Unveiling the Molecular Footprint: Proteome-Based Biomarkers for Alzheimer’s Disease

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
1.1. Importance of Biomarkers in AD Diagnosis and Management
1.2. Role of Proteomic Identifying Biomarkers
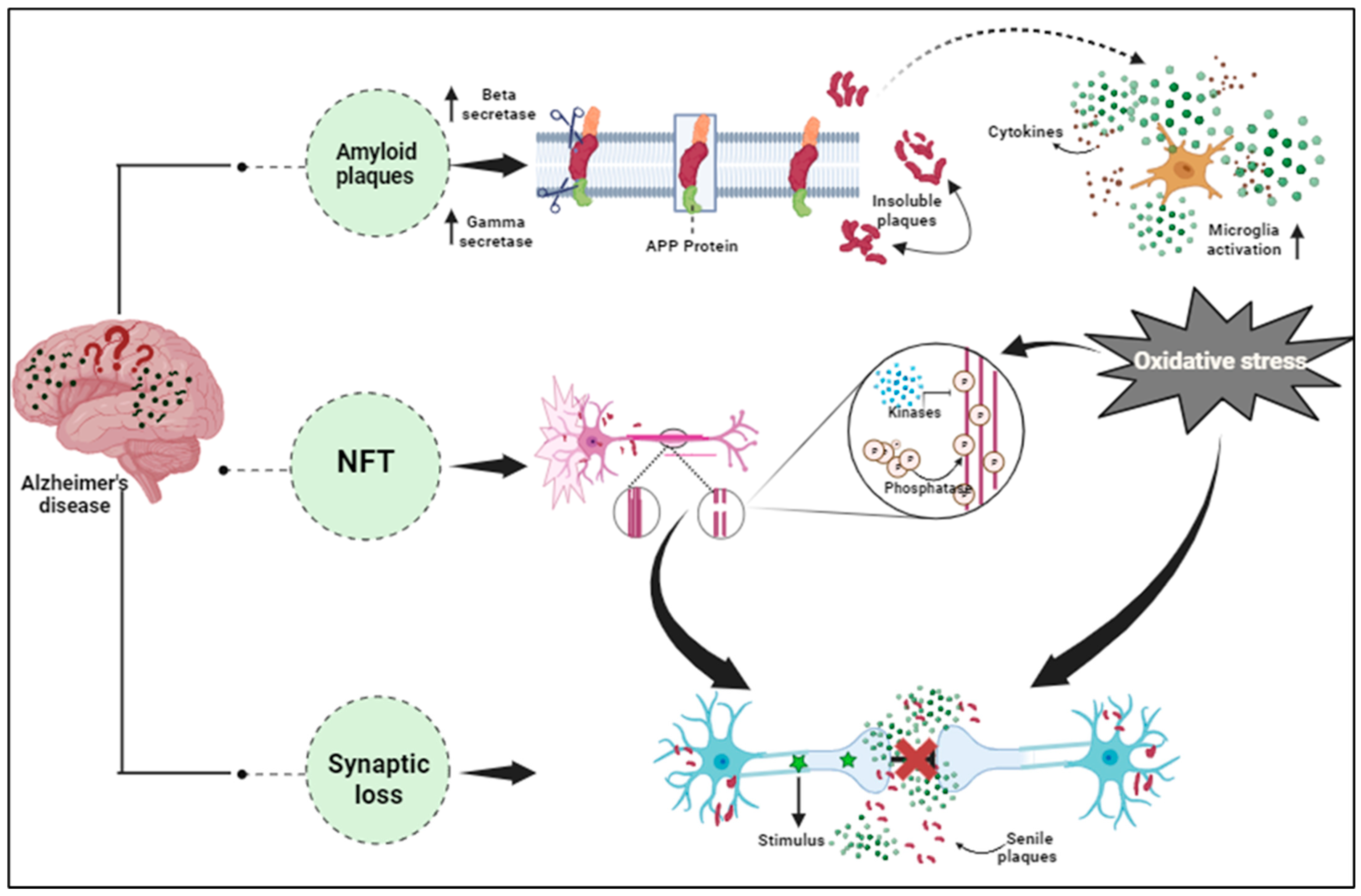
2. Alzheimer’s Disease: Pathophysiology and Proteomic Approaches
2.1. Overview of AD: Pathology and Molecular Mechanism
2.2. Introduction to Proteomics and Its Application in AD Research
2.3. Challenges in Identifying Reliable Biomarkers
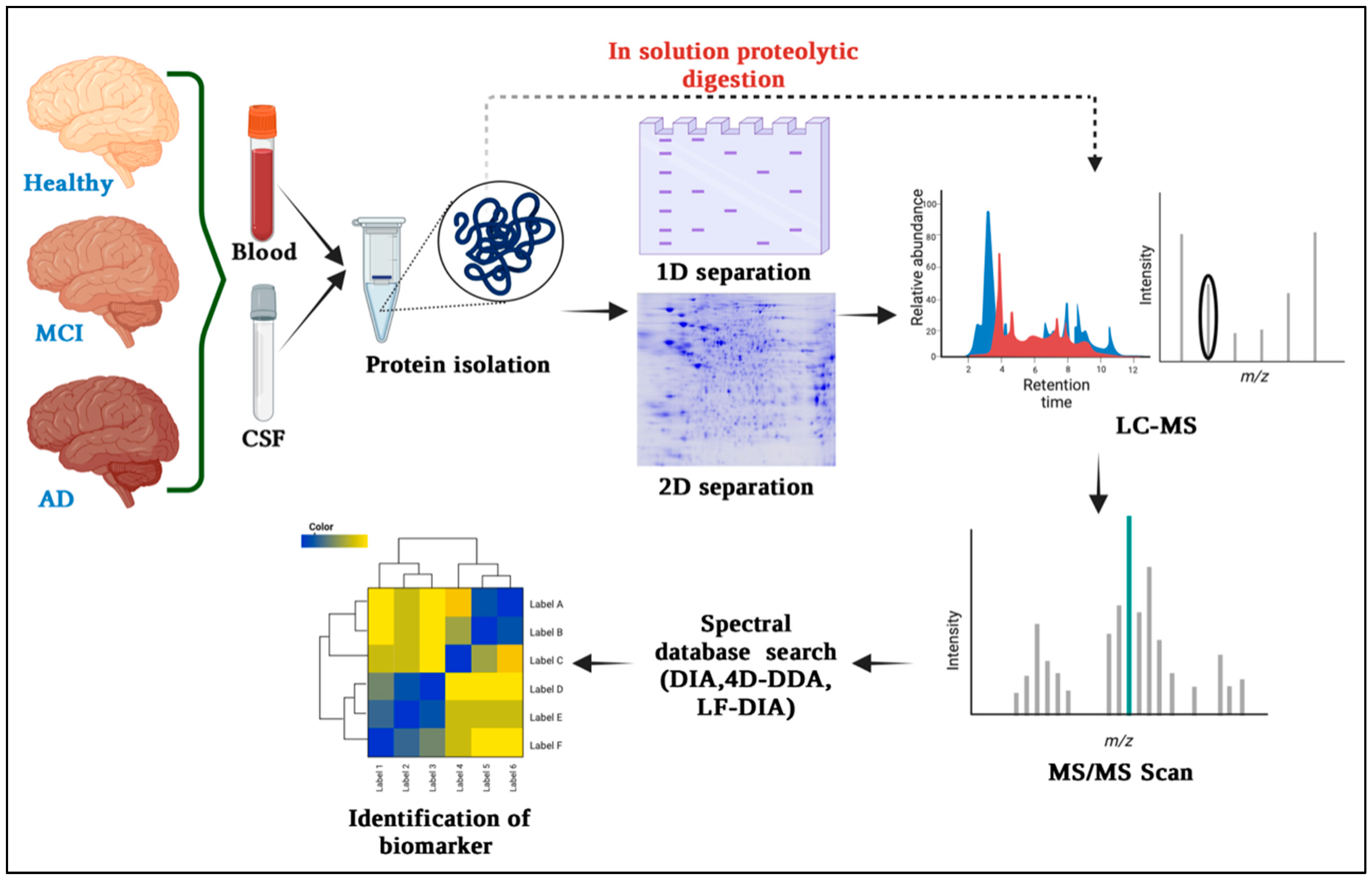
3. Proteomics Techniques for Biomarker Discovery in Alzheimer’s Disease
3.1. Gel-Based Quantitative Technique and Differential Proteomics
3.2. Mass Spectrometry (MS) for Protein Identification and Quantification
3.3. Protein Microarrays and Antibody-Based Techniques
3.4. Advancements in High-Throughput Proteomics Technologies
4. Candidate Proteome-Based Biomarkers for Alzheimer’s Disease
4.1. Amyloid-Beta (Aβ) Peptides and Tau Protein
4.2. Apolipoprotein E (APOE)
4.3. Clusterin (CLU) and Other Chaperone Proteins
4.4. Inflammatory Markers and Complement Proteins
4.5. Other Potential Proteomic Biomarkers for AD
5. Validation and Clinical Utility of Proteome-Based Biomarkers
- Sample accumulation;
- Parallelism;
- Range finding;
- Relative precision and accuracy;
- Specificity and stability.
5.1. Challenges in the Validation of Proteomic Biomarkers
