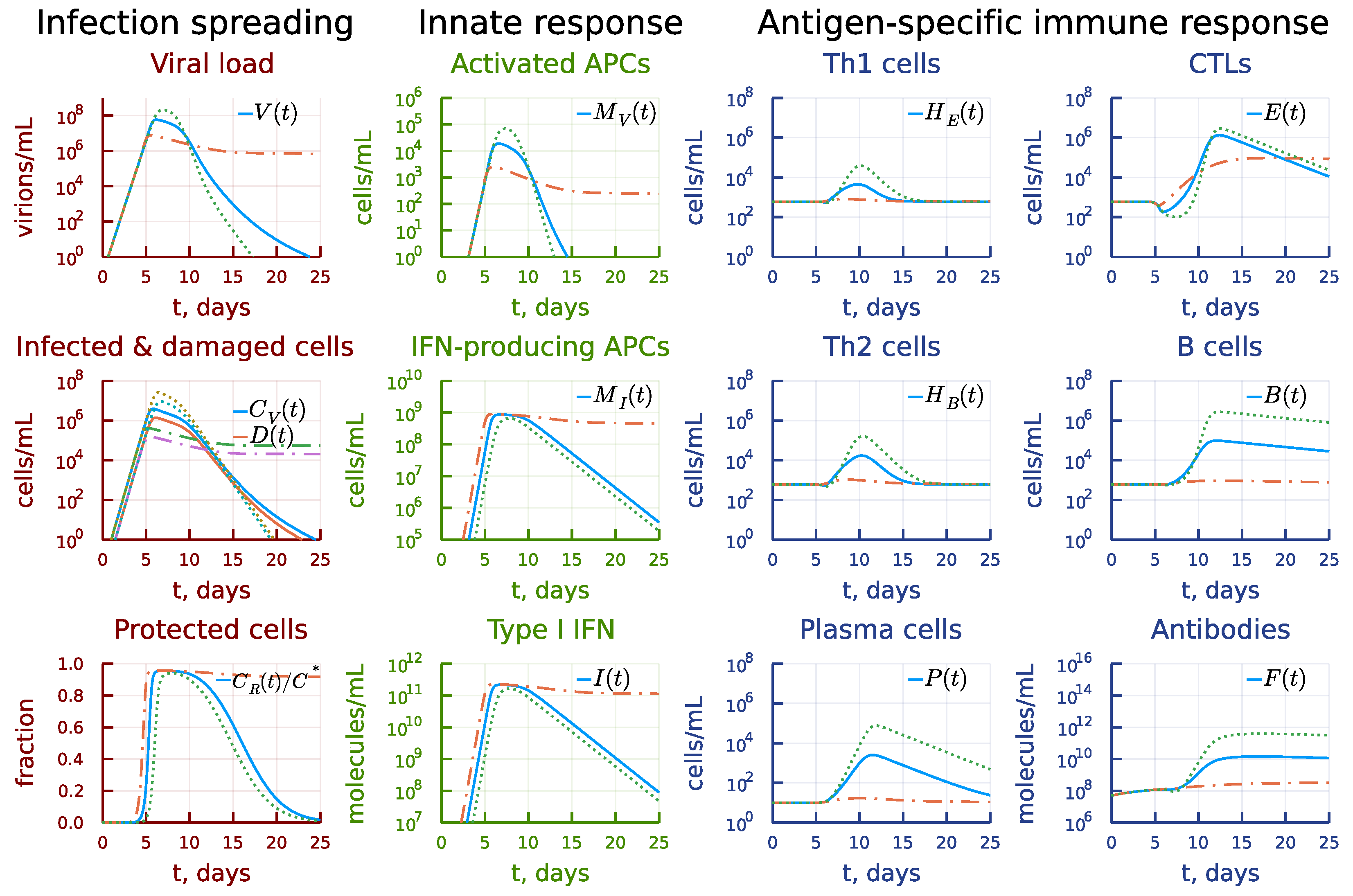
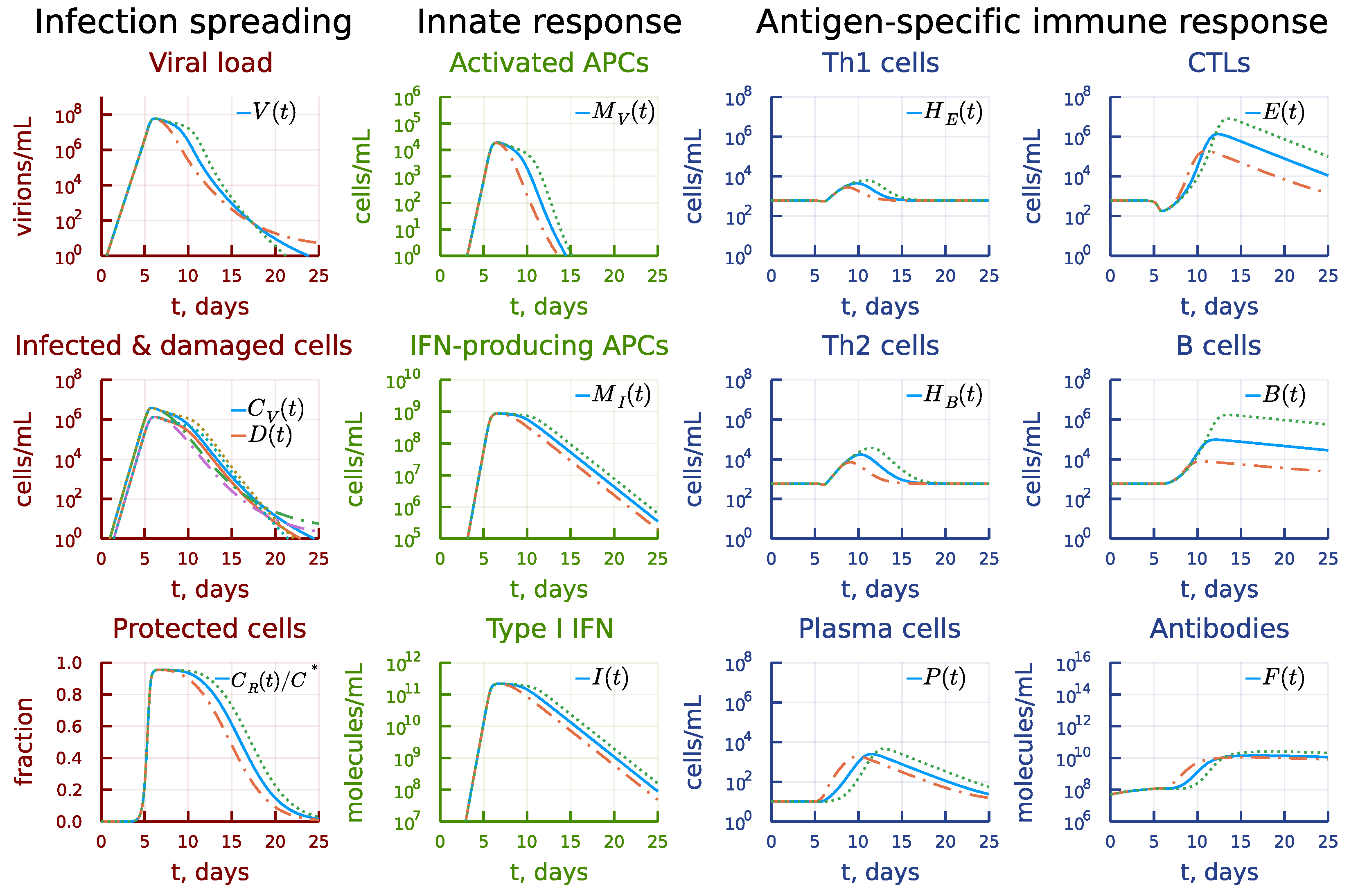
3.7. Kinetic Mechanisms of Long COVID-19 Pathogenesis
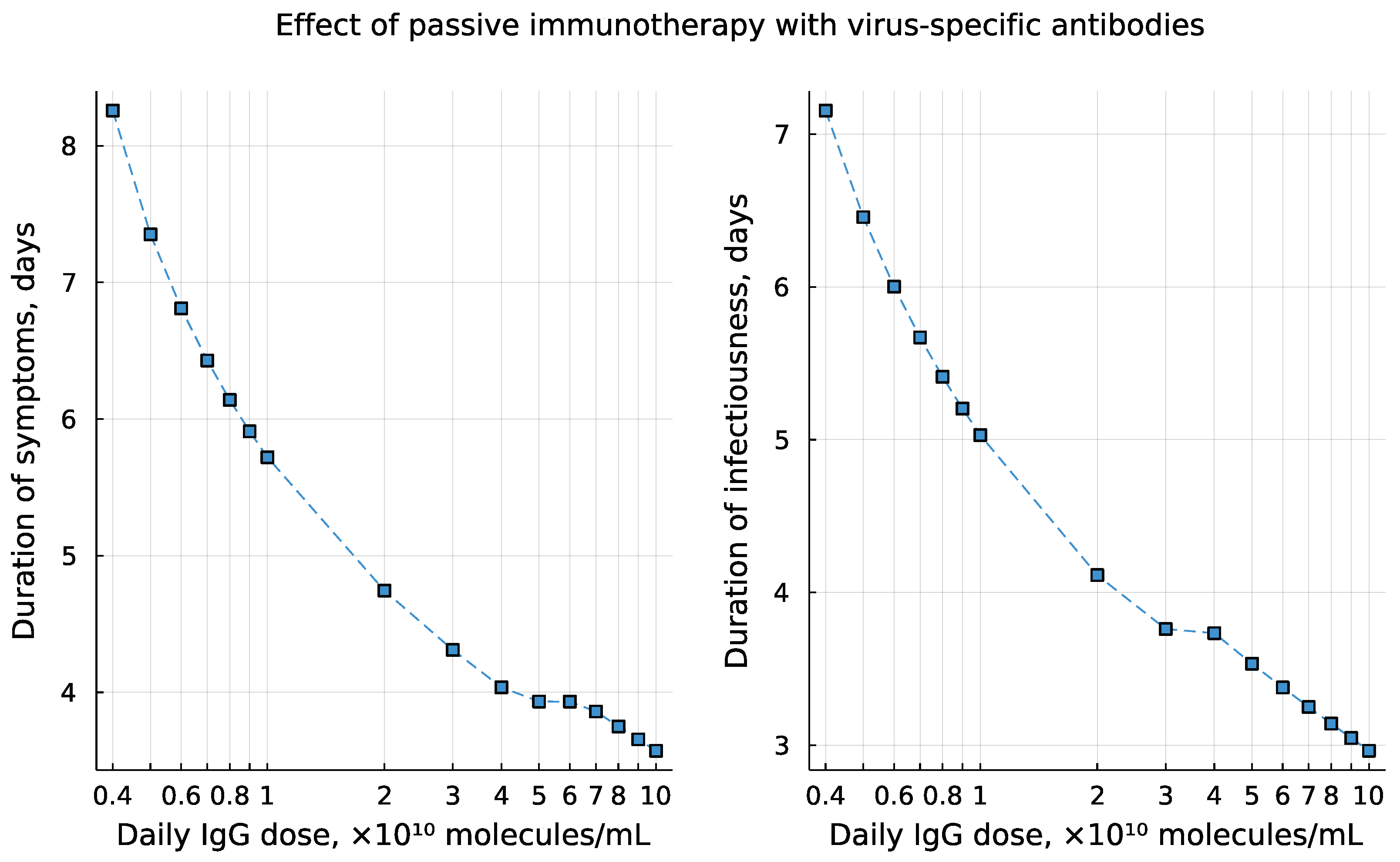
3.8. Individual’s Infectiousness
3.9. Day-by-Day Use of the Model
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus |
| ODE | Ordinary differential equations |
| COVID-19 | Infectious disease caused by SARS-CoV-2 |
| IFN-I | Type I interferon |
References
- Bar-On, Y.M.; Flamholz, A.; Phillips, R.; Milo, R. SARS-CoV-2 (COVID-19) by the numbers. eLife 2020, 9, e57309. [Google Scholar] [CrossRef] [PubMed]
- Ostaszewski, M.; Niarakis, A.; Mazein, A.; Kuperstein, I.; Phair, R.; Orta-Resendiz, A.; Singh, V.; Aghamiri, S.S.; Acencio, M.L.; Glaab, E.; et al. COVID19 Disease Map, a computational knowledge repository of virus-host interaction mechanisms. Mol. Syst. Biol. 2021, 17, e10387, Erratum in Mol Syst Biol. 2021, 17, e10851. [Google Scholar] [CrossRef]
- Du, S.Q.; Yuan, W. Mathematical modeling of interaction between innate and adaptive immune responses in COVID-19 and implications for viral pathogenesis. J. Med. Virol. 2020, 92, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Pan, Y.; Wang, Q.; Miao, H.; Brown, A.N.; Rong, L. Modeling the viral dynamics of SARS-CoV-2 infection. Math. Biosci. 2020, 328, 108438. [Google Scholar] [CrossRef]
- Hernandez-Vargas, E.A.; Velasco-Hernandez, J.X. In-host Mathematical Modelling of COVID-19 in Humans. Annu. Rev. Control 2020, 50, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Chimal-Eguia, J.C. Mathematical Model of Antiviral Immune Response against the COVID-19 Virus. Mathematics 2021, 9, 1356. [Google Scholar] [CrossRef]
- Rodriguez, T.; Dobrovolny, H.M. Estimation of viral kinetics model parameters in young and aged SARS-CoV-2 infected macaques. R. Soc. Open Sci. 2021, 8, 202345. [Google Scholar] [CrossRef]
- Sadria, M.; Layton, A.T. Modeling within-Host SARS-CoV-2 Infection Dynamics and Potential Treatments. Viruses 2021, 13, 1141. [Google Scholar] [CrossRef]
- Fatehi, F.; Bingham, R.J.; Dykeman, E.C.; Stockley, P.G.; Twarock, R. Comparing antiviral strategies against COVID-19 via multiscale within-host modelling. R. Soc. Open Sci. 2021, 8, 210082. [Google Scholar] [CrossRef]
