Next Generation Sequencing Based Forward Genetic Approaches for Identification and Mapping of Causal Mutations in Crop Plants: A Comprehensive Review

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Mutation Breeding for Crop Improvement
3. Need of Identification and Mapping of Causal Mutations
4. Concept of Mapping, Sequencing, Resequencing, and Mapping by Sequencing
5. Role of NGS in Detection and Mapping of Mutated Genes/Locus
6. NGS Based Forward Genetics for Identification and Mapping of Causal Mutations
6.1. SHOREmap
6.2. NGM (Next-Generation Mapping)
6.3. dCARE (Deep CAndidate RE-Sequencing)
6.4. MutMap Approach
6.5. MutMap+ Approach
6.6. MutMap-Gap
6.7. RNA Sequencing (RNA Seq) Based Mapping
6.8. QTL-Seq Approach
6.9. Exome Capture Approach
6.10. NIKS (Needle in the k-Stack) Approach
6.11. MutChromSeq (Mutant Chromosome Sequencing) Approach
6.12. MutRenSeq Approach
6.13. SIMM (Simultaneous Identification of Multiple Causal Mutations)
6.14. TACCA (Targeted Chromosome-Based Cloning via Long-Range Assembly)
6.15. AgRenSeq (Association Genetics with R-Gene Enrichment Sequencing) Approach
6.16. LNISKS (Longer Needle in a Scanter K-Stack) Approach
7. Bioinformatics Tools/Software/Pipelines Used in NGS Based Forward Genetic Screen for Mutation Identification and Mapping
7.1. MAQGene
7.2. GenomeMapper
7.3. MASS (Mapping and Assembly with Short Sequences)
7.4. Next-Generation Mapping (NGM)
- i.
- SNP data from F2 mapping population: This involves getting sequence data from sequencer, cleaning, and pre-processing of sequence data. Uploading and filtering of SNP data to website.
- ii.
- Localization of SNPs: Localization of mutants to Arabidopsis chromosome is done by identifying non-recombinant (less heterozygosity) area within genomic region with mutations.
- iii.
- Segregating SNPs based on their variation to reference genome.
- iv.
- Localization and annotation of causal SNP by fine mapped region.
7.5. The SNPTrack Tool
7.6. CloudMap
7.7. CandiSNP
7.8. A SIMPLE Pipeline
7.9. artMAP
8. Limitations and Way Ahead
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chaudhary, J.; Deshmukh, R.; Sonah, H. Mutagenesis approaches and their role in crop improvement. Plants 2019, 8, 467. [Google Scholar] [CrossRef] [Green Version]
- Cassells, A.C.; Doyel, B.M. Genetic engineering and mutation breeding for tolerance to abiotic and biotic stresses. Bulg. J. Plant Physiol. 2003, SI, 52–82. [Google Scholar]
- Suprasanna, P.; Jain, S.M. Mutant Resources and Mutagenomics in crop plants. Emir. J. Food Agric. 2017, 29, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Oladosu, Y.; Rafii, Y.; Abdullah, N.; Hussin, G.; Ramli, A.; Rahim, H.A.; Miah, G.; Usman, M. Principle and application of plant mutagenesis in crop improvement: A review. Biotechnol. Biotechnol. Equip. 2016, 30, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Kumawat, S.; Rana, N.; Bansal, R.; Vishwakarma, G.; Mehetre, S.T.; Das, B.K.; Kumar, M.; Yadav, S.K.; Sonah, H.; Sharma, T.R.; et al. Expanding avenue of fast neutron mediated mutagenesis for crop improvement. Plants 2019, 8, 164. [Google Scholar] [CrossRef] [Green Version]
- Jankowicz-Cieslak, J.; Mba, C.; Till, B.J. Mutagenesis for crop breeding and functional genomics. In Biotechnologies for Plant Mutation Breeding; Jankowicz-Cieslak, J., Tai, T., Kumlehn, J., Till, B., Eds.; Joint FAO/IAEA Division of Nuclear Techniques in Food and Agriculture International Atomic Energy Agency: Vienna, Austria, 2017; pp. 3–18. [Google Scholar]
- Pereira, R.; Oliveira, J.; Sousa, M. Bioinformatics and computational tools for next-generation sequencing analysis in clinical genetics. J. Clin. Med. 2020, 9, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneeberger, K. Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat. Rev. Genet. 2014, 15, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Lukowitz, W.; Gillmor, C.S.; Scheible, W. Positional Cloning in Arabidopsis. Why It Feels Good to Have a Genome Initiative Working for You. Plant Physiol. 2000, 123, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson-Sanchez, D.; Lup, S.D.; Sarmiento-Manus, R.; Ponce, M.R.; Micol, J.L. Next-generation forward genetic screens: Using simulated data to improve the design of mapping-by-sequencing experiments in Arabidopsis. Nucleic Acids Res. 2019, 47. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, K.; Ossowski, S.; Lanz, C.; Juul, T.; Petersen, A.H.; Nielsen, K.L.; Jorgensen, J.E.; Weigel, D.; Andersen, S.U. SHOREmap: Simultaneous mapping and mutation identification by deep sequencing. Nat. Methods 2009, 6, 550–551. [Google Scholar] [CrossRef]
- Austin, R.S.; Vidaurre, D.; Stamatiou, G.; Breit, R.; Provart, N.J.; Bonetta, D. Next-generation mapping of Arabidopsis genes. Plant J. 2011, 67, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, B.; James, G.V.; Konrad, K.; Schneeberger, K.; Turck, F. Fast Isogenic mapping-by-sequencing of ethyl methanesulfonate-induced mutant bulks. Plant Physiol. 2012, 160, 591–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekih, R.; Takagi, H.; Tamiru, M.; Abe, A.; Natsume, S.; Yaegashi, H.; Sharma, S.; Sharma, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap+: Genetic Mapping and Mutant Identification without Crossing in Rice. PLoS ONE 2013, 8, e68529. [Google Scholar] [CrossRef]
- Takagi, H.; Uemura, A.; Yaegashi, H.; Tamiru, M.; Abe, A.; Mitsuoka, C.; Utsushi, H.; Natsume, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap-Gap: Whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene pii. New Phytol. 2013, 200, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.C.; Obholzer, N.D.; Shah, A.N.; Megason, S.G.; Moens, C.B. RNA-seq-based mapping and candidate identification of mutations from forward genetic screens. Genome Res. 2013, 23, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Mascher, M.; Richmond, T.A.; Gerhardt, D.J.; Himmelbach, A.; Clissold, L.; Sampath, D.; Ayling, S.; Steuernagel, B.; Pfeifer, M.; D’Ascenzo, M.; et al. Barley whole exome capture: A tool for genomic research in the genus Hordeum and beyond. Plant J. 2013, 76, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Henry, I.M.; Nagalakshmi, U.; Lieberman, M.C.; Ngo, K.J.; Krasileva, K.V.; Vasquez-Gross, H.; Akhunova, A.; Akhunov, E.; Dubcovsky, J.; Tai, T.H.; et al. Efficient genome-wide detection and cataloging of EMS-induced mutations using exome capture and next-generation sequencing. Plant Cell 2014, 26, 1382–1397. [Google Scholar] [CrossRef] [Green Version]
