Identification of BELL Transcription Factors Involved in Nodule Initiation and Development in the Legumes Pisum sativum and Medicago truncatula


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
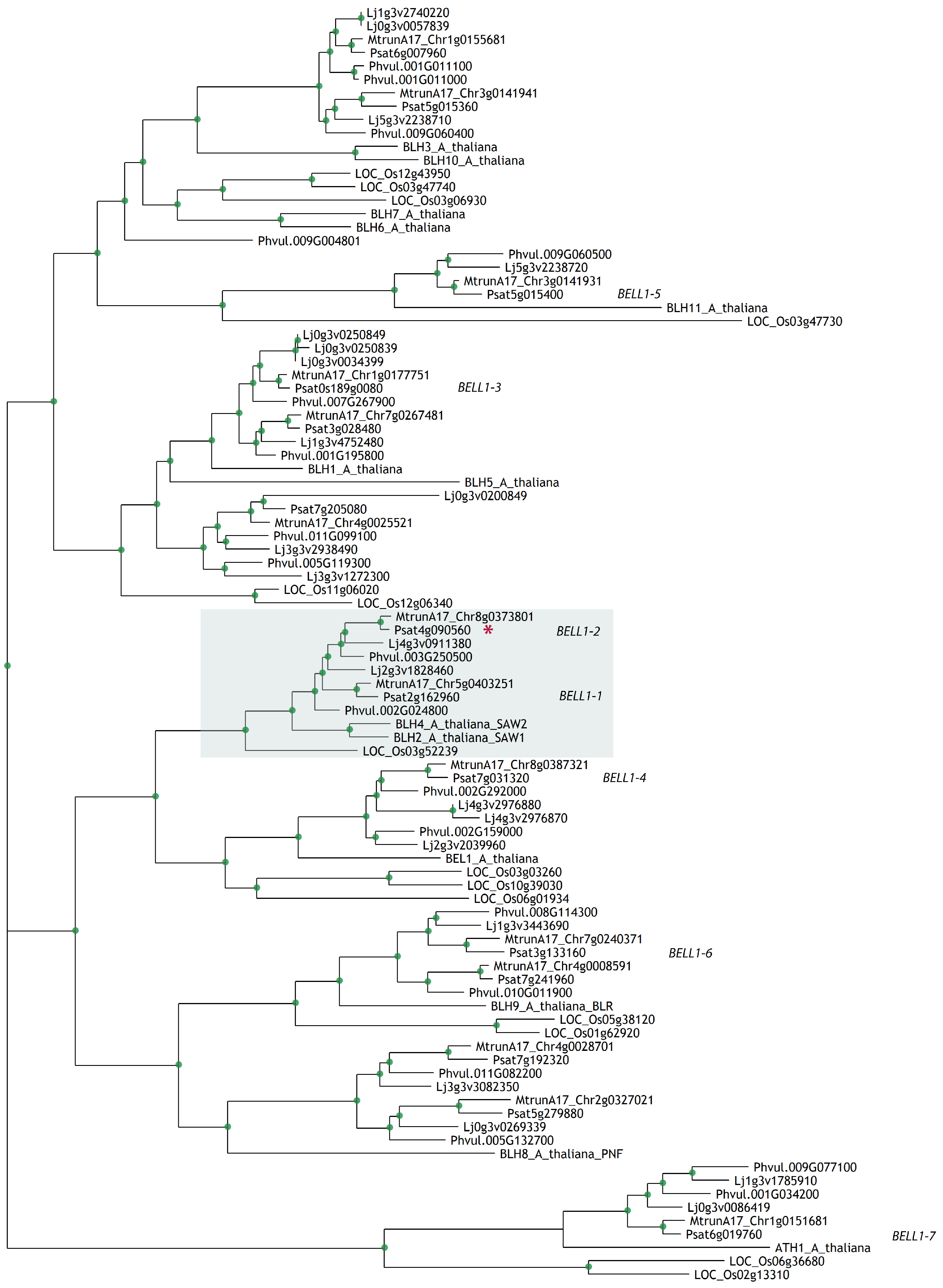
2.1. Diversity of BEL-Like Genes in Legumes
2.2. Search for BELL Genes Regulated by NIN Transcription Factor during Symbiosis Development
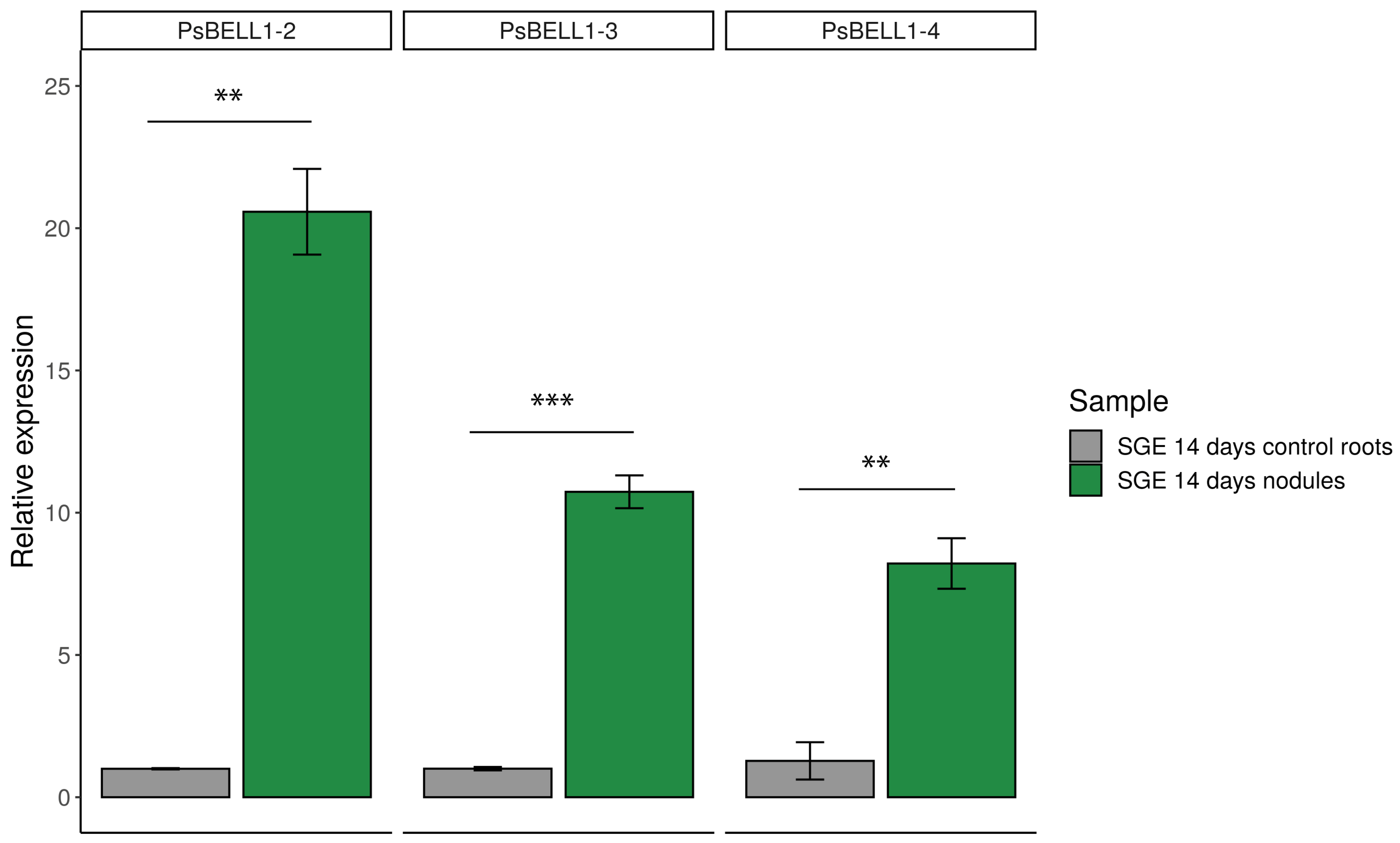
2.3. Expression of PsBELL1-2, PsBEL L1-3, and PsBELL1-4 Genes in Pea SGENod−-1 (Psnin-1) Mutant
2.4. Analysis of Interaction between PsBELL and PsKNOX Regulators during Nodulation
2.5. Analysis of Interaction between the Most Important Gibberellin Network Regulators, DELLA Proteins, and BELL Transcription Factors
3. Discussion
4. Materials and Methods
4.1. Phylogenetic Reconstruction
4.2. RNA-Seq Quantification and Statistical Analysis
4.3. Bacterial Strains and Inoculation
4.4. Plant Material and Growth Conditions
4.5. RNA Extraction, cDNA Synthesis, and Quantitative Reverse Transcription PCR (RT-qPCR)
4.6. RT-qPCR Data Analysis
4.7. Cloning of PsDELLA1, PsKNOX3, and PsBELL1 Genes for Yeast Transformation
4.8. Yeast Two-Hybrid Assay (GAL4 Transcription Factor-Based Assay)
4.9. Protein Synthesis in E. coli and Co-Immunoprecipitation Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jin, Y.; Liu, H.; Luo, D.; Yu, N.; Dong, W.; Wang, C.; Zhang, X.; Dai, H.; Yang, J.; Wang, E. DELLA proteins are common components of symbiotic rhizobial and mycorrhizal signalling pathways. Nat. Commun. 2016, 7, 12433. [Google Scholar] [CrossRef]
- Fonouni-Farde, C.; Tan, S.; Baudin, M.; Brault, M.; Wen, J.; Mysore, K.S.; Niebel, A.; Frugier, F.; Diet, A. DELLA-mediated gibberellin signalling regulates Nod factor signalling and rhizobial infection. Nat. Commun. 2016, 7, 12636. [Google Scholar] [CrossRef] [PubMed]