- Voutouri, C.; Nikmaneshi, M.R.; Hardin, C.C.; Patel, A.B.; Verma, A.; Khandekar, M.J.; Dutta, S.; Stylianopoulos, T.; Munn, L.L.; Jain, R.K. In silico dynamics of COVID-19 phenotypes for optimizing clinical management. Proc. Natl. Acad. Sci. USA 2021, 118, e2021642118. [Google Scholar] [CrossRef]
- Blanco-Rodríguez, R.; Du, X.; Hernández-Vargas, E. Computational simulations to dissect the cell immune response dynamics for severe and critical cases of SARS-CoV-2 infection. Comput. Methods Programs Biomed. 2021, 211, 106412. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I. Within Host Dynamics of SARS-CoV-2 in Humans: Modeling Immune Responses and Antiviral Treatments. SN Comput. Sci. 2021, 2, 482. [Google Scholar] [CrossRef]
- Jenner, A.L.; Aogo, R.A.; Alfonso, S.; Crowe, V.; Deng, X.; Smith, A.P.; Morel, P.A.; Davis, C.L.; Smith, A.M.; Craig, M. COVID-19 virtual patient cohort suggests immune mechanisms driving disease outcomes. PLoS Pathog. 2021, 17, e1009753. [Google Scholar] [CrossRef] [PubMed]
- Mochan, E.; Sego, T.J.; Gaona, L.; Rial, E.; Ermentrout, G.B. Compartmental Model Suggests Importance of Innate Immune Response to COVID-19 Infection in Rhesus Macaques. Bull. Math. Biol. 2021, 83, 79. [Google Scholar] [CrossRef] [PubMed]
- Mondal, J.; Samui, P.; Chatterjee, A.N. Dynamical demeanour of SARS-CoV-2 virus undergoing immune response mechanism in COVID-19 pandemic. Eur. Phys. J. Spec. Top. 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rana, P.; Chauhan, S.; Mubayi, A. Burden of cytokines storm on prognosis of SARS-CoV-2 infection through immune response: Dynamic analysis and optimal control with immunomodulatory therapy. Eur. Phys. J. Spec. Top. 2022, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Marzban, S.; Han, R.; Juhász, N.; Röst, G. A hybrid PDE-ABM model for viral dynamics with application to SARS-CoV-2 and influenza. R. Soc. Open Sci. 2021, 8, 210787. [Google Scholar] [CrossRef]
- Afonyushkin, V.N.; Akberdin, I.R.; Kozlova, Y.N.; Schukin, I.A.; Mironova, T.E.; Bobikova, A.S.; Cherepushkina, V.S.; Donchenko, N.A.; Poletaeva, Y.E.; Kolpakov, F.A. Multicompartmental Mathematical Model of SARS-CoV-2 Distribution in Human Organs and Their Treatment. Mathematics 2022, 10, 1925. [Google Scholar] [CrossRef]
- Getz, M.; Wang, Y.; An, G.; Asthana, M.; Becker, A.; Cockrell, C.; Collier, N.; Craig, M.; Davis, C.L.; Faeder, J.R.; et al. Iterative community-driven development of a SARS-CoV-2 tissue simulator. bioRxiv 2021. [Google Scholar] [CrossRef]