- Nordstrom, K.; Albani, M.; James, G.; Gutjahr, C.; Hartwig, B.; Turck, F.; Paszkowski, U.; Coupland, G.; Schneeberger, K. Mutation identification by direct comparison of whole-genome sequencing data from mutant and wild-type individuals using k-mers. Nat. Biotechnol. 2013, 31, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martin, J.; Steuernagel, B.; Ghosh, S.; Herren, G.; Hurni, S.; Adamski, N.; Vrana, J.; Kubalakova, M.; Krattinger, S.G.; Wicker, T.; et al. Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol. 2016, 17, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steuernagel, B.; Periyannan, S.; Hernández-Pinzón, I.; Witek, K.; Rouse, M.N.; Yu, G.; Hatta, A.; Ayliffe, M.; Bariana, H.; Jones, J.D.G.; et al. Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat. Biotechnol. 2016, 34, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Chen, Z.; Lu, J.; Xu, C.; Xie, G.; Li, Y.; Deng, X.W.; He, H.; Tang, X. Simultaneous Identification of Multiple Causal Mutations in Rice. Front. Plant Sci. 2017, 7, 2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thind, A.K.; Wicker, T.; Simkova, H.; Fossati, D.; Moullet, O.; Brabant, C.; Vrana, J.; Doležel, J.; Krattinger, S.G. Rapid cloning of genes in hexaploid wheat using cultivar-specific long-range chromosome assembly. Nat. Biotechnol. 2017, 35, 793–796. [Google Scholar] [CrossRef]
- Arora, S.; Steuernagel, B.; Gaurav, K.; Chandramohan, S.; Long, Y.; Matny, O. Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat. Biotechnol. 2019, 37, 139–143. [Google Scholar] [CrossRef]
- Suchecki, R.; Sandhu, A.; Deschamps, S.; Llaca, V.; Wolters, P.; Watson-Haigh, N.S.; Pallotta, M.; Whitford, R.; Baumann, U. LNISKS: Reference-free mutation identification for large and complex crop genomes. bioRxiv 2019. [Google Scholar] [CrossRef]
- Javorka, P.; Raxwal, V.K.; Najvarek, J.; Riha, K. artMAP: A user-friendly tool for mapping ethyl methanesulfonate-induced mutations in Arabidopsis. Plant Direct. 2019, 3, e00146. [Google Scholar] [CrossRef] [Green Version]
- Bigelow, H.; Doitsidou, M.; Sarin, S.; Hobert, O. A software tool, MAQGene, facilitating C. elegans whole genome sequence analysis for mutant identification. Nat. Methods 2009, 6, 549. [Google Scholar] [CrossRef] [Green Version]
- Schneeberger, K.; Hagmann, J.; Ossowski, J.; Warthmann, N.; Gesing, S.; Kohlbacher, O.; Weigel, D. Simultaneous alignment of short reads against multiple genomes. Genome Biol. 2009, 10, R98. [Google Scholar] [CrossRef] [Green Version]
- Cuperus, J.T.; Montgomery, T.A.; Fahlgren, N.; Burke, R.T.; Townsend, T.; Sullivan, C.M.; Carrington, J.C. Identification of MIR390a precursor processing-defective mutants in Arabidopsis by direct genome sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 466–471. [Google Scholar] [CrossRef] [Green Version]
- Leshchiner, I.; Alexa, K.; Kelsey, P.; Adzhubei, I.; Austin-Tse, C.A.; Cooney, J.D.; Anderson, H.; King, M.J.; Stottmann, R.W.; Garnaas, M.K.; et al. Mutation mapping and identification by whole genome sequencing. Genome Res. 2012, 22, 1541–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minevich, G.; Park, D.S.; Blankenberg, D.; Poole, R.J.; Hobert, O. CloudMap: A cloud-based pipeline for analysis of mutant genome sequences. Genetics 2012, 192, 1249–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etherington, G.J.; Monaghan, J.; Zipfel, C.; Maclean, D. Mapping mutations in plant genomes with the user-friendly web application CandiSNP. Plant Methods 2014, 10, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachsman, G.; Jennifer, L.M.; Manuel, V.; Philip, N.B. A SIMPLE Pipeline for Mapping Point Mutations. Plant Physiol. 2017, 174, 1307–1313. [Google Scholar] [CrossRef] [Green Version]
- Kharkwal, M.C.; Shu, Q.Y. The role of induced mutations in world food security. In Induced Plant Mutations in the Genomics Era, Proceedings of the International Joint FAO/IAEA Symposium IAEA, Vienna, Austria, 8–11 June 2009; Food and Agriculture Organization of the United Nations: Rome, Italy, 2009; pp. 33–38. [Google Scholar]
- Forster, B.P.; Shu, Q.Y. Plant Mutagenesis in Crop Improvement: Basic Terms and Applications. In Plant Mutation Breeding and Biotechnology; Shu, Q.Y., Forster, B.P., Nakagawa, H., Eds.; Joint FAO/IAEA Division of Nuclear Techniques in Food and Agriculture International Atomic Energy Agency: Vienna, Austria, 2011; pp. 9–71. [Google Scholar]
- Freisleben, R.A.; Lein, A. Moglichkeiten und praktischeDurchführung der Mutationszüchtung. Kuhn-Arhiv 1944, 60, 211–222. [Google Scholar]
- FAO/IAEA, Mutant Variety Database. Available online: https://mvd.iaea.org/ (accessed on 25 June 2020).
- Sharma, D.; Das, B.K.; Sahu, P.K.; Tiwari, A.K.; Baghel, S.; Sao, R.; Singh, S.; Kumar, V. Improvement of Traditional Farmers’ Varieties of Rice through Radiation Induced Mutation Breeding; IGKV Publications: Raipur, India, 2019; pp. 1–48. [Google Scholar]
- Sharma, D.; Sahu, P.K.; Das, B.K. BARC-IGKV MoU: A Unique Model of Mutual Collaboration towards Welfare of Farmers’ and Nation; IGKV Publications: Raipur, India, 2020; pp. 1–65. [Google Scholar]
- Sharma, D.; Das, B.K.; Kumar, V.; Tiwari, A.K.; Sahu, P.K.; Singh, S.; Baghel, S. Identification of semi-dwarf and high yielding mutants in Dubraj rice variety of Chhattisgarh through gamma ray based induced mutagenesis. Int. J. Genet. 2017, 9, 298–303. [Google Scholar]
- Martienssen, R.A. Functional genomics: Probing plant gene function and expression with transposons. Proc. Natl. Acad. Sci. USA 1998, 95, 2021–2026. [Google Scholar] [CrossRef] [Green Version]
- Papworth, C.; Bauer, J.C.; Braman, J. QuikChange site-directed mutagenesis. Strategies 1996, 9, 3–4. [Google Scholar]
- Hemsley, A.; Arnheim, N.; Toney, M.D.; Cortopassi, G.; Galas, D.J. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 1989, 17, 6545–6551. [Google Scholar] [CrossRef] [Green Version]
- Serguei, P.; Mayalagu, S.; De, Y.; Yang, W.C.; Kumaran, M.; Sundaresan, V. Analysis of Flanking Sequences from Dissociation Insertion Lines: A Database for Reverse Genetics in Arabidopsis. Plant Cell. 1999, 11, 2263–2270. [Google Scholar]
- Takagi, H.; Tamiru, M.; Abe, A.; Yoshida, K.; Uemura, A.; Yaegashi, H.; Obara, T.; Oikawa, K.; Utsushi, H.; Kanzaki, E.; et al. MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat. Biotechnol. 2015, 33, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Sturtevant, A.H. The Linear Arrangement of Six Sex-Linked Factors in Drosophila, as shown by their mode of Association. J. Exp. Zool. 1913, 14, 43–59. [Google Scholar] [CrossRef]
- Mohan, M.; Nair, S.; Bhagwat, A.; Krishna, T.G.; Yano, M.; Bhatia, C.R.; Sasaki, T. Genome mapping, molecular markers and marker-assisted selection in crop plants. Mol. Breed. 1997, 3, 87–103. [Google Scholar] [CrossRef]
- Collard, B.C.Y.; Jahufer, M.Z.Z.; Brouwer, J.B.; Pang, E.C.K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar] [CrossRef]