5.2. Clinical Studies and Diagnostic Accuracy of Proteomic Biomarkers
5.3. Monitoring Disease Progression and Response to Treatment
5.4. Future Prospects and Integration with Other Diagnostic Approaches
6. Bioinformatics and Data Analysis in Proteomics for AD Biomarkers
- (i)
- Pre-mass spectrometry (MS) analysis;
- (ii)
- Acquisition of MS data;
- (iii)
- Post-MS Bioinformatics data processing.
- (a)
- Protein Identification;
- (b)
- Protein Quantification;
- (c)
- Statistical Classification;
- (d)
- Differential Expression;
- (e)
- Network/Pathway Analysis;
- (f)
- Integration of Multi-Omics;
- (g)
- Hypotheses Generation.
6.1. Data Pre-Processing and Quality Control
6.2. Statistical Analysis and Identification of Significant Biomarkers
- (a)
- They should be notably modified in the diseased patients as compared to the control group;
- (b)
- Assessment of the diagnostic attributes of biomarkers;
- (c)
- Comparison between the contemporary diagnostic tests and the concerned biomarkers;
- (d)
- Evaluation of the quality of biomarkers, for instance, assessing rapidity, invasiveness, cost, technical challenges, et cetera.
6.2.1. Decision Matrix
6.2.2. Likelihood Ratios
6.2.3. ROC Curve
6.3. Pathway Analysis and Functional Annotation
- (a)
- Recognizing the non-coding parts of protein chains;
- (b)
- Carrying out gene prediction;
- (c)
- Affixing biological information to this gathered data.