- Alexandre, M.; Marlin, R.; Prague, M.; Coleon, S.; Kahlaoui, N.; Cardinaud, S.; Naninck, T.; Delache, B.; Surenaud, M.; Galhaut, M.; et al. Modelling the response to vaccine in non-human primates to define SARS-CoV-2 mechanistic correlates of protection. eLife 2022, 11, e75427. [Google Scholar] [CrossRef]
- Ke, R.; Zitzmann, C.; Ho, D.D.; Ribeiro, R.M.; Perelson, A.S. In vivo kinetics of SARS-CoV-2 infection and its relationship with a person’s infectiousness. Proc. Natl. Acad. Sci. USA 2021, 118, e2111477118. [Google Scholar] [CrossRef] [PubMed]
- Grossman, Z.; Paul, W.E. Dynamic tuning of lymphocytes: Physiological basis, mechanisms, and function. Annu. Rev. Immunol. 2015, 33, 677–713. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, G.A.; Romanyukha, A.A. Mathematical model of antiviral immune response. III. Influenza A virus infection. J. Theor. Biol. 1994, 167, 323–360. [Google Scholar] [CrossRef]
- Bocharov, G.; Grebennikov, D.; Argilaguet, J.; Meyerhans, A. Examining the cooperativity mode of antibody and CD8+ T cell immune responses for vaccinology. Trends Immunol. 2021, 42, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Killingley, B.; Mann, A.J.; Kalinova, M.; Boyers, A.; Goonawardane, N.; Zhou, J.; Lindsell, K.; Hare, S.S.; Brown, J.; Frise, R.; et al. Safety, tolerability and viral kinetics during SARS-CoV-2 human challenge in young adults. Nat. Med. 2022, 28, 1031–1041. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e14. [Google Scholar] [CrossRef]
- Mettelman, R.C.; Allen, E.K.; Thomas, P.G. Mucosal immune responses to infection and vaccination in the respiratory tract. Immunity 2022, 55, 749–780. [Google Scholar] [CrossRef]
- Wiech, M.; Chroscicki, P.; Swatler, J.; Stepnik, D.; De Biasi, S.; Hampel, M.; Brewinska-Olchowik, M.; Maliszewska, A.; Sklinda, K.; Durlik, M.; et al. Remodeling of T Cell Dynamics During Long COVID Is Dependent on Severity of SARS-CoV-2 Infection. Front. Immunol. 2022, 13, 886431. [Google Scholar] [CrossRef]
- Zuin, J.; Fogar, P.; Musso, G.; Padoan, A.; Piva, E.; Pelloso, M.; Tosato, F.; Cattelan, A.; Basso, D.; Plebani, M. T Cell Senescence by Extensive Phenotyping: An Emerging Feature of COVID-19 Severity. Lab. Med. 2022, lmac048. [Google Scholar] [CrossRef]