- Bürglin, T.R.; Affolter, M. Homeodomain proteins: An update. Chromosoma 2016, 125, 497–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamant, O.; Pautot, V. Plant development: A TALE story. C. R. Biol. 2010, 333, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Kushalappa, K.; Godt, D.; Pidkowich, M.S.; Pastorelli, S.; Hepworth, S.R.; Haughn, G.W. The Arabidopsis BEL1-LIKE HOMEODOMAIN proteins SAW1 and SAW2 act redundantly to regulate KNOX expression spatially in leaf margins. Plant Cell 2007, 19, 2719–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; You, S.; Taylor-Teeples, M.; Li, W.L.; Schuetz, M.; Brady, S.M.; Douglas, C.J. BEL1-LIKE HOMEODOMAIN6 and KNOTTED ARABIDOPSIS THALIANA7 interact and regulate secondary cell wall formation via repression of REVOLUTA. Plant Cell 2014, 26, 4843–4861. [Google Scholar] [CrossRef] [Green Version]
- Jasinski, S.; Piazza, P.; Craft, J.; Hay, A.; Woolley, L.; Rieu, I.; Phillips, A.; Hedden, P.; Tsiantis, M. KNOX action in Arabidopsis is mediated by coordinate regulation of cytokinin and gibberellin activities. Curr. Biol. 2005, 15, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Dean, G.; Casson, S.; Lindsey, K. KNAT6 gene of Arabidopsis is expressed in roots and is required for correct lateral root formation. Plant Mol. Biol. 2004, 54, 71–84. [Google Scholar] [CrossRef]
- Kim, J.Y.; Rim, Y.; Wang, J.; Jackson, D. A novel cell-to-cell trafficking assay indicates that the KNOX homeodomain is necessary and sufficient for intercellular protein and mRNA trafficking. Genes Dev. 2005, 19, 788–793. [Google Scholar] [CrossRef] [Green Version]
- Souček, P.; Klíma, P.; Reková, A.; Brzobohatý, B. Involvement of hormones and KNOXI genes in early Arabidopsis seedling development. J. Exp. Bot. 2007, 58, 3797–3810. [Google Scholar] [CrossRef]
- Souček, P.; Hanáček, P.; Mazura, P.; Reinöhl, V. Interaction among BREVIPEDICELLUS, BLH6 and auxin in roots of Arabidopsis thaliana. Russ. J. Plant Physiol. 2017, 64, 386–397. [Google Scholar] [CrossRef]
- Di Giacomo, E.; Laffont, C.; Sciarra, F.; Iannelli, M.A.; Frugier, F.; Frugis, G. KNAT3/4/5-like class 2 KNOX transcription factors are involved in Medicago truncatula symbiotic nodule organ development. New Phytol. 