6.4. Integration of Multi-Omics Data for Systems-Level Understanding
7. Discussion
8. Limitations and Future Directions in AD Biomarker Research
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Cummings, J.L.; Dekosky, S.T.; Barberger-Gateau, P.; Delacourte, A.; Frisoni, G.; Fox, N.C.; Galasko, D.; et al. Revising the definition of Alzheimer’s disease: A new lexicon. Lancet Neurol. 2010, 9, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.d.C.; ElAkkad, E.; Gong, C.; Liu, F.; Tanaka, T.; Kudo, T.; Tatebayashi, Y.; Pei, J.; Wang, J.; Khatoon, S.; et al. Inge Grundke-Iqbal, Ph.D. (1937–2012): The discoverer of the abnormal hyperphosphorylation of tau in Alzheimer’s disease. J. Mol. Neurosci. 2013, 49, 430–435. [Google Scholar] [CrossRef]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-β/Fyn-induced synaptic, network and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Xu, J.; Chatterjee, M.; Baguley, T.D.; Brouillette, J.; Kurup, P.; Ghosh, D.; Kanyo, J.; Zhang, Y.; Seyb, K.; Ononenyi, C.; et al. Inhibitor of the tyrosine phosphatase STEP reverses cognitive deficits in a mouse model of Alzheimer’s disease. PLoS Biol. 2014, 12, e1001923. [Google Scholar] [CrossRef]
- Basir, R.; Hasballah, K.; Jabbarzare, M.; Gam, L.H.; Abdul Majid, A.M.; Yam, M.F.; Moklas, M.A.; Othman, F.; Che Norma, M.T.; Zalinah, A.; et al. Modulation of interleukin-18 release produced positive outcomes on parasitaemia development and cytokines production during malaria in mice. Trop. Biomed. 2012, 29, 405–421. [Google Scholar]
- Srinivas, P.R.; Verma, M.; Zhao, Y.; Srivastava, S. Proteomics for cancer biomarker discovery. Clin. Chem. 2002, 48, 1160–1169. [Google Scholar]
- Amiri-Dashatan, N.; Koushki, M.; Abbaszadeh, H.A.; Rostami-Nejad, M.; Rezaei-Tavirani, M. Proteomics Applications in Health: Biomarker and Drug Discovery and Food Industry. Iran. J. Pharm. Res. 2018, 17, 1523–1536. [Google Scholar]
- Eyjolfsdottir, H.; Eriksdotter, M.; Linderoth, B.; Lind, G.; Juliusson, B.; Kusk, P.; Almkvist, O.; Andreasen, N.; Blennow, K.; Ferreira, D.; et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: Application of a second-generation encapsulated cell biodelivery device. Alzheimer’s Res. Ther. 2016, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Mirmosayyeb, O.; Tanhaei, A.; Sohrabi, H.R.; Martins, R.N.; Tanhaei, M.; Najafi, M.A.; Safaei, A.; Meamar, R. Possible Role of Common Spices as a Preventive and Therapeutic Agent for Alzheimer’s Disease. Int. J. Prev. Med. 2017, 8, 5. [Google Scholar] [PubMed]
- Zali, H.; Zamanian-Azodi, M.; Rezaei Tavirani, M.; Akbar-Zadeh Baghban, A. Protein Drug Targets of Lavandula angustifolia on treatment of Rat Alzheimer’s Disease. Iran. J. Pharm. Res. 2015, 14, 291–302. [Google Scholar] [PubMed]
- Karbalaei, R.; Allahyari, M.; Rezaei-Tavirani, M.; Asadzadeh-Aghdaei, H.; Zali, M.R. Protein-protein interaction analysis of Alzheimer’s disease and NAFLD based on systems biology methods unhide common ancestor pathways. Gastroenterol. Hepatol. Bed Bench 2018, 11, 27–33. [Google Scholar] [PubMed]
- Lei, P.; Ayton, S.; Bush, A.I. The essential elements of Alzheimer’s disease. J. Biol. Chem. 2021, 296, 100105. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Golembiewski, J.A.; Zeisel, J. Salutogenic Approaches to Dementia Care. In The Handbook of Salutogenesis, 2nd ed.; Springer: Cham, Switzerland, 2022; pp. 513–532. [Google Scholar]
- Goate, A.; Hardy, J. Twenty years of Alzheimer’s disease-causing mutations. J. Neurochem. 2012, 1, 3–8. [Google Scholar] [CrossRef]
- Xia, D.; Watanabe, H.; Wu, B.; Lee, S.H.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y.; Shen, J.; Kelleher, R.J. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 2015, 85, 967–981. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940.e7. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Masters, C.L.; Multhaup, G.; Simms, G.; Pottgiesser, J.; Martins, R.N.; Beyreuther, K. Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985, 4, 2757–2763. [Google Scholar] [CrossRef] [PubMed]
- Goldgaber, D.; Lerman, M.I.; McBride, O.W.; Saffiotti, U.; Gajdusek, D.C. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 1987, 235, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Kumar, D.; Aditi, S.; Lalit, S. A Comprehensive Review of Alzheimer’s Association with Related Proteins: Pathological Role and Therapeutic Significance. Curr. Neuropharmacol. 2020, 18, 674–695. [Google Scholar] [CrossRef]
- Hodes, R.J.; Buckholtz, N. Accelerating Medicines Partnership: Alzheimer’s Disease (AMP-AD) Knowledge Portal Aids Alzheimer’s Drug Discovery through Open Data Sharing. Expert Opin. Ther. Targets 2016, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 106, 700. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Lv, X.; Fan, Z.; Zhang, M.; Wang, H.; Yu, X. Association of Odor Identification Ability with Amyloid-β and Tau Burden: A Systematic Review and Meta-Analysis. Front. Neurosci. 2020, 14, 586330. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Petzold, A.; Bennett, J.L.; Berven, F.S.; Brundin, L.; Comabella, M.; Franciotta, D.; Frederiksen, J.L.; Fleming, J.O.; Furlan, R.; et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology 2009, 73, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Verwey, N.A.; van der Flier, W.M.; Blennow, K.; Clark, C.; Sokolow, S.; De Deyn, P.P.; Galasko, D.; Hampel, H.; Hartmann, T.; Kapaki, E.; et al. A worldwide multicentre comparison of assays for cerebrospinal fluid biomarkers in Alzheimer’s disease. Ann. Clin. Biochem. 2009, 46 Pt 3, 235–240. [Google Scholar] [CrossRef]