- Cheemarla, N.R.; Watkins, T.A.; Mihaylova, V.T.; Wang, B.; Zhao, D.; Wang, G.; Landry, M.L.; Foxman, E.F. Dynamic innate immune response determines susceptibility to SARS-CoV-2 infection and early replication kinetics. J. Exp. Med. 2021, 218, e20210583. [Google Scholar] [CrossRef]
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.L.; Chia, A.; Smith, G.J.D.; et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021, 34, 108728. [Google Scholar] [CrossRef] [PubMed]
- Beule, A.G. Physiology and Pathophysiology of Respiratory Mucosa of the Nose and the Paranasal Sinuses. GMS Curr. Top. Otorhinolaryngol.—Head Neck Surg. 2010, 9, Doc07. [Google Scholar] [CrossRef] [PubMed]
- Ritthidej, G.C. Nasal Delivery of Peptides and Proteins with Chitosan and Related Mucoadhesive Polymers. In Peptide and Protein Delivery; Elsevier: Amsterdam, The Netherlands, 2011; pp. 47–68. ISBN 9780123849359. [Google Scholar]
- Zinkernagel, R.M.; Hengartner, H. On immunity against infections and vaccines: Credo 2004. Scand. J. Immunol. 2004, 60, 9–13, Erratum in Scand. J. Immunol. 2004, 60, 327. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Weaver, C. Janeway’s Immunobiology, 9th ed.; Garland Science, Taylor and Francis Group, LLC: New York, NY, USA, 2017; 924p, ISBN 978-0-8153-4505-3. [Google Scholar]
- Deem, M.W.; Hejazi, P. Theoretical aspects of immunity. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 247–276. [Google Scholar] [CrossRef] [PubMed]
- Perelson, A.S.; Weisbuch, G. Immunology for physicists. Rev. Mod. Phys. 1997, 69, 1219–1268. [Google Scholar] [CrossRef]
- Cosgrove, J.; Hustin, L.S.P.; de Boer, R.J.; Perié, L. Hematopoiesis in numbers. Trends Immunol. 2021, 42, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Grebennikov, D.; Kholodareva, E.; Sazonov, I.; Karsonova, A.; Meyerhans, A.; Bocharov, G. Intracellular Life Cycle Kinetics of SARS-CoV-2 Predicted Using Mathematical Modelling. Viruses 2021, 13, 1735. [Google Scholar] [CrossRef]
- Sazonov, I.; Grebennikov, D.; Meyerhans, A.; Bocharov, G. Sensitivity of SARS-CoV-2 Life Cycle to IFN Effects and ACE2 Binding Unveiled with a Stochastic Model. Viruses 2022, 14, 403. [Google Scholar] [CrossRef]