- Paterson, A.H.; Lander, E.S.; Hewitt, J.D.; Peterson, S.; Lincoln, S.E.; Tanksley, S.D. Resolution of quantitative traits into Mendelian factors by using a complete linkage map of restriction fragment length polymorphisms. Nature 1988, 335, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Somers, D.J.; Isaac, P.; Edwards, K. A high-density microsatellite consensus map of bread wheat (Triticum aestivum). Theor. Appl. Genet. 2004, 109, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Milczarski, P.; Bolibok-Bragoszewska, H.; Myskow, B.; Stojalowski, S.; Heller-Uszynska, K.; Goralska, M.; Bragoszewski, P.; Uszynski, G.; Kilian, A.; Rakoczy-Trojanowska, M. A high density consensus map of rye (Secale cereale L.) based on DArT markers. PLoS ONE 2011, 6, e2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harushima, Y.; Yano, M.; Shomura, A.; Sato, M.; Shimano, T.; Kuboki, Y.; Yamamoto, T.; Lin, S.Y.; Antonio, B.A.; Parco, A.; et al. A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics 1998, 148, 479–494. [Google Scholar]
- Gupta, S.K.; Souframanien, J.; Gopalakrishna, T. Construction of a genetic linkage map in blackgram [Vigna mungo(L.) Hepper] based on molecular markers and comparative studies. Genome 2008, 51, 628–637. [Google Scholar] [CrossRef]
- Verma, S.; Gupta, S.; Bandhiwal, N.; Kumar, T.; Bharadwaj, C.; Bhatia, S. High-density linkage map construction and mapping of seed trait QTLs in chickpea (Cicer arietinum L.) using Genotyping-by-Sequencing (GBS). Sci. Rep. 2015, 5, 17512. [Google Scholar] [CrossRef] [Green Version]
- Muchero, W.; Ndeye, N.D.; Bhat, P.R.; Fenton, R.D.; Wanamaker, S.; Pottorff, M.; Hearne, S.; Cisse, N.; Fatokun, C.; Ehlers, J.D.; et al. A consensus genetic map of cowpea [Vigna unguiculata (L.) Walp.] and synteny based on EST derived SNPs. Proc. Natl. Acad. Sci. USA 2009, 106, 18159–18164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.; Jenkins, J.; Jia, G.; Hyten, D.L.; Pantalone, V.; Jackson, S.A.; Schmutz, J.; Cregan, P.B. Construction of high resolution genetic linkage maps to improve the soybean genome sequence assembly Glyma1.01. BMC Genom. 2016, 17, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yu, K.; Li, H.; Peng, Q.; Chen, F.; Zhang, W.; Chen, S.; Hu, M.; Zhang, J. High-density SNP map construction and qtl identification for the apetalous character in Brassica napus L. Front. Plant Sci. 2015, 6, 1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.H.; Zhang, S.Z.; Miao, H.R.; Cui, F.G.; Shen, Y.; Yang, W.Q.; Xu, T.T.; Chen, N.; Chi, X.Y.; Zhang, Z.M.; et al. High-Density Genetic Map Construction and Identification of QTLs Controlling Oleic and Linoleic Acid in Peanut using SLAF-seq and SSRs. Sci. Rep. 2018, 8, 5479. [Google Scholar] [CrossRef] [PubMed]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis—A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Xu, F.; Li, Q.; Wang, G.; Liu, N.; Gong, Y.; Li, L.; Chen, Z.H.; Xu, S. QTL mapping combined with bulked segregant analysis identify SNP markers linked to leaf shape traits in Pisum sativum using SLAF sequencing. Front. Genet. 2018, 9, 615. [Google Scholar] [CrossRef] [Green Version]
- Govindaraj, P.; Arumugachamy, S.; Maheswaran, M. Bulked segregant analysis to detect main effect QTL associated with grain quality parameters in Basmati 370/ASD 16 cross in rice Oryza sativa L) using SSR markers. Euphytica 2005, 144, 61–68. [Google Scholar] [CrossRef]
- Gupta, S.K.; Charpe, A.; Prabhu, K.V.; Haque, Q.M.R. Identification and validation of molecular markers linked to the leaf rust resistance gene Lr19 in wheat. Theor. Appl. Genet. 2006, 113, 1027–1036. [Google Scholar] [CrossRef]
- Muylle, H.; Baert, J.; Van Bockstaele, E.; Moerkerke, B.; Goetghebeur, E.; Roldán-Ruiz, I. Identification of molecular markers linked with crown rust (Puccinia coronata f. sp. lolii) resistance in perennial ryegrass (Lolium perenne) using AFLP markers and a bulked segregant approach. Euphytica 2005, 143, 135–144. [Google Scholar] [CrossRef]
- Salunkhe, A.S.; Poornima, R.; Prince, K.S.; Kanagaraj, P.; Sheeba, J.A.; Amudha, K.; Suji, K.K.; Senthil, A.; Babu, R.C. Fine mapping QTL for drought resistance traits in rice (Oryza sativa L.) using bulk segregant analysis. Mol. Biotechnol. 2011, 49, 90–95. [Google Scholar] [CrossRef]
- Huang, C.; Cui, Y.; Weng, C.; Zabel, P.; Lindhout, P. Development of diagnostic PCR markers closely linked to the tomato powdery mildew resistance gene Ol-1 on chromosome 6 of tomato. Theor. Appl. Genet. 2000, 101, 918–924. [Google Scholar] [CrossRef]
- Wang, G.L.; Warren, R.; Innes, G.; Osborne, B.; Baker, B.; Ronald, P.C. Construction of an Arabidopsis BAC library and isolation of clones hybridizing with disease-resistance, gene-like sequences. Plant Mol. Biol. Rep. 1996, 14, 107–114. [Google Scholar] [CrossRef]
- Nilmalgoda, S.D.; Cloutier, S.; Walichnowski, A.Z. Construction and characterization of a bacterial artificial chromosome (BAC) library of hexaploid wheat (Triticum aestivum L.) and validation of genome coverage using locus-specific primers. Genome 2003, 46, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.J.; Thompson, H.; Edwards, D.; de Saizien, A.; Sparks, C.; Thompson, J.A.; Greenland, A.J.; Eyers, M.; Schuch, W. Construction and characterisation of a yeast artificial chromosome library containing three haploid maize genome equivalents. Plant Mol. Biol. 1992, 19, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Umehara, Y.; Inagaki, A.; Tanoue, H.; Yasukochi, Y.; Nagamura, Y.; Saji, S.; Otsuki, Y.; Fujimura, T.; Kurata, N.; Minobe, Y. Construction and characterization of a rice YAC library for physical mapping. Mol. Breed. 1995, 1, 79–89. [Google Scholar] [CrossRef]
- Tomkins, J.P.; Peterson, D.G.; Yang, T.J.; Main, D.; Wilkins, T.A.; Paterson, A.H.; Wing, R.A. Development of genomic resources for cotton (Gossypium hirsutum L.): BAC library construction, preliminary STC analysis, and identification of clones associated with fiber development. Mol. Breed. 2001, 8, 255–261. [Google Scholar] [CrossRef]
- Park, J.Y.; Koo, D.H.; Hong, C.P.; Lee, S.J.; Jeon, J.W.; Lee, S.H.; Yun, P.Y.; Park, B.S.; Kim, H.R.; Bang, J.W.; et al. Physical mapping and microsynteny of Brassica rapa ssp. pekinensisgenome corresponding to a 222 kb gene-rich region of Arabidopsis chromosome-4 and partially duplicated on chromosome 5. Mol. Genet. Genom. 2005, 274, 579–588. [Google Scholar] [CrossRef]
- Nam, Y.W.; Penmetsa, R.V.; Endre, G.; Uribe, P.; Kim, D.; Cool, D.R. Construction of a bacterial artificial chromosome library of Medicago truncatula and identification of clones containing ethylene-response gene. Theor. Appl. Genet. 1999, 98, 638–646. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Maxam, A.M.; Gilbert, W.A. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef] [Green Version]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef] [Green Version]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.A.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, A.N.; Schlueter, J.; Spooner, D.M. Applications of next-generation sequencing in plant biology. Am. J. Bot. 2012, 99, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thudi, M.; Li, Y.; Jackson, S.A.; May, G.D.; Varshney, R.K. Current state-of-art of sequencing technologies for plant genomics research. Brief. Funct. Genom. 2012, 11, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R. A decade’s perspective on DNA sequencing technology. Nature 2011, 470, 198–203. [Google Scholar] [CrossRef]