- Garfin, D.E. One-dimensional gel electrophoresis. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1990; Volume 182, pp. 425–441. [Google Scholar]
- Lee, P.Y.; Saraygord-Afshari, N.; Low, T.Y. The evolution of two-dimensional gel electrophoresis-from proteomics to emerging alternative applications. J. Chromatogr. A 2020, 1615, 460763. [Google Scholar] [CrossRef]
- Shevchenko, G.; Konzer, A.; Musunuri, S.; Bergquist, J. Neuroproteomics tools in clinical practice. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2015, 1854, 705–717. [Google Scholar] [CrossRef]
- Friedman, D.B.; Hoving, S.; Westermeier, R. Isoelectric focusing and two-dimensional gel electrophoresis. Methods Enzymol. 2009, 463, 515–540. [Google Scholar]
- Santoni, V.; Molloy, M.; Rabilloud, T. Membrane proteins and proteomics: Un amour impossible? Electrophor. Int. J. 2000, 21, 1054–1070. [Google Scholar] [CrossRef]
- Rabilloud, T. Use of thiourea to increase the solubility of membrane proteins in two-dimensional electrophoresis. Electrophoresis 1998, 19, 758–760. [Google Scholar] [CrossRef]
- Yan, J.X.; Kett, W.C.; Herbert, B.R.; Gooley, A.A.; Packer, N.H.; Williams, K.L. Identification and quantitation of cysteine in proteins separated by gel electrophoresis. J. Chromatogr. A 1998, 813, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Görg, A.; Klaus, A.; Lück, C.; Weiland, F.; Weiss, W. Two-dimensional electrophoresis with immobilized pH gradients for proteome analysis. A Lab. Man. 2007, 1855, 125–129. [Google Scholar]
- Craft, G.E.; Chen, A.; Nairn, A.C. Recent advances in quantitative neuroproteomics. Methods 2013, 61, 186–218. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Erwin, M.S.; Morgan, D.; Roberts, D.C.; Vrana, K.E.; Freeman, W.M. Persistent proteomic alterations in the medial prefrontal cortex with abstinence from cocaine self-administration. Proteom. Clin. Appl. 2009, 3, 462. [Google Scholar] [CrossRef]
- Naseri, N.N.; Ergel, B.; Kharel, P.; Na, Y.; Huang, Q.; Huang, R.; Dolzhanskaya, N.; Burré, J.; Velinov, M.T.; Sharma, M. Aggregation of mutant cysteine string protein-α via Fe–S cluster binding is mitigated by iron chelators. Nat. Struct. Mol. Biol. 2020, 27, 192–201. [Google Scholar] [CrossRef]
- Freeman, W.M.; Hemby, S.E. Proteomics for protein expression profiling in neuroscience. Neurochem. Res. 2004, 29, 1065–1081. [Google Scholar] [CrossRef]
- Baggerman, G.; Vierstraete, E.; De Loof, A.; Schoofs, L. Gel-based versus gel-free proteomics: A review. Comb. Chem. High Throughput Screen. 2005, 8, 669–677. [Google Scholar] [CrossRef]
- Thiede, B.; Höhenwarter, W.; Krah, A.; Mattow, J.; Schmid, M.; Schmidt, F.; Jungblut, P.R. Peptide mass fingerprinting. Methods 2005, 35, 237–247. [Google Scholar] [CrossRef]
- Háda, V.; Bagdi, A.; Bihari, Z.; Timári, S.B.; Fizil, Á.; Szántay, C., Jr. Recent advancements, challenges, and practical considerations in the mass spectrometry-based analytics of protein biotherapeutics: A viewpoint from the biosimilar industry. J. Pharm. Biomed. Anal. 2018, 161, 214–238. [Google Scholar] [CrossRef]
- Stump, M.J.; Fleming, R.C.; Gong, W.H.; Jaber, A.J.; Jones, J.J.; Surber, C.W.; Wilkins, C.L. Matrix-assisted laser desorption mass spectrometry. Appl. Spectrosc. Rev. 2002, 37, 275–303. [Google Scholar] [CrossRef]
- Domon, B.; Aebersold, R. Mass spectrometry and protein analysis. Science 2006, 312, 212–217. [Google Scholar] [CrossRef]
- Blackstock, W.P.; Weir, M.P. Proteomics: Quantitative and physical mapping of cellular proteins. Trends Biotechnol. 1999, 17, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.; Müller, M.; Appel, R.D. Automated protein identification by tandem mass spectrometry: Issues and strategies. Mass Spectrom. Rev. 2006, 25, 235–254. [Google Scholar] [CrossRef]
- Villar-Garea, A.; Griese, M.; Imhof, A. Biomarker discovery from body fluids using mass spectrometry. J. Chromatogr. 2007, 849, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Palubeckaitė, I. Analysis of Three-Dimensional Cell Cultures Using Mass Spectrometry Imaging; Sheffield Hallam University: Sheffield, UK, 2018. [Google Scholar]
- Grela, A.; Turek, A.; Piekoszewski, W. Application of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) in Alzheimer’s disease. Clin. Chem. Lab. Med. 2012, 50, 1297–1304. [Google Scholar] [CrossRef]
- Korolainen, M. Proteomic Analysis of Post-Translationally Modified Proteins in Alzheimer’s Disease (Alzheimerin Taudin Post-Translationaalisesti Muunneltujen Proteiinien Kartoittaminen Proteomiikan Avulla); Kuopion Yliopisto: Kuopio, Finland, 2006. [Google Scholar]
- Sobek, J.; Bartscherer, K.; Jacob, A.; Hoheisel, J.D.; Angenendt, P. Microarray technology as a universal tool for high-throughput analysis of biological systems. Comb. Chem. High Throughput Screen. 2006, 9, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Hemby, S.E.; Lee, V.M.; Eberwine, J.H.; Trojanowski, J.Q. Expression profile of transcripts in Alzheimer’s disease tangle-bearing CA1 neurons. Ann. Neurol. 2000, 48, 77–87. [Google Scholar] [CrossRef]
- Housley, W.J.; Pitt, D.; Hafler, D.A. Biomarkers in multiple sclerosis. Clin. Immunol. 2015, 161, 51–58. [Google Scholar] [CrossRef]
- Ho, L.; Fivecoat, H.; Wang, J.; Pasinetti, G.M. Alzheimer’s disease biomarker discovery in symptomatic and asymptomatic patients: Experimental approaches and future clinical applications. Exp. Gerontol. 2010, 45, 15–22. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 1–22. [Google Scholar]