- Kim, K.S.; Ejima, K.; Iwanami, S.; Fujita, Y.; Ohashi, H.; Koizumi, Y.; Asai, Y.; Nakaoka, S.; Watashi, K.; Aihara, K.; et al. A quantitative model used to compare within-host SARS-CoV-2, MERS-CoV, and SARS-CoV dynamics provides insights into the pathogenesis and treatment of SARS-CoV-2. PLoS Biol. 2021, 19, e3001128. [Google Scholar] [CrossRef]
- Marino, S.; Hogue, I.B.; Ray, C.J.; Kirschner, D.E. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J. Theor. Biol. 2008, 254, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef] [PubMed]
- Skevaki, C.; Karsonova, A.; Karaulov, A.; Fomina, D.; Xie, M.; Chinthrajah, S.; Nadeau, K.C.; Renz, H. SARS-CoV-2 infection and COVID-19 in asthmatics: A complex relationship. Nat. Rev. Immunol. 2021, 21, 202–203. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Yang, J.; Deng, H.; Chen, D.; Yang, X.; Tang, Z.H. Depletion and Dysfunction of Dendritic Cells: Understanding SARS-CoV-2 Infection. Front. Immunol. 2022, 13, 843342. [Google Scholar] [CrossRef] [PubMed]
- Farhangnia, P.; Dehrouyeh, S.; Safdarian, A.R.; Farahani, S.V.; Gorgani, M.; Rezaei, N.; Akbarpour, M.; Delbandi, A.A. Recent advances in passive immunotherapies for COVID-19: The Evidence-Based approaches and clinical trials. Int. Immunopharmacol. 2022, 109, 108786. [Google Scholar] [CrossRef] [PubMed]
- Deere, J.D.; Carroll, T.D.; Dutra, J.; Fritts, L.; Sammak, R.L.; Yee, J.L.; Olstad, K.J.; Reader, J.R.; Kistler, A.; Kamm, J.; et al. SARS-CoV-2 Infection of Rhesus Macaques Treated Early with Human COVID-19 Convalescent Plasma. Microbiol. Spectr. 2021, 9, e0139721. [Google Scholar] [CrossRef]
- Xiang, H.R.; Cheng, X.; Li, Y.; Luo, W.W.; Zhang, Q.Z.; Peng, W.X. Efficacy of IVIG (intravenous immunoglobulin) for corona virus disease 2019 (COVID-19): A meta-analysis. Int. Immunopharmacol. 2021, 96, 107732. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.A.; Habiballah, S.B.; Platt, C.D.; Geha, R.S.; Chou, J.S.; McDonald, D.R. Immunoglobulins in the treatment of COVID-19 infection: Proceed with caution! Clin. Immunol. 2020, 216, 108459. [Google Scholar] [CrossRef]
- Salehi, M.; Barkhori Mehni, M.; Akbarian, M.; Fattah Ghazi, S.; Khajavi Rad, N.; Moradi Moghaddam, O.; Jamali Moghaddam, S.; Hosseinzadeh Emam, M.; Abtahi, S.H.; Moradi, M.; et al. The outcome of using intravenous immunoglobulin (IVIG) in critically ill COVID-19 patients’: A retrospective, multi-centric cohort study. Eur. J. Med. Res. 2022, 27, 18. [Google Scholar] [CrossRef]