2017, 213, 822–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azarakhsh, M.; Kirienko, A.N.; Zhukov, V.A.; Lebedeva, M.A.; Dolgikh, E.A.; Lutova, L.A. KNOTTED1-LIKE HOMEOBOX 3: A new regulator of symbiotic nodule development. J. Exp. Bot. 2015, 66, 7181–7195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azarakhsh, M.; Rumjantsev, A.; Lebedeva, M.; Lutova, L. Cytokinin biosynthesis genes expressed during nodule organogenesis are directly regulated by the KNOX3 protein in Medicago truncatula. PLoS ONE 2020, 15, e0232352. [Google Scholar] [CrossRef]
- Dolgikh, A.V.; Kirienko, A.N.; Tikhonovich, I.A.; Foo, E.; Dolgikh, E.A. The DELLA proteins influence the expression of cytokinin biosynthesis and response genes during nodulation. Front. Plant Sci. 2019, 10, 432. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Pecrix, Y.; Staton, S.E.; Sallet, E.; Lelandais-Brière, C.; Moreau, S.; Carrère, S.; Blein, T.; Jardinaud, M.F.; Latrasse, D.; Zouine, M.; et al. Whole-genome landscape of Medicago truncatula symbiotic genes. Nat. Plants 2018, 4, 1017–1025. [Google Scholar] [CrossRef]
- Kreplak, J.; Madoui, M.A.; Cápal, P.; Novák, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef]
- Soyano, T.; Hirakawa, H.; Sato, S.; Hayashi, M.; Kawaguchi, M. NODULE INCEPTION creates a long-distance negative feedback loop involved in homeostatic regulation of nodule organ production. Proc. Natl. Acad. Sci. USA 2014, 111, 14607–14612. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, Y.; Wang, X.; Pei, S.; Kong, Y.; Hu, R.; Zhou, G. Transcription Factors BLH2 and BLH4 Regulate Demethylesterification of Homogalacturonan in Seed Mucilage. Plant Physiol. 2020, 183, 96–111. [Google Scholar] [CrossRef] [Green Version]
- Tsyganova, A.V.; Seliverstova, E.V.; Brewin, N.J.; Tsyganov, V.E. Comparative analysis of remodelling of the plant–microbe interface in Pisum sativum and Medicago truncatula symbiotic nodules. Protoplasma 2019, 256, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Tsyganova, A.V.; Ivanova, K.A.; Tsyganov, V.E. Histological and ultrastructural nodule organization of the pea (Pisum sativum) mutant SGEFix−-5 in the Sym33 gene encoding the transcription factor PsCYCLOPS/PsIPD3. Ecol. Genet. 2019, 17, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Tsyganov, V.E.; Morzhina, E.V.; Stefanov, S.Y.; Borisov, A.Y.; Lebsky, V.K.; Tikhonovich, I.A. The pea (Pisum sativum L.) genes sym33 and sym40 control infection thread formation and root nodule function. Mol. Gen. Genet. 