- Levites, Y.; Das, P.; Price, R.W.; Rochette, M.J.; Kostura, L.A.; McGowan, E.M.; Murphy, M.P.; Golde, T.E. Anti-Aβ42- and anti-Aβ40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2006, 116, 193–201. [Google Scholar] [CrossRef]
- Seubert, P.; Oltersdorf, T.; Lee, M.G.; Barbour, R.; Blomquist, C.; Davis, D.L.; Bryant, K.; Fritz, L.C.; Galasko, D.; Thal, L.J.; et al. Secretion of β-amyloid precursor protein cleaved at the amino terminus of the β-amyloid peptide. Nature 1993, 361, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Hou, H.; Giunta, B.; Mori, T.; Wang, Y.J.; Fernandez, F.; Weggen, S.; Araki, W.; Obregon, D.; Tan, J. Autoreactive-Aβ antibodies promote APP β-secretase processing. J. Neurochem. 2012, 120, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Baghallab, I.; Reyes-Ruiz, J.M.; Abulnaja, K.; Huwait, E.; Glabe, C. Epitomic characterization of the specificity of the anti-amyloid Aβ monoclonal antibodies 6E10 and 4G8. J. Alzheimer’s Dis. 2018, 66, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Hartley, D.M.; Condron, M.M.; Selkoe, D.J.; Teplow, D.B. In vitro studies of amyloid β-protein fibril assembly and toxicity provide clues to the aetiology of Flemish variant (Ala692→ Gly) Alzheimer’s disease. Biochem. J. 2001, 355, 869–877. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease–linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, J.S.; Wang, X.; Wang, J.; Bao, F.X.; Pang, S.Y.; Yin, F.; Hu, H.G.; Peng, X.L.; Sun, W.M.; et al. Administration of Amyloid-β 42 Oligomer-Specific Monoclonal Antibody Improved Memory Performance in SAMP8 Mice. J. Alzheimer’s Dis. 2011, 23, 551–561. [Google Scholar] [CrossRef]
- Kayed, R.; Canto, I.; Breydo, L.; Rasool, S.; Lukacsovich, T.; Wu, J.; Albay, R.; Pensalfini, A.; Yeung, S.; Head, E.; et al. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 2010, 5, 57. [Google Scholar] [CrossRef]
- Lambert, M.P.; Velasco, P.T.; Chang, L.; Viola, K.L.; Fernandez, S.; Lacor, P.N.; Khuon, D.; Gong, Y.; Bigio, E.H.; Shaw, P.; et al. Monoclonal antibodies that target pathological assemblies of Aβ. J. Neurochem. 2007, 100, 23–35. [Google Scholar] [CrossRef]
- Isas, J.M.; Luibl, V.; Johnson, L.V.; Kayed, R.; Wetzel, R.; Glabe, C.G.; Langen, R.; Chen, J. Soluble and mature amyloid fibrils in drusen deposits. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Chebli, J.; Rahmati, M.; Lashley, T.; Edeman, B.; Oldfors, A.; Zetterberg, H.; Abramsson, A. The localization of amyloid precursor protein to ependymal cilia in vertebrates and its role in ciliogenesis and brain development in zebrafish. Sci. Rep. 2021, 11, 19115. [Google Scholar] [CrossRef] [PubMed]
- García-Ayllón, M.S.; Lopez-Font, I.; Boix, C.P.; Fortea, J.; Sánchez-Valle, R.; Lleó, A.; Molinuevo, J.L.; Zetterberg, H.; Blennow, K.; Sáez-Valero, J. C-terminal fragments of the amyloid precursor protein in cerebrospinal fluid as potential biomarkers for Alzheimer disease. Sci. Rep. 2017, 7, 2477. [Google Scholar] [CrossRef] [PubMed]
- Jingwu, Z.; Vandenbark, A.A.; Jacobs, M.P.; Offner, H.; Raus, J.C. Murine monoclonal anti-myelin basic protein (MBP) antibodies inhibit proliferation and cytotoxicity of MBP-specific human T cell clones. J. Neuroimmunol. 1989, 24, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Jeong, S.; Kim, J.; Ju, S.; Im, E.; Heo, G.; Park, S.; Yoo, J.W.; Lee, J.; Yoon, I.S.; et al. N-Acetylserotonin is an oxidation-responsive activator of Nrf2 ameliorating colitis in rats. J. Pineal Res. 2023, 74, e12835. [Google Scholar] [CrossRef]
- Pryor, N.E.; Moss, M.A.; Hestekin, C.N. Unraveling the early events of amyloid-β protein (Aβ) aggregation: Techniques for the determination of Aβ aggregate size. Int. J. Mol. Sci. 2012, 13, 3038–3072. [Google Scholar] [CrossRef]
- Puig Gomà-Camps, E. Structural Characterization of Amyloid Beta Oligomers with Functional Links Associated to Alzheimer’s Disease. 2019. Available online: https://dialnet.unirioja.es/servlet/tesis?codigo=250722 (accessed on 29 August 2023).
- Miyoshi, E.; Bilousova, T.; Melnik, M.; Fakhrutdinov, D.; Poon, W.W.; Vinters, H.V.; Miller, C.A.; Corrada, M.; Kawas, C.; Bohannan, R.; et al. Exosomal tau with seeding activity is released from Alzheimer’s disease synapses, and seeding potential is associated with amyloid beta. Lab. Investig. 2021, 101, 1605–1617. [Google Scholar] [CrossRef]
- Ying, Z.; Xin, W.; Jin-Sheng, H.; Fu-Xiang, B.; Wei-Min, S.; Xin-Xian, D.; Xiao-Bo, W.; Yi-Qin, L.; Xian-Xian, Z.; Hong-Gang, H.; et al. Preparation and characterization of a monoclonal antibody with high affinity for soluble Aβ oligomers. Hybridoma 2009, 28, 349–354. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef]
- Yang, Y.W.; Hsu, K.C.; Wei, C.Y.; Tzeng, R.C.; Chiu, P.Y. Operational Determination of Subjective Cognitive Decline, Mild Cognitive Impairment, and Dementia Using Sum of Boxes of the Clinical Dementia Rating Scale. Front. Aging Neurosci. 2021, 13, 705782. [Google Scholar] [CrossRef]
- Pham, T.K.; Buczek, W.A.; Mead, R.J.; Shaw, P.J.; Collins, M.O. Proteomic approaches to study cysteine oxidation: Applications in neurodegenerative diseases. Front. Mol. Neurosci. 2021, 14, 678837. [Google Scholar] [CrossRef] [PubMed]