- Gedil, M.; Ferguson, M.; Girma, G.; Gisel, A.; Stavolone, L.; Rabbi, I. Perspectives on the application of next generation sequencing to the improvement of Africa’s staple food crops. In Next Generation Sequencing—Advances, Applications and Challenges; Kulski, J., Ed.; InTech Open Limited: London, UK, 2016; pp. 287–321. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2010, 10, 57–63. [Google Scholar] [CrossRef]
- Billoud, B.; Le Bail, A.; Charrier, B. A stochastic ID nearest-neighbour automaton models early development of the brown alga Ectocarpus siliculosus. Funct. Plant Biol. 2008, 35, 1014–1024. [Google Scholar] [CrossRef]
- Sarin, S.; Prabhu, S.; O’Meara, M.M.; Pe’er, I.; Hobert, O. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat. Methods 2008, 5, 865–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.; Quinlan, A.R.; Peckham, H.E.; Makowsky, K.; Tao, W.; Woolf, B.; Shen, L.; Donahue, W.F.; Tusneem, N.; Stromberg, M.P.; et al. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008, 18, 1638–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doitsidou, M.; Jarriault, S.; Poole, R.J. Next-Generation Sequencing-based Approaches for Mutation Mapping and identification in Caenorhabditis elegans. Genetics 2016, 204, 451–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, R.; Patel, R.K.; Tyagi, A.K.; Jain, M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011, 18, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.A.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Henry, R.J. Next-generation sequencing for understanding and accelerating crop domestication. Brief. Funct. Genom. 2012, 11, 51–56. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Tabata, R.; Kamiya, T.; Shigenobu, S.; Yamaguchi, K.; Yamada, M.; Hasebe, M.; Fujiwara, T.; Sawa, S. Identification of an EMS-induced causal mutation in a gene required for boron-mediated root development by low-coverage genome re-sequencing in Arabidopsis. Plant Signal Behav. 2013, 8, e22534. [Google Scholar] [CrossRef] [Green Version]
- Salvi, S.; Tuberosa, R. To clone or not to clone plant QTLs: Present and future challenges. Trends Plant Sci. 2005, 10, 297–304. [Google Scholar] [CrossRef]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-throughput genotyping by whole genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef] [Green Version]
- Spindel, J.; Wright, M.; Chen, C.; Cobb, J.; Gage, J.; Harrington, S.; Lorieux, M.; Ahmadi, N.; McCouch, S. Bridging the genotyping gap: Using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor. Appl. Genet. 2013, 126, 2699–2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.A.; Mascher, M.; Buluc, A.; Barry, K.; Georganas, E.; Session, A.; Strnadova, V.; Jenkins, J.; Sehgal, S.; Oliker, L.; et al. A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome. Genome Biol. 2015, 16, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.L.; Grondin, A.; Courtois, B.; Gantet, P. Next-Generation Sequencing Accelerates Crop Gene Discovery. Trends Plant Sci. 2019, 24, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed] [Green Version]
- Gelli, M.; Mitchell, S.E.; Liu, K.; Clemente, T.E.; Weeks, D.P.; Zhang, C.; Holding, D.R.; Dweikat, I.M. Mapping QTLs and association of differentially expressed gene transcripts for multiple agronomic traits under different nitrogen levels in sorghum. BMC Plant Biol. 2016, 16, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Tang, D.; Hu, C.; Qu, J.; Liu, J. Combining meta-QTL with RNA-seq data toidentify candidate genes ofkernel row number trait in maize. Maydica 2016, 61, 9. [Google Scholar]
- Qi, X.; Li, M.; Xie, M.; Liu, X.; Ni, M.; Shao, G.; Song, C.; Yim, A.K.; Tao, Y.; Wong, F.; et al. Identification of a novel salt tolerance gene in wildsoybean by whole-genome sequencing. Nat. Commun. 2014, 5, 4340. [Google Scholar] [CrossRef] [Green Version]
- Majewski, J.; Pastinen, T. The study of eQTL variations by RNA-seq: From SNPs to phenotypes. Trends Genet. 2011, 27, 72–79. [Google Scholar] [CrossRef]
- Westra, H.; Franke, L. From genome to function by studying eQTLs. Biochim. Biophys. Acta 2014, 1842, 1896–1902. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Schneeberger, K. SHOREmap v3.0: Fast and accurate identification of causal mutations from forward genetic screens. Methods Mol. Biol. 2015, 1284, 381–395. [Google Scholar]
- Wang, H.; Zhang, Y.; Sun, L.; Xu, P.; Tu, R.; Meng, S.; Wu, W.; Anis, G.B.; Hussain, K.; Riaz, A.; et al. WB1, a Regulator of Endosperm Development in Rice, Is Identified by a Modified MutMap Method. Int. J. Mol. Sci. 2018, 19, 2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Bayer, P.E.; Batley, J.; Edwards, D. Improvements in genomic technologies: Application to crop genomics. Trends Biotechnol. 2017, 35, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Xu, R.; Duan, P.; Yu, H.; Luo, Y.; Li, Y. Genetic analysis and identification of candidate genes for two spotted-leaf mutants (spl101 and spl102) in rice. Hereditas 2017, 39, 346–353. [Google Scholar] [PubMed]
- Deng, L.; Qin, P.; Liu, Z.; Wang, G.; Chen, W.; Tong, J.; Xiao, L.; Tu, B.; Sun, Y.; Yan, W.; et al. Characterization and fine-mapping of a novel premature leaf senescence mutant yellow leaf and dwarf 1 in rice. Plant Physiol. Biochem. 2017, 111, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.G.; Guo, L.; Yang, G.T.; Qin, P.; Fan, C.L.; Peng, Y.L.; Yan, W.; He, H.; Li, S.G. Genetic analysis of dense and erect panicle-2 allele DEP2-1388 and its application in hybrid rice breeding. Hereditas 2016, 38, 72–81. [Google Scholar]
- Cao, Z.Z.; Lin, X.Y.; Yang, Y.J.; Guan, M.Y.; Xu, P.; Chen, M.X. Gene identification and transcriptome analysis of low cadmium accumulation rice mutant (lcd1) in response to cadmium stress using MutMap and RNA-seq. BMC Plant Biol. 2019, 19, 250. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, M.; He, D.; Wang, K.; Yang, P. Mutations on ent-kaurene oxidase 1 encoding gene attenuate its enzyme activity of catalyzing the reaction from ent-kaurene to ent-kaurenoic acid and lead to delayed germination in rice. PLoS Genet. 2020, 16, e1008562. [Google Scholar] [CrossRef]
- Chen, J.; Huang, X.Y.; Salt, D.E.; Zhao, F.J. Mutation in OsCADT1 enhances cadmium tolerance and enriches selenium in rice grain. New Phytol. 2020, 226, 838–850. [Google Scholar] [CrossRef]