1998, 259, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Altmann, M.; Altmann, S.; Rodriguez, P.A.; Weller, B.; Elorduy Vergara, L.; Palme, J.; Marín-de la Rosa, N.; Sauer, M.; Wenig, M.; Villaécija-Aguilar, J.A.; et al. Extensive signal integration by the phytohormone protein network. Nature 2020, 583, 271–276. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Löytynoja, A. Phylogeny-Aware Alignment with PRANK BT—Multiple Sequence Alignment Methods; Russell, D.J., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 155–170. ISBN 978-1-62703-646-7. [Google Scholar]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Schiessl, K.; Lilley, J.; Lee, T.; Tamvakis, I.; Kohlen, W.; Bailey, P.; Thomas, A.; Luptak, J.; Karunakaran, R.; Carpenter, M.; et al. Nodule inception recruits the lateral root the lateral root developmental program for symbiotic nodule organogenesis in Medicago truncatula. Curr. Biol. 2019, 29, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenn, A.R.; Poole, P.S.; Hudman, J.F. Succinate uptake by free-living and bacteroid forms of Rhizobium leguminosarum. Microbiology 1980, 119, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Safronova, V.I.; Novikova, N.I. Comparison of two methods for root nodule bacteria preservation: Lyophilization and liquid nitrogen freezing. J. Microbiol. Methods 1996, 24, 231–237. [Google Scholar] [CrossRef]
- Van Brussel, A.; Planqué, K.; Quispel, A. The wall of Rhizobium leguminosarum in bacteroid and free-living forms. J. Gen. Microbiol. 1977, 101, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Kosterin, O.E.; Rozov, S.M. Mapping of the new mutation blb and the problem of integrity of linkage group I. Pisum Genet. 1993, 25, 27–31. [Google Scholar]
- Tsyganov, V.E.; Voroshilova, V.A.; Kukalev, A.S.; Azarova, T.S.; Yakobi, L.M.; Borisov, A.Y.; Tikhonovich, I.A. Pisum sativum L. genes sym14 and sym35 control infection thread growth initiation during the development of symbiotic nodules. Russ. J. Genet. 1999, 35, 284–291. [Google Scholar]