- García-Santamarina, S.; Boronat, S.; Domènech, A.; Ayté, J.; Molina, H.; Hidalgo, E. Monitoring in vivo reversible cysteine oxidation in proteins using ICAT and mass spectrometry. Nat. Protoc. 2014, 9, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Brenneman, R.; Becker, K.G.; Gucek, M.; Cole, R.N.; Maudsley, S. iTRAQ analysis of complex proteome alterations in 3xTgAD Alzheimer’s mice: Understanding the interface between physiology and disease. PLoS ONE 2008, 3, e2750. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, E.; Borner, G.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Murphy, S.; Zweyer, M.; Sabir, H.; Swandulla, D.; Ohlendieck, K. The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle. Proteomes 2021, 9, 9. [Google Scholar] [CrossRef]
- Baazaoui, N.; Iqbal, K. Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound. Biomolecules 2022, 12, 1409. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Ossenkoppele, R.; Kvartsberg, H.; Brinkmalm, A.; Mattsson-Carlgren, N.; Stomrud, E.; Smith, R.; Zetterberg, H.; Blennow, K.; et al. Untangling the association of amyloid-β and tau with synaptic and axonal loss in Alzheimer’s disease. Brain 2021, 144, 310–324. [Google Scholar] [CrossRef]
- Li, Y.; Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Weiner, M.W.; Shaw, L.M.; Masters, C.L.; Fowler, C.J.; Trojanowski, J.Q.; et al. Validation of plasma amyloid-β 42/40 for detecting Alzheimer disease amyloid plaques. Neurology 2022, 98, e688–e699. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Lehmann, S.; Otto, M.; Zetterberg, H.; Lewczuk, P. Advantages and disadvantages of the use of the CSF Amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimer’s Res. Ther. 2019, 11, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Guerra, A.; Walker, D.G. Amyloid beta and tau as Alzheimer’s disease blood biomarkers: Promise from new technologies. Neurol. Ther. 2017, 6, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef]
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63. [Google Scholar] [CrossRef]
- Kotze, M.J.; Lückhoff, H.K.; Brand, T.; Pretorius, J.; van Rensburg, S.J. Apolipoprotein E ε-4 as a genetic determinant of Alzheimer’s disease heterogeneity. Degener. Neurol. Neuromuscul. Dis. 2015, 5, 9–18. [Google Scholar]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef]
- Kurz, C.; Walker, L.; Rauchmann, B.S.; Perneczky, R. Dysfunction of the blood-brain barrier in Alzheimer’s disease: Evidence from human studies. Neuropathol. Appl. Neurobiol. 2022, 48, e12782. [Google Scholar] [CrossRef]
- Milinkeviciute, G.; Green, K.N. Clusterin/apolipoprotein J, its isoforms, and Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1167886. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, J.H.; Xu, W.; Hou, X.H.; Li, J.Q.; Yu, J.T.; Tan, L.; Chi, S.; Alzheimer’s Disease Neuroimaging Initiative. The associations of cerebrospinal fluid ApoE and biomarkers of Alzheimer’s disease: Exploring interactions with sex. Front. Neurosci. 2021, 15, 633576. [Google Scholar] [CrossRef] [PubMed]
- Lennol, M.P.; Sánchez-Domínguez, I.; Cuchillo-Ibañez, I.; Camporesi, E.; Brinkmalm, G.; Alcolea, D.; Fortea, J.; Lleó, A.; Soria, G.; Aguado, F.; et al. Apolipoprotein E imbalance in the cerebrospinal fluid of Alzheimer’s disease patients. Alzheimer’s Res. Ther. 2022, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.; Tremblay, M.È. Chronic stress as a risk factor for Alzheimer’s disease: Roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobio Stress 2018, 9, 9–21. [Google Scholar] [CrossRef]
- Veitch, D.P.; Weiner, M.W.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Green, R.C.; Harvey, D.; Jack, C.R., Jr.; Jagust, W.; Morris, J.C.; et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s Dement. 2019, 15, 106–152. [Google Scholar] [CrossRef]
- Yuste-Checa, P.; Bracher, A.; Hartl, F.U. The chaperone Clusterin in neurodegeneration-friend or foe? Bioessays 2022, 44, e2100287. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Begum, M.M.; Islam, M.S.; Behl, T.; Ashraf, G.M. Exploring the Role of CLU in the Pathogenesis of Alzheimer’s Disease. Neurotox. Res. 2021, 39, 108–2119. [Google Scholar] [CrossRef]
- Li, K.W.; Ganz, A.B.; Smit, A.B. Proteomics of neurodegenerative diseases: Analysis of human post-mortem brain. J. Neurochem. 2019, 151, 435–445. [Google Scholar] [CrossRef]
- Han, S.; Nho, K.; Lee, Y. Alternative Splicing Regulation of an Alzheimer’s Risk Variant in CLU. Int. J. Mol. Sci. 2020, 21, 7079. [Google Scholar] [CrossRef]
- Fareed, M.M.; Qasmi, M.; Aziz, S.; Völker, E.; Förster, C.Y.; Shityakov, S. The Role of Clusterin Transporter in the Pathogenesis of Alzheimer’s Disease at the Blood–Brain Barrier Interface: A Systematic Review. Biomolecules 2022, 12, 1452. [Google Scholar] [CrossRef]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons From Other Pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Yuste-Checa, P.; Trinkaus, V.A.; Riera-Tur, I.; Imamoglu, R.; Schaller, T.F.; Wang, H.; Dudanova, I.; Hipp, M.S.; Bracher, A.; Hartl, F.U. The extracellular chaperone Clusterin enhances Tau aggregate seeding in a cellular model. Nat. Commun. 2021, 12, 4863. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, J.; Kucharska, E.; Garcez, M.L.; Rodrigues, M.S.; Quevedo, J.; Moreno-Gonzalez, I.; Budni, J. Inflammatory cascade in Alzheimer’s disease pathogenesis: A review of experimental findings. Cells 2021, 28, 2581. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.R.; Touchard, S.; Leckey, C.; O’Hagan, C.; Nevado-Holgado, A.J.; Barkhof, F.; Bertram, L.; Blin, O.; Bos, I.; Dobricic, V.; et al. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimer’s. Dement. 2019, 15, 776–787. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef]