- Sathe, A.P.; Su, X.; Chen, Z.; Chen, T.; Wei, X.; Tang, S.; Zhang, X.; Wu, J. Identification and characterization of a spotted-leaf mutant spl40 with enhanced bacterial blight resistance in rice. Rice 2019, 12, 68. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Q.; Zhang, Y.; Chen, Y.; Yu, N.; Cao, Y.; Zhan, X.; Cheng, S.; Cao, L. LMM24 Encodes Receptor-Like Cytoplasmic Kinase 109, Which Regulates Cell Death and Defense Responses in Rice. Int. J. Mol. Sci. 2019, 20, 3243. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Ma, X.; Zhao, S.; Tang, Y.; Liu, F.; Gu, P.; Fu, Y.; Zhu, Z.; Cai, H.; Sun, C.; et al. The APETALA2-Like Transcription Factor SUPERNUMERARY BRACT Controls Rice Seed Shattering and Seed Size. Plant Cell 2019, 31, 17–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Li, J.; Chen, S.; Heng, Y.; Chen, Z.; Yang, J.; Zhou, K.; Pei, J.; He, H.; Deng, X.W.; et al. Poaceae-specific MS1 encodes a phospholipid-binding protein for male fertility in bread wheat. Proc. Natl. Acad. Sci. USA 2017, 114, 12614–12619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Jiang, Y.; Laza, H.; Payton, P.; Ware, D.; Xin, Z. Identification of the First Nuclear Male Sterility Gene (Male-sterile 9) in Sorghum. Plant Genome 2019, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Burow, G.; Gladman, N.; Martinez, V.A.; Chen, J.; Burke, J.; Ware, D.; Xin, Z. Efficient identification of causal mutations through sequencing of bulked F2 from two allelic bloomless mutants of Sorghum bicolor. Front. Plant Sci. 2018, 8, 2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Jin, F.; Zaman, Q.U.; Ding, B.; Hao, M.; Wang, Y.; Huang, Y.; Wells, R.; Dong, Y.; Hu, Q. Identification of Bna.IAA7.C05 as allelic gene for dwarf mutant generated from tissue culture in oilseed rape. BMC Plant Biol. 2019, 19, 500. [Google Scholar] [CrossRef]
- Tran, Q.H.; Bui, N.H.; Kappel, C.; Dau, N.T.N.; Nguyen, L.T.; Tran, T.T.; Khanh, T.D.; Trung, K.H.; Lenhard, M.; Vi, S.L. Mapping-by-Sequencing via MutMap Identifies a Mutation in ZmCLE7 Underlying Fasciation in a Newly Developed EMS Mutant Population in an Elite Tropical Maize Inbred. Genes 2020, 11, 281. [Google Scholar] [CrossRef] [Green Version]
- Klein, H.; Xiao, Y.; Conklin, P.A.; Govindarajulu, R.; Kelly, J.A.; Scanlon, M.J.; Whipple, C.J.; Bartlett, M. Bulked-Segregant Analysis Coupled to Whole Genome Sequencing (BSA-Seq) for Rapid Gene Cloning in Maize. G3 2018, 8, 3583–3592. [Google Scholar] [CrossRef] [Green Version]
- Amin, G.M.; Kong, K.; Sharmin, R.A.; Kong, J.; Bhat, J.A.; Zhao, T. Characterization and Rapid Gene-Mapping of Leaf Lesion Mimic Phenotype of spl-1 Mutant in Soybean (Glycine max (L.) Merr.). Int. J. Mol. Sci. 2019, 20, 2193. [Google Scholar] [CrossRef] [Green Version]
- Takagi, H.; Abe, A.; Uemura, A.; Oikawa, K.; Utsushi, H.; Yaegashi, H.; Kikuchi, H.; Shimizu, M.; Abe, Y.; Kanzaki, H.; et al. Rice blast resistance gene Pii is controlled by a pair of NBS-LRR genes Pii-1 and Pii-2. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Nakata, M.; Miyashita, T.; Kimura, R.; Nakata, Y.; Takagi, H.; Kuroda, M.; Yamaguchi, T.; Umemoto, T.; Yamakawa, H. MutMapPlus identified novel mutant alleles of a rice starch branching enzyme II b gene for fine-tuning of cooked rice texture. Plant Biotechnol. 2018, 16, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Imamura, T.; Takagi, H.; Miyazato, A.; Ohki, S.; Mizukoshi, H.; Mori, M. Isolation and characterization of the betalain biosynthesis gene involved in hypocotyl pigmentation of the allotetraploid Chenopodium quinoa. Biochem. Biophys. Res. Commun. 2018, 496, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Xiao, Q.; Li, W.; Luo, T.; Yuan, G.; He, Z.; Liu, M.; Li, Q.; Xu, P.; Zhu, J.; et al. OsLAP6/OsPKS1, an orthologue of Arabidopsis PKSA/LAP6, is critical for proper pollen exine formation. Rice 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Dracatos, P.M.; Bartos, J.; Elmansour, H.; Singh, D.; Karafiatova, M.; Zhang, P.; Steuernagel, B.; Svacina, R.; Cobbin, J.C.A.; Clark, B.; et al. The Coiled-Coil NLR Rph1, confers leaf rust resistance in barley cultivar sudan. Plant Physiol. 2019, 179, 1362–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchal, C.; Zhang, J.; Zhang, P.; Fenwick, P.; Steuernagel, B.; Adamski, N.M.; Boyd, L.; McIntosh, R.; Wulff, B.; Berry, S.; et al. BED-domain-containing immune receptors confer diverse resistance spectra to yellow rust. Nat. Plants 2018, 4, 662–668. [Google Scholar] [CrossRef]
- Mo, Y.; Howell, T.; Vasquez-Gross, H.; de Haro, L.A.; Dubcovsky, J.; Pearce, S. Mapping causal mutations by exome sequencing in a wheat TILLING population: A tall mutant case study. Mol. Genet. Genom. 2018, 293, 463–477. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Iqbal, M.A.; Till, B.J.; Rahman, M. Identification of induced mutations in hexaploid wheat genome using exome capture assay. PLoS ONE 2018, 13, e0201918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saintenac, C.; Jiang, D.; Akhunov, E.D. Targeted analysis of nucleotide and copy number variation by exon capture in allotetraploid wheat genome. Genome Biol. 2011, 12, R88. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Upadhyaya, H.D.; Bajaj, D.; Kujur, A.; Badoni, S.; Kumar, V.; Tripathi, S.; Gowda, C.L.L.; Sharma, S.; Singh, S.; et al. Deploying QTL-seq for rapid delineation of a potential candidate gene underlying major trait-associated QTL in chickpea. DNA Res. 2015, 22, 193–203. [Google Scholar] [CrossRef]
- Lu, H.; Lin, T.; Klein, J.; Wang, S.; Qi, J.; Zhou, Q.; Sun, J.; Zhang, Z.; Weng, Y.; Huang, S. QTL-seq identifies an early flowering QTL located near flowering locus T. in cucumber. Theor. Appl. Genet. 2014, 127, 1491–1499. [Google Scholar] [CrossRef]
- Yaobin, Q.; Peng, C.; Yichen, C.; Yue, F.; Derun, H.; Tingxu, H.; Xianjun, S.; Jiezheng, Y. QTL-Seq identified a major QTL for grain length and weight in rice using near isogenic F2 population. Rice Sci. 2018, 25, 121–131. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Guo, N.; Zhang, Y.; Bu, Y.; Zhao, J.; Xing, H. Combining QTL-seq and linkage mapping to fine map a wild soybean allele characteristic of greater plant height. BMC Genom. 2018, 19, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, A.; Fu, Y.; Michael, V.; Meru, G. QTL-seq for identification of loci associated with resistance to Phytophthora crown rot in squash. Sci. Rep. 2020, 10, 5326. [Google Scholar] [CrossRef] [PubMed]
- Edae, E.A.; Rouse, M.N. Bulked segregant analysis RNA-seq (BSR-Seq) validated a stem resistance locus in Aegilops umbellulata, a wild relative of wheat. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishijima, R.; Yoshida, K.; Sakaguchi, K.; Yoshimura, S.I.; Sato, K.; Takumi, S. RNA sequencing-based bulked segregant analysis facilitates efficient D-genome marker development for a specific chromosomal region of synthetic hexaploid wheat. Int. J. Mol. Sci. 2018, 19, 3749. [Google Scholar] [CrossRef] [Green Version]