- Raoult, D.; Zumla, A.; Locatelli, F.; Ippolito, G.; Kroemer, G. Coronavirus infections: Epidemiological, clinical and immunological features and hypotheses. Cell Stress 2020, 4, 66–75. [Google Scholar] [CrossRef]
- Speranza, E.; Purushotham, J.N.; Port, J.R.; Schwarz, B.; Flagg, M.; Williamson, B.N.; Feldmann, F.; Singh, M.; Pérez-Pérez, L.; Sturdevant, G.L.; et al. Age-related differences in immune dynamics during SARS-CoV-2 infection in rhesus macaques. Life Sci. Alliance 2022, 5, e202101314. [Google Scholar] [CrossRef]
- Cao, S.; Zhang, Q.; Song, L.; Xiao, M.; Chen, Y.; Wang, D.; Li, M.; Hu, J.; Lin, L.; Zheng, Y.; et al. Dysregulation of Innate and Adaptive Immune Responses in Asymptomatic SARS-CoV-2 Infection with Delayed Viral Clearance. Int. J. Biol. Sci. 2022, 18, 4648–4657. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Clues to long COVID. Science 2022, 376, 1261–1265. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, D.; Chen, D.G.; Ng, R.H.; Wang, K.; Choi, J.; Li, S.; Hong, S.; Zhang, R.; Xie, J.; et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 2022, 185, 881–895.e20. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Deeks, S.G. Early clues regarding the pathogenesis of long-COVID. Trends Immunol. 2022, 43, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Manthiram, K.; Xu, Q.; Milanez-Almeida, P.; Martins, A.; Radtke, A.; Hoehn, K.; Chen, J.; Liu, C.; Tang, J.; Grubbs, G.; et al. Robust, persistent adaptive immune responses to SARS-CoV-2 in the oropharyngeal lymphoid tissue of children. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Wadman, M.; Couzin-Frankel, J.; Kaiser, J.; Matacic, C. A rampage through the body. Science 2020, 368, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Pertsev, N.; Loginov, K.; Lukashev, A.; Vakulenko, Y. Stochastic Modeling of Dynamics of the Spread of COVID-19 Infection Taking Into Account the Heterogeneity of Population According To Immunological, Clinical and Epidemiological Criteria. Math. Biol. Bioinform. 2022, 17, 43–81. (In Russian) [Google Scholar] [CrossRef]
- Simoneau, C.R.; Ott, M. Modeling Multi-organ Infection by SARS-CoV-2 Using Stem Cell Technology. Cell Stem Cell 2020, 27, 859–868. [Google Scholar] [CrossRef]