- Van Brussel, A.A.N.; Tak, T.; Wetselaar, A.; Pees, E.; Wijffelman, C.A. Small leguminosae as test plants for nodulation of Rhizobium leguminosarum and other rhizobia and agrobacteria harbouring a leguminosarum sym-plasmid. Plant Sci. Lett. 1982, 27, 317–325. [Google Scholar] [CrossRef]
- Dolgikh, E.A.; Kusakin, P.G.; Kitaeva, A.B.; Tsyganova, A.V.; Kirienko, A.N.; Leppyanen, I.V.; Dolgikh, A.V.; Ilina, E.L.; Demchenko, K.N.; Tikhonovich, I.A.; et al. Mutational analysis indicates that abnormalities in rhizobial infection and subsequent plant cell and bacteroid differentiation in pea (Pisum sativum) nodules coincide with abnormal cytokinin responses and localization. Ann. Bot. 2020, 125, 905–923. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- James, P.; Halladay, J.; Craig, E.A. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 1996, 144, 1425–1436. [Google Scholar]
- Gietz, R.D.; Schiestl, R.H. Frozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Alves-Carvalho, S.; Aubert, G.; Carrère, S.; Cruaud, C.; Brochot, A.L.; Jacquin, F.; Klein, A.; Martin, C.; Boucherot, K.; Kreplak, J.; et al. Full length de novo assembly of RNA-seq data in pea (Pisum sativum L.) provides a gene expression atlas and gives insights into root nodulation in this species. Plant J. 2015, 84, 1–19. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolgikh, A.V.; Rudaya, E.S.; Dolgikh, E.A. Identification of BELL Transcription Factors Involved in Nodule Initiation and Development in the Legumes Pisum sativum and Medicago truncatula. Plants 2020, 9, 1808. https://doi.org/10.3390/plants9121808
Dolgikh AV, Rudaya ES, Dolgikh EA. Identification of BELL Transcription Factors Involved in Nodule Initiation and Development in the Legumes Pisum sativum and Medicago truncatula. Plants. 2020; 9(12):1808. https://doi.org/10.3390/plants9121808
Chicago/Turabian StyleDolgikh, Alexandra V., Elizaveta S. Rudaya, and Elena A. Dolgikh. 2020. "Identification of BELL Transcription Factors Involved in Nodule Initiation and Development in the Legumes Pisum sativum and Medicago truncatula" Plants 9, no. 12: 1808. https://doi.org/10.3390/plants9121808