- Thakur, V.; Wanchana, S. Gene Discovery by Forward Genetic Approach in the Era of High-Throughput Sequencing. OMICS-Based Approaches in Plant Biotechnology. In OMICS-BasedApproaches in Plant Biotechnology; Banerjee, R., Kumar, G.V., Kumar, S.P.J., Eds.; Scrivener Publishing LLC: Beverly, MA, USA, 2019; pp. 75–90. [Google Scholar]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Khan, A.W.; Saxena, R.K.; Kumar, V.; Kale, S.M.; Sinha, P.; Chitikineni, A.; Pazhamala, L.T.; Garg, V.; Sharma, M.; et al. Next-generation sequencing for identification of candidate genes for Fusarium wilt and sterility mosaic disease in pigeonpea (Cajanus cajan). Plant Biotechnol. J. 2016, 14, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.P.M. Discovering new biology through sequencing of RNA. Plant Physiol. 2015, 169, 1524–1531. [Google Scholar]
- Giovannoni, J.J.; Wing, R.A.; Ganal, M.W.; Tanksley, S.D. Isolation of molecular markers from specific chromosomal intervals using DNA pools from existing mapping populations. Nucleic Acids Res. 1991, 19, 6553–6558. [Google Scholar] [CrossRef] [Green Version]
- Illa-Berenguer, E.; Van Houten, J.; Huang, Z.; Van der Knaap, E. Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor. Appl. Genet. 2015, 128, 1329–1342. [Google Scholar] [CrossRef]
- Singh, V.K.; Khan, A.W.; Jaganathan, D.; Thudi, M.; Roorkiwal, M.; Takagi, H.; Garg, V.; Kumar, V.; Chitikineni, A.; Gaur, P.M.; et al. QTL-seq for rapid identification of candidate genes for 100-seed weight and root/total plant dry weight ratio under rainfed conditions in chickpea. Plant Biotechnol. J. 2016, 14, 2110–2119. [Google Scholar] [CrossRef]
- Clevenger, J.; Chu, Y.; Chavarro, C.; Botton, S.; Culbreath, A.; Isleib, T.G.; Holbrook, C.C.; Ozias-Akins, P. Mapping late leaf spot resistance in peanut (Arachis hypogaea) using QTL-seq reveals markers for marker-assisted selection. Front. Plant Sci. 2018, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Branham, S.E. QTL-seq and marker development for resistance to Fusarium oxysporum f. sp. niveum race 1 in cultivated watermelon. Mol. Breed. 2018, 38, 139. [Google Scholar] [CrossRef]
- Fall, L.A.; Clevenger, J.; McGregor, C. Assay development and marker validation for marker assisted selection of Fusarium oxysporum f. sp. niveum race 1 in watermelon. Mol. Breed. 2018, 38, 130. [Google Scholar] [CrossRef]
- Shu, J.; Liu, Y.; Zhang, L.; Li, Z.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H. QTL-seq for rapid identification of candidate genes for flowering time in broccoli × cabbage. Theor. Appl. Genet. 2018, 131, 917–928. [Google Scholar] [CrossRef]
- Branham, S.E.; Farnham, M.W. Identification of heat tolerance loci in broccoli through bulked segregant analysis using whole genome resequencing. Euphytica 2019, 215, 34. [Google Scholar] [CrossRef]
- Warr, A.; Robert, C.; Hume, D.; Archibald, A.; Deeb, N.; Watson, M. Exome sequencing: Current and future perspectives. G3 Genes Genomes Genet. 2015, 5, 1543–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasileva, K.V.; Vasquez-Gross, H.A.; Howell, T.; Bailey, P.; Paraiso, F.; Clissold, L.; Simmonds, J.; Ramirez-Gonzalez, R.H.; Wang, X.; Borrill, P.; et al. Uncovering hidden variation in polyploid wheat. Proc. Natl. Acad. Sci. USA 2017, 114, E913–E921. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.S.; Nakasugi, K.; Doran, R.L.; Millar, A.A.; Waterhouse, P.M. Facile mutant identification via a single parental backcross method and application of whole genome sequencing based mapping pipelines. Front. Plant Sci. 2013, 4, 362. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Gaikwad, K. From Genomes to GENE-omes: Exome Sequencing Concept and Applications in Crop Improvement. Front. Plant Sci. 2017, 8, 2164. [Google Scholar] [CrossRef] [Green Version]
- Marcais, G.; Kingsford, C.A. Fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Feuillet, C.; Travella, S.; Stein, N.; Albar, L.; Nublat, A.; Keller, B. Map-based isolation of the leaf rust disease resistance gene Lr10 from the hexaploid wheat (Triticum aestivum L.) genome. Proc. Natl. Acad. Sci. USA 2003, 100, 15253–15258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowicz, P.D. Constructing gene enriched plant genomic libraries using methylation filtration technology. Methods Mol. Biol. 2003, 236, 21–36. [Google Scholar] [PubMed]
- Shagina, I.; Bogdanova, E.; Mamedov, I.Z.; Lebedev, Y.; Lukyanov, S.; Shagin, D. Normalization of genomic DNA using duplex-specific nuclease. BioTechniques 2010, 48, 455–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oono, Y.; Kobayashi, F.; Kawahara, Y.; Yazawa, T.; Handa, H.; Itoh, T.; Matsumoto, T. Characterization of the wheat (Triticum aestivum L.) transcriptome by de novo assembly for the discovery of phosphate starvation-responsive genes: Gene expression in pi-stressed wheat. BMC Genom. 2013, 214, 77. [Google Scholar]
- Steuernagel, B.; Vrana, J.; Karafiatova, M.; Wulff, B.B.H.; Dolezel, J. Rapid gene isolation using MutChromSeq. In Wheat Rust Diseases: Methods in Molecular Biology; Periyannan, S., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1659, pp. 231–243. [Google Scholar]
- Jupe, F.; Chen, X.; Verweij, W.; Witek, K.; Jones, J.D.; Hein, I. Genomic DNA library preparation for resistance gene enrichment and sequencing (RenSeq) in plants. Methods Mol. Biol. 2014, 1127, 291–303. [Google Scholar]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie-2. Nat. Methods. 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Li, Y.; Fang, X.; Yang, H.; Wang, J.; Kristiansen, K.; Wang, J. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009, 19, 1124–1132. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Gorsi, B.; Su, Y.C.; Yost, H.J. MMAPPR: Mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 2013, 23, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Lochlainn, S.O.; Amoah, S.; Graham, N.S.; Alamer, K.; Rios, J.J.; Kurup, S.; Stoute, A.; Hammond, J.P.; Ostergaard, L.; King, G.J.; et al. High resolution melt (HRM) analysis is an efficient tool to genotype EMS mutant sincomplex crop genomes. Plant Methods 2011, 7, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Yan, W.; Chang, Z.; Xu, Y.; Luo, M.; Xu, C.; Chen, Z. OsMYB80 regulates anther development and pollen fertility by targeting multiple biological pathways. Plant Cell Physiol. 2020, 61, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J. A new mapping strategy. Nat. Plants 2019, 5, 245. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, D.; Bohra, A.; Thudi, M.; Varshney, R.K. Fine mapping and gene cloning in the post-NGS era: Advances and prospects. Theor. Appl. Genet. 2020, 133, 1791–1810. [Google Scholar] [CrossRef] [Green Version]
- Candela, H.; Casanova-Sez, R.; Micol, J.L. Getting started in mapping-by-sequencing. J. Integr. Plant Biol. 2015, 57, 606–612. [Google Scholar] [CrossRef]
- Jordan, K.W.; Wang, S.; Lun, Y.; Gardiner, L.J.; MacLachlan, R.; Hucl, P.; Wiebe, K.; Wong, D.; Forrest, K.L.; Sharpe, A.G.; et al. A haplotype map of allohexaploid wheat reveals distinct patterns of selection on homoeologous genomes. Genome Biol. 2015, 16, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chiodini, R.; Badr, A.; Zhang, G. The impact of next-generation sequencing on genomics. J. Genet. Genom. 2011, 38, 95–109. [Google Scholar] [CrossRef] [Green Version]