- Zinkernagel, R.M.; Hengartner, H.; Stitz, L. On the role of viruses in the evolution of immune responses. Br. Med. Bull. 1985, 41, 92–97. [Google Scholar] [CrossRef] [Green Version]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter, Units | Range, Initial Guess | Estimate | |
|---|---|---|---|
| Concentration of APCs, cells/mL | |||
| Concentration of IFN-producing APCs, cells/mL | |||
| Concentration of SARS-CoV-2 specific Th1 cells, cells/mL | 600 | ||
| Concentration of SARS-CoV-2 specific Th2 cells, cells/mL | 600 | ||
| Concentration of SARS-CoV-2 specific CTLs, cells/mL | 600 | ||
| Concentration of SARS-CoV-2 specific B cells, cells/mL | 600 | ||
| Concentration of SARS-CoV-2 specific plasma cells, cells/mL | 10 | ||
| Concentration of SARS-CoV-2 specific antibodies, molecules/mL | |||
| Concentration of epithelial cells, cells/mL | |||
| Rate of stimulated state loss for APCs, day−1 | |||
| Rate of activated state loss for Th1 cells, day−1 | 1 | ||
| Rate of activated state loss for Th2 cells, day−1 | 1 | ||
| Rate of natural death for CTLs, day−1 | |||
| Rate of natural death for B cells, day−1 | |||
| Rate of natural death for plasma cells, day−1 | |||
| Rate of natural death for antibodies, day−1 | |||
| Duration of Th1 cell division cycle, days | |||
| Duration of Th2 cell division cycle, days | |||
| Duration of CTL division cycle, days | |||
| Duration of B cell division cycle, days | |||
| Duration of B cell differentiation into plasma cells, days | |||
| Number of Th1 cells created during division cycle | 4 | ||
| Number of Th2 cells created during division cycle | 4 | ||
| Number of CTLs created during division cycle | 2 | ||
| Number of B cells in clone created by series of 1 or 2 divisions | 3 | ||
| Number of plasma cells in clone created by series of 1 or 2 divisions | 1 | ||
| Rate of IgG production per plasma cell, molecules/cell/day | |||
| Rate of Th1 cells stimulation, (cells/mL)−1day−1 | , | ||
| Rate of Th2 cells stimulation, (cells/mL)−1day−1 | , | ||
| Rate of CTL stimulation, (cells/mL)−2day−1 | , | ||
| Rate of B cell stimulation, (cells/mL)−2day−1 | , | ||
| Rate of plasma cell stimulation, (cells/mL)−2day−1 | , | ||
| Rate of Th1 cells suppression, (cells/mL)−2day−1 | |||
| Rate of Th2 cells suppression, (cells/mL)−2day−1 | |||
| Rate of APC stimulation, (cells/mL)−1day−1 | , | ||
| Rate of IgG binding to SARS-CoV-2, (virions/mL)−1day−1 | , | ||