- Chavarría, C.; Ivagnes, R.; Souza, J.M. Extracellular Alpha-Synuclein: Mechanisms for Glial Cell Internalization and Activation. Biomolecules 2022, 12, 655. [Google Scholar] [CrossRef]
- Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 7898. [Google Scholar] [CrossRef]
- Rather, M.A.; Khan, A.; Alshahrani, S.; Rashid, H.; Qadri, M.; Rashid, S.; Alsaffar, R.M.; Kamal, M.A.; Rehman, M.U. Inflammation and Alzheimer’s Disease: Mechanisms and Therapeutic Implications by Natural Products. Mediat. Inflamm. 2021, 2021, 9982954. [Google Scholar] [CrossRef]
- Shen, X.N.; Niu, L.D.; Wang, Y.J.; Cao, X.P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.T. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598. [Google Scholar] [CrossRef]
- Shah, A.; Kishore, U.; Shastri, A. Complement System in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 13647. [Google Scholar] [CrossRef] [PubMed]
- Agliardi, C.; Guerini, F.R.; Meloni, M.; Clerici, M. Alpha-synuclein as a biomarker in Parkinson’s disease: Focus on neural derived extracelluar vesicles. Neural Reg. Res. 2022, 17, 1503. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Sengupta, U.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L.; Lasagna-Reeves, C.A.; Gerson, J.E.; Paulucci-Holthauzen, A.A.; Krishnamurthy, S.; Farhed, M.; Jackson, G.R.; Kayed, R. Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol. Psychiatry 2015, 78, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Shim, K.H.; Kang, M.J.; Youn, Y.C.; An, S.S.A.; Kim, S. Alpha-synuclein: A pathological factor with Aβ and tau and biomarker in Alzheimer’s disease. Alzheimer’s Res. Ther. 2022, 14, 201. [Google Scholar] [CrossRef] [PubMed]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.M.; Ryan, P.; Rudrawar, S.; Quinn, R.J.; Zhang, H.Y.; Mellick, G.D. Advances in the development of imaging probes and aggregation inhibitors for alpha-synuclein. Acta Pharmacol. Sin. 2020, 41, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vieitez, E.; Nielsen, H.M. Associations Between APOE Variants, Tau and α-Synuclein. Adv. Exp. Med. Biol. 2019, 1184, 177–186. [Google Scholar]
- Butterfield, D.A. Phosphoproteomics of Alzheimer disease brain: Insights into altered brain protein regulation of critical neuronal functions and their contributions to subsequent cognitive loss. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2031–2039. [Google Scholar] [CrossRef]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Washburn, M.P.; Wolters, D.; Yates, J.R. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R., 3rd. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Pääbo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [Google Scholar] [CrossRef]
- Xu, P.; Duong, D.M.; Peng, J. Systematical Optimization of Reverse-Phase Chromatography for Shotgun Proteomics. J. Proteome Res. 2009, 8, 3944–3950. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef]
- Ting, L.; Rad, R.; Gygi, S.P.; Haas, W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8, 937–940. [Google Scholar] [CrossRef]
- Akbani, R.; Becker, K.F.; Carragher, N.; Goldstein, T.; de Koning, L.; Korf, U.; Liotta, L.; Mills, G.B.; Nishizuka, S.S.; Pawlak, M.; et al. Realizing the promise of reverse phase protein arrays for clinical, translational, and basic research: A workshop report: The RPPA (Reverse Phase Protein Array) society. Mol. Cell. Proteom. 2014, 13, 1625–1643. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Gozal, Y.M.; Duong, D.M.; Gearing, M.; Cheng, D.; Hanfelt, J.J.; Funderburk, C.; Peng, J.; Lah, J.J.; Levey, A.I. Proteomics analysis reveals novel components in the detergent-insoluble subproteome in Alzheimer’s disease. J. Proteome Res. 2009, 8, 5069–5079. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.M.; Seyfried, N.T.; Dammer, E.B.; Duong, D.; Yi, H.; Gearing, M.; Troncoso, J.C.; Mufson, E.J.; Thambisetty, M.; Levey, A.I.; et al. U1 small nuclear ribonucleoproteins (snRNPs) aggregate in Alzheimer’s disease due to autosomal dominant genetic mutations and trisomy 21. Mol. Neurodegener. 2014, 9, 15. [Google Scholar] [CrossRef]
- Raj, T.; Li, Y.I.; Wong, G.; Humphrey, J.; Wang, M.; Ramdhani, S.; Wang, Y.C.; Ng, B.; Gupta, I.; Haroutunian, V.; et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 2018, 50, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Shang, Y.; Xu, X.; Dong, Z.; Zhang, Y.; Du, Z.; Lu, X.; Zhang, T. Presenilin 1 mutation likely contributes to U1 small nuclear RNA dysregulation and Alzheimer’s disease-like symptoms. Neurobiol. Aging 2021, 100, 1–10. [Google Scholar] [CrossRef]
- Cheng, D.; Hoogenraad, C.C.; Rush, J.; Ramm, E.; Schlager, M.A.; Duong, D.M.; Xu, P.; Wijayawardana, S.R.; Hanfelt, J.; Nakagawa, T.; et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteom. 2006, 5, 1158–1170. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D.; Castegna, A. Proteomics in Alzheimer’s disease: Insights into potential mechanisms of neurodegeneration. J. Neurochem. 2003, 86, 1313–1327. [Google Scholar] [CrossRef]