- Weigel, D.; Mott, R. The 1001 Genomes Project for Arabidopsis thaliana. Genome Biol. 2009, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Ossowski, S.; Schneeberger, K.; Clark, R.M.; Lanz, C.; Warthmann, N.; Weigel, D. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 2008, 18, 2024–2033. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Han, M.H.; Fedoroff, N. The RNA-binding proteins HYL1 and SE promote accurate in vitro processing of pri-miRNA by DCL1. Proc. Natl. Acad. Sci. USA 2008, 105, 9970–9975. [Google Scholar] [CrossRef] [Green Version]
- Hiraguri, A.; Itoh, R.; Kondo, N.; Nomura, Y.; Aizawa, D.; Murai, Y.; Koiwa, H.; Seki, M.H.; Shinozaki, K.; Fukuhara, T. Specific interactions between Dicer-like proteins and HYL1/DRB family dsRNA-binding proteins in Arabidopsis thaliana. Plant Mol. Biol. 2005, 57, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Kelly, R.; Zhou, G.; Turner, S.A.; Ding, D.; Harris, S.C.; Hong, H.; Fang, H.; Tong, W. SNPTrack: An integrated bioinformatics system for genetic association studies. Hum. Genom. 2012, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marjoram, L.; Alvers, A.; Deerhake, M.E.; Bagwell, J.; Mankiewicz, J.; Cocchiaro, J.L.; Beerman, R.W.; Willer, J.; Sumigray, K.D.; Katsanis, N.; et al. Epigenetic control of intestinal barrier function and inflammation in zebrafish. Proc. Natl. Acad. Sci. USA 2015, 112, 2770–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberman, L.M.; Sparks, E.E.; Moreno-Risueno, M.A.; Petricka, J.J.; Benfey, P.N. MYB36 regulates the transition from proliferation to differentiation in the Arabidopsis root. Proc. Natl. Acad. Sci. USA 2015, 112, 12099–12104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; le Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Wambugu, P.; Ndjiondjop, M.N.; Furtado, A.; Henry, R. Sequencing of bulks of segregants allows dissection of genetic control of amylose content in rice. Plant Biotechnol. J. 2018, 16, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Dhiman, R.; Garibay, A.; Mock, H.P.; Leister, D.; Kleine, T. Cellulose defects in the Arabidopsis secondary cell wall promote early chloroplast development. Plant J. 2020, 101, 156–170. [Google Scholar] [CrossRef]
- Voelkerding, K.V.; Dames, S.; Durtschi, J.D. Next generation sequencing for clinical diagnostics—Principles and application to targeted resequencing for hypertrophic cardiomyopathy. J. Mol. Diagn. 2010, 12, 539–551. [Google Scholar] [CrossRef]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2011, 8, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.D.; Kim, J.B. Frequency and Spectrum of Radiation-Induced Mutations Revealed by Whole-Genome Sequencing Analyses of Plants. Quantum Beam Sci. 2019, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Stahlberg, A.; Paul, M.; Krzyzanowski, P.M.; Jackson, J.B.; Egyud, M.; Stein, L.; Godfrey, T.E. Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection, in liquid biopsies using sequencing. Nucleic Acids Res. 2016, 44. [Google Scholar] [CrossRef] [PubMed]
- Stahlberg, A.; Krzyzanowski, P.M.; Egyud, M.; Filges, S.; Stein, L.; Godfrey, T.E. Simple multiplexed PCR-based barcoding of DNA for ultrasensitive mutation detection by next-generation sequencing. Nat. Protoc. 2017, 12, 664–682. [Google Scholar] [CrossRef]
- Monson-Miller, J.; Sanchez-Mendez, D.C.; Fass, J.; Henry, I.M.; Tai, T.H.; Comai, L. Reference genome-independent assessment of mutation density using restriction enzyme-phased sequencing. BMC Genom. 2012, 13, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlk, D.; Repkova, J. Application of Next-Generation Sequencing in Plant Breeding. Czech J. Genet. Plant Breed. 2017, 53, 89–96. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
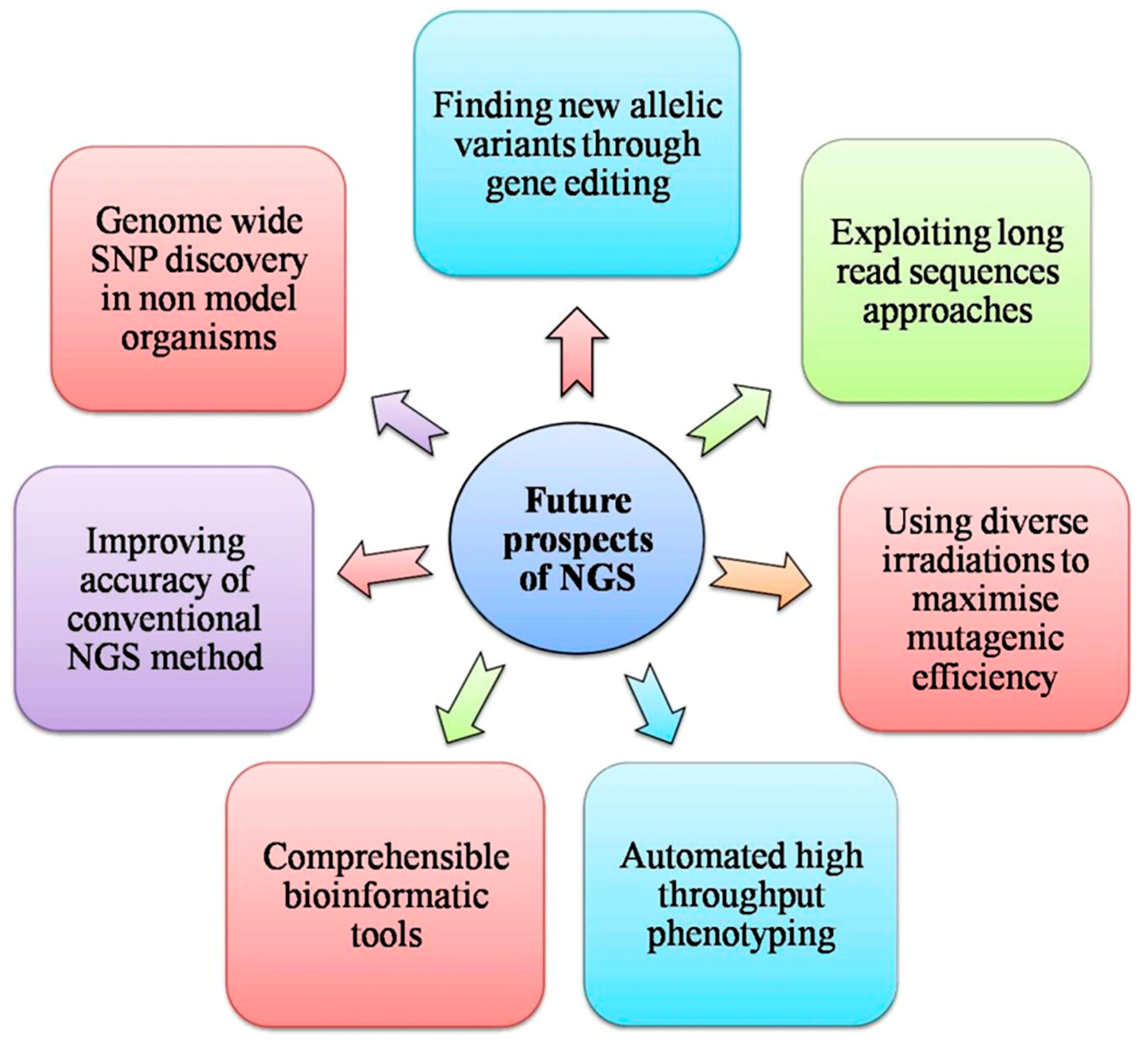{kind=link}
| S. No. | Top 10 Countries Who Has Developed the Highest Number of Crop Mutants | Top 10 Crops Having Highest Number of Crop Mutants | ||
|---|---|---|---|---|
| Country | Number of Mutants | Crops | Number of Mutants | |
| 1 | China | 810 | Rice | 833 |
| 2 | Japan | 479 | Barley | 305 |
| 3 | India | 341 | Chrysanthemum | 285 |
| 4 | Russian Federation | 216 | Wheat | 264 |
| 5 | The Netherlands | 176 | Soybean | 175 |
| 6 | Germany | 171 | Maize | 89 |
| 7 | United States | 139 | Groundnut | 78 |
| 8 | Bulgaria | 76 | Rose | 67 |
| 9 | Bangladesh | 75 | Common bean | 57 |
| 10 | Viet Nam | 58 | Cotton | 48 |
| S. No. | NGS Based Techniques/Approaches | Name of the Gene(s) | Trait(s) | Crop/Species | Strategy Followed | Population Used | Sequencing Platform | Depth (×) | References |
|---|---|---|---|---|---|---|---|---|---|
| 1 | MutMap | OsCAO1 | Pale green leaf | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using an Illumina GAIIx sequencer | >12× | [13] |
| 2 | MutMap | OsRR22 | Salt tolerance | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina GAIIx or Illumina HiSeq2500 | - | [44] |
| 3 | MutMap | Os04t0413500(WB1) | White-belly endosperm | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F 2:3 | Whole-genome sequencing using Illumina HiSeq2500 | 30× | [108] |
| 4 | MutMap | 08SG2/OsBAK1 | Small grain (sg2) | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing | - | [109] |
| 5 | MutMap | OsEDR1 gene | Spotted-leaf mutants (spl101 and spl102) | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing | - | [110] |
| 6 | MutMap | LOC_Os06g29380 | Yellow leaf and dwarf 1 (yld1) | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing | - | [111] |
| 7 | MutMap | DEP2-1388 | Erect panicle (R1338) | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing | - | [112] |
| 8 | MutMap | OsNRAMP5 | Low Cadmium accumulation (lcd1) | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina HiSeq4000 | - | [113] |
| 9 | MutMap | ent-kaurene oxidase 1 (OsKO1) | Delayed seed germination | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina HiSeq 2000 | - | [114] |
| 10 | MutMap | OsCADT1 | Enhanced cadmium tolerance and selenium enriched grain | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina HiSeq4000 | 40× | [115] |
| 11 | MutMap | Os05G0312000 | Spotted-leaf mutant (spl40) | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | High throughput sequencing | 25× | [116] |
| 12 | MutMap | OsRLCK109 (LMM24) | Lesion mimic | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina HiSeq | 50× | [117] |
| 13 | MutMap | SUPERNUMERARY BRACT(SNB) | Loss of shattering | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using llumina HiSeq2500 | - | [118] |
| 14 | MutMap | MS1 | Male Sterility 1 (Ms1) | Wheat (Triticum aestivum L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina HiSeq 2500 | - | [119] |
| 15 | MutMap | Sobic.002G221000 (Ms9) | Nuclear male sterility | Sorghum (Sorghum bicolor) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina X-10 | 15× | [120] |