| Rate of epithelial cell infection with SARS-CoV-2, (cells/mL)−1day−1 | , | ||
| Rate of infected epithelial cell damage by CTLs, (virions/mL)−1day−1 | , | ||
| Rate of CTL death due to lytic interactions with infected cells, (cells/mL)−1day−1 | |||
| Rate of infected cell damage due to SARS-CoV-2 cytopathicity, day−1 | |||
| Rate of epithelial cell regeneration, day−1 | 4 | ||
| Rate of SARS-CoV-2 virions secretion per infected epithelial cell, day−1 | , 130 | 144 | |
| Rate of SARS-CoV-2 absorption by epithelial cell, (cells/mL)−1day−1 | , | ||
| Rate of nonspecific SARS-CoV-2 elimination, day−1 | , | 4 | |
| Rate of SARS-CoV-2 neutralization by specific IgG, (virions/mL)−1day−1 | , | ||
| Parameter for inflammation-based enhancement of IgG effect | , 1000 | 2628 | |
| Parameter for inflammation-based enhancement of CTL effect | , 1000 | 1407 | |
| Rate of induction of IFN-producing state in APCs, (cells/mL)−1day−1 | , | ||
| Rate of IFN-producing state loss by APCs, day−1 | |||
| Rate of IFN production per IFN-producing cells, molecules/cell/day | 6000 | ||
| Type I IFN clearance rate, day−1 | 24 | ||
| Rate of IFN binding with epithelial cells, (cells/mL)−1day−1 | , | ||
| Rate of virus-resistant state induction in epithelial cells, (cells/mL)−1day−1 | , | ||
| Rate of virus-resistant state loss in epithelial cells, day−1 | 1 |
| Parameter | ||||||||||
| Variation | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grebennikov, D.; Karsonova, A.; Loguinova, M.; Casella, V.; Meyerhans, A.; Bocharov, G. Predicting the Kinetic Coordination of Immune Response Dynamics in SARS-CoV-2 Infection: Implications for Disease Pathogenesis. Mathematics 2022, 10, 3154. https://doi.org/10.3390/math10173154
Grebennikov D, Karsonova A, Loguinova M, Casella V, Meyerhans A, Bocharov G. Predicting the Kinetic Coordination of Immune Response Dynamics in SARS-CoV-2 Infection: Implications for Disease Pathogenesis. Mathematics. 2022; 10(17):3154. https://doi.org/10.3390/math10173154
Chicago/Turabian StyleGrebennikov, Dmitry, Antonina Karsonova, Marina Loguinova, Valentina Casella, Andreas Meyerhans, and Gennady Bocharov. 2022. "Predicting the Kinetic Coordination of Immune Response Dynamics in SARS-CoV-2 Infection: Implications for Disease Pathogenesis" Mathematics 10, no. 17: 3154. https://doi.org/10.3390/math10173154