- Seyfried, N.T.; Dammer, E.B.; Swarup, V.; Nandakumar, D.; Duong, D.M.; Yin, L.; Deng, Q.; Nguyen, T.; Hales, C.M.; Wingo, T.; et al. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst. 2017, 4, 60–72. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Yin, L.; Thambisetty, M.; Troncoso, J.C.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener. 2018, 13, 52. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Y.; Li, Y.; Bai, B.; Wang, X.; Tan, H.; Liu, T.; Beach, T.G.; Peng, J.; Wu, Z. Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J. Proteome Res. 2015, 14, 829–838. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Wingo, A.P.; Liu, Y.; Gerasimov, E.S.; Gockley, J.; Logsdon, B.A.; Duong, D.M.; Dammer, E.B.; Robins, C.; Beach, T.G.; Reiman, E.M.; et al. Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nat. Genet. 2021, 53, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Esteve, P.; Rueda-Carrasco, J.; Inés Mateo, M.; Martin-Bermejo, M.J.; Draffin, J.; Pereyra, G.; Sandonís, Á.; Crespo, I.; Moreno, I.; Aso, E.; et al. Elevated levels of Secreted-Frizzled-Related-Protein 1 contribute to Alzheimer’s disease pathogenesis. Nat. Neurosci. 2019, 22, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, T.; Renier, N.; Bettayeb, K.; Greengard, P.; Tessier-Lavigne, M.; Flajolet, M. Three-Dimensional Study of Alzheimer’s Disease Hallmarks Using the iDISCO Clearing Method. Cell Rep. 2016, 16, 1138–1152. [Google Scholar] [CrossRef]
- Spilman, P.R.; Corset, V.; Gorostiza, O.; Poksay, K.S.; Galvan, V.; Zhang, J.; Rao, R.; Peters-Libeu, C.; Vincelette, J.; McGeehan, A.; et al. Netrin-1 Interrupts Amyloid-beta Amplification, Increases sAbetaPPalpha in vitro and in vivo, and Improves Cognition in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 52, 223–242. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. The Brain Hepatocyte Growth Factor/c-Met Receptor System: A New Target for the Treatment of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 985–1000. [Google Scholar] [CrossRef]
- Zheng, H.; Jia, L.; Liu, C.C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.F.; Fryer, J.D.; Wang, X.; Zhang, Y.W.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef]
- Lutz, B.M.; Peng, J. Deep profiling of the aggregated proteome in Alzheimer’s disease: From pathology to disease mechanisms. Proteomes 2018, 6, 46. [Google Scholar] [CrossRef]
- Zhang, D.F.; Fan, Y.; Xu, M.; Wang, G.; Wang, D.; Li, J.; Kong, L.L.; Zhou, H.; Luo, R.; Bi, R.; et al. Complement C7 is a novel risk gene for Alzheimer’s disease in Han Chinese. Natl. Sci. Rev. 2019, 6, 257–274. [Google Scholar] [CrossRef]
- Popov, I.A.; Starodubtseva, N.L.; Indeikina, M.I.; Kostyukevich, Y.I.; Kononikhin, A.S.; Nikolaeva, M.I.; Kukaev, E.N.; Kozin, S.A.; Makarov, A.A.; Nikolaev, E.N. Mass spectrometric identification of posttranslational modifications in transthyretin from human blood. Mol. Biol. 2013, 47, 885–893. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P. Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopathol. 2018, 40, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Litvinchuk, A.; Wan, Y.W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337–1353. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Biomarker | Location | Molecular Function | Role in AD |
|---|---|---|---|
| Amyloids | Arterial wall in brain | Helps in hormone release and plays a role in forming melanin, which helps protect the skin from sun damage. | Induces mitochondrial oxidative stress |
| p-Tau | Neurons, somato-dendritic compartments | Stabilizes neuronal microtubules and promotes axonal outgrowth | Accelerates the fibrillization of α-syn |
| Alpha-synuclein | In the axon terminals of presynaptic neurons | Role in neurotransmission at the synapse, calcium homeostasis, mitochondrial function, and gene regulation. | Induces the formation of Aβ oligomers; induces tau aggregation |
| Recognition Motif | Nature of Antibody | Name of Antibody | Reference |
|---|---|---|---|
| Aβ1–16 | Monoclonal | Ab9 | [65] |
| Aβ1–16 | Monoclonal | 6C6 | [66] |
| Aβ1–17 | Monoclonal | 6E10 | [67] |
| Aβ17–24 | Monoclonal | 4G8 | [68] |
| Aβ31–40 | Monoclonal | 2G3 | [69] |
| Aβ1–40, C-terminal | Monoclonal | BA-27 | [70] |
| Aβ1–42, C-terminal | Monoclonal | BC-05 | [71] |
| Amyloid oligomers | Monoclonal | A8 | [72] |
| Amyloid oligomers | Monoclonal | A11 | [73] |
| Amyloid oligomers | Monoclonal | NU-4 | [74] |
| Amyloid fibrils | Polyclonal | OC | [75] |
| Anti-amyloid beta precursor protein | Monoclonal | Y188 | [76] |
| Anti-APP | Monoclonal | A8717 | [77] |
| Anti-myelin basic protein | Monoclonal | MBP | [78] |
| Anti-kelch-like ECH-associated protein 1 | Monoclonal | KEAP1 | [79] |
| Sr. No. | Purpose of the Tools | Tools and Softwares |
|---|---|---|
| 1. | Identification of Data | Mascot |
| SEQUEST | ||
| OMSSA | ||
| X!TANDEM | ||
| 2. | Quantification of Data | MSQuant |
| Progenesis | ||
| 3. | Function and Localization | TMHMM |
| Signal P | ||
| PSORTb | ||
| 4. | Pathway and Network Analysis | STRING |
| Cytoscape | ||
| KEGG | ||
| 5. | Integration of Multi-Omics Data | iCluster |
| TransPro | ||
| PARADIGM | ||
| LRAcluster | ||
| PSDF | ||
| BCC | ||
| MDI |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, M.; Dhariwal, R.; Patil, N.; Ojha, S.; Tendulkar, R.; Tendulkar, M.; Dhanda, P.S.; Yadav, A.; Kaushik, P. Unveiling the Molecular Footprint: Proteome-Based Biomarkers for Alzheimer’s Disease. Proteomes 2023, 11, 33. https://doi.org/10.3390/proteomes11040033
Jain M, Dhariwal R, Patil N, Ojha S, Tendulkar R, Tendulkar M, Dhanda PS, Yadav A, Kaushik P. Unveiling the Molecular Footprint: Proteome-Based Biomarkers for Alzheimer’s Disease. Proteomes. 2023; 11(4):33. https://doi.org/10.3390/proteomes11040033
Chicago/Turabian StyleJain, Mukul, Rupal Dhariwal, Nil Patil, Sandhya Ojha, Reshma Tendulkar, Mugdha Tendulkar, Parmdeep Singh Dhanda, Alpa Yadav, and Prashant Kaushik. 2023. "Unveiling the Molecular Footprint: Proteome-Based Biomarkers for Alzheimer’s Disease" Proteomes 11, no. 4: 33. https://doi.org/10.3390/proteomes11040033