| 16 | MutMap | Sobic.001G228100 (GDSL-like lipase/acylhydrolase) | Devoid of epi-cuticular wax (EW) | Sorghum (Sorghum bicolor) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina X-10 | 15× | [121] |
| 17 | MutMap | Bna.IAA7.C05 | Dwarfism | Oilseed rape (Brassica napus) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina HiSeq X 10 | - | [122] |
| 18 | MutMap | ZmCLE7 | Fasciated-ear mutant | Maize (Zea mays L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina HiSeq platform | 20× | [123] |
| 19 | MutMap | Zm00001d028818 (Dek1) | Very narrow sheath (vns) | Maize (Zea mays L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina Hi-Seq 2500 | - | [124] |
| 20 | MutMap | Glyma.04g242300 | Spotted leaf-1 (spl-1) | Soybean (Glycine max L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina HiSeq 2500 | - | [125] |
| 21 | MutMap Gap | Os09t0327600-01 (Pii-1) | Susceptibility to rice blast fungus (M. Oryzae) | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina GAIIx sequencer | - | [16] |
| 22 | MutMap Gap | NLR gene (Pii-2) | Susceptibility to rice blast fungus (M. Oryzae) | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | BC1F2 | Whole-genome sequencing using Illumina NextSeq500 | - | [126] |
| 23 | MutMap+ | SNP variants | Leaf colouration | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | M3 population | Whole-genome sequencing using an Illumina GAIIx sequencer | - | [15] |
| 24 | MutMap+ | Starch branching enzyme IIb (BEIIb) gene | Starch gelatinization property | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | M3 population | Illumina Whole-genome sequencing sequencer | >10× | [127] |
| 25 | MutMap+ | CqCYP76AD1-1 | Green hypocotyl mutant (ghy) | Chenopodium quinoa | Mapping by sequencing through BSA | M3 population | Illumina Whole-genome sequencing | - | [128] |
| 26 | MutMap+ | OsLAP6/OsPKS1 | Sterility | Rice (Oryza sativa L.) | Mapping by sequencing through BSA | M3 population | Illumina Whole-genome sequencing | - | [129] |
| 27 | MutChromSeq | Eceriferum-q | Resistant to wax covered leaf sheath | Barley (Hordeum vulgare L.) | Gene cloning by chromosome flow sorting and sequencing | M3 population | Illumina HiSeq2000 platform | 27× | [22] |
| 28 | MutChromSeq | Rph1 | Leaf rust resistance | Barley (Hordeum vulgare L.) | Gene cloning by chromosome flow sorting and sequencing and mutational genomics | DH, RIL and M4 Population | Illumina HiSeq in rapid run mode | 18–30× | [130] |
| 29 | MutChromSeq | Pm2 | Powdery mildew resistance | Wheat (Triticum aestivum L.) | Gene cloning by chromosome flow sorting and sequencing and mutational genomics | M3 population | Illumina HiSeq2000 platform | 35× | [22] |
| 30 | MutRenSeq | Sr22 and Sr45 | Stem rust resistance | Wheat (Triticum aestivum L.) | Exome sequencing of R-gene complements (NB-LRR sequence) and mutational genomics | M3 population | Illumina MiSeq or HiSeq platforms at TGAC | - | [23] |
| 31 | MutRenSeq | Yr7, Yr5 and YrSP | Yellow rust resistance | Wheat (Triticum aestivum L.) | Exome sequencing of R-gene complements (BED-NLR sequence) and mutational genomics | M5 population | Illumina MiSeq or HiSeq platforms | - | [131] |
| 32 | NIKS (needle in the k-stack) | OsCAO1 gene | Pale green leaves and semidwarfism | Rice (Oryza sativa L.) | Identification of frequencies of short subsequences (k-mers) within WGS data of two populations | M3 population | Illumina MiSeq or HiSeq platforms | 51× and 105× | [21] |
| 33 | NIKS (needle in the k-stack) | floral defective 1 (fde1) | Floral homeotic defects | Arabis alpina | Identification of frequencies of short subsequences (k-mers) within WGS data of two populations | M3 population | Illumina MiSeq or HiSeq platforms | 51× and 105× | [21] |
| 34 | LNISKS (longer needle in a scanter k-stack) | ms5 gene | Genic male sterility | Wheat (Triticum aestivum L.) | Identification of frequencies of short subsequences (k-mers) within WGS data of two populations with custom k-filter | BC1F2 | Illumina HiSeq2500 platform | 19× and 23× | [27] |
| 35 | TACCA (Targeted chromosome based via Long-range assembly) | Lr22a | Leaf rust resistance | Wheat (Triticum aestivum L.) | Chromosome sorting, followed by long range chromosome sequencing.Proximity ligation of in-vitroreconstituted chromatin (Chicago) | F2 for mapping and M2 for further validation. | Illumina HiSeq 2500 | 30× | [25] |
| 36 | NGM (Next Generation Mapping) | fph-1, fph-2, mur11-1 | Flupoxam hypersensitive, MURUS-11 both genes involved in cell wall composition. | Arabidopsis (Arabidopsis thaliana) | Short read sequencing of F2 bulks followed by SNP identification in regions of low recombination. | F2 bulks | Illumina GA IIx | 30× or higher | [12] |
| 37 | Exome Capture | Rht-B1 | Height in wheat plants. | Tetraploid Wheat (Triticum turgidum) | The whole complement of exons (coding regions) can be enriched and sequenced using an exome capture approach to reduce the number of bases sequenced leading to lower assay costs. | M4 or stable mutants | Illumina HiSeq 3000 | 20× | [132] |
| 38 | Exome Capture | Chimeric allele of Lr21 | Leaf and Yellow rust | Hexaploid Wheat (Triticum aestivum L.) | The whole complement of exons (coding regions) can be enriched and sequenced using an exome capture approach. | M4 or stable mutants. | Illumina HiSeq 2000 | >20× | [133] |
| 39 | Exome Capture | SNP variants | - | Barley and Wheat (Triticum aestivum L.) | Target sequences derived from full-length cDNA or RNA-Seq contigs are aligned against the Morex assembly. | Wild types and improved cultivars | Single HiSeq2000 lane | 20× | [19] |
| 40 | Exome Capture | SNP variants | - | Allotetraploid Wheat (Triticum turgidum) | Target sequences derived from full-length cDNA or RNA-Seq contigs are aligned against the Morex assembly. | Wild types and improved cultivars | Single HiSeq2000 lane | 20× | [134] |
| 41 | QTL-Seq | Nortai qPi-nor1(t) qPHS3-2 | Partial resistance to the fungal rice blast disease and seedling vigor | Rice (Oryza sativa L.) | QTL identification by combining bulked-segregant analysis and whole-genome resequencing | RIL and F2 | Illumina Genome Analyzer IIx | >6× | [18] |
| 42 | QTL-Seq | SW QTL (CaqSW1.1) | 100-seed weight QTL | Chickpea (Cicer arietinum L.) | NGS-based whole-genome QTL-seq strategy | F4 mapping population | Illumina HiSeq2000 Sequencer | 91–93% coverage | [135] |
| 43 | QTL-seq | Ef1.1 | Early flowering QTL | Cucumber (Cucumis sativus L.) | NGS-based whole-genome QTL-seq strategy | F2 and BC1F2 | Illumina Genome Analyzer IIx machine | 8× | [136] |
| 44 | QTL-seq | qTGW5.3 | Grain size and weight | Rice (Oryza sativa L.) | Bulk segregant analysis and whole genome resequencing | F2 (NIL-F2) | HiSeqXTen (Illumina Sequencer) | 30× | [137] |
| QTL-seq | Glyma.13 g249400 | Plant Height | Soybean (Glycine max L.) | Bulk segregant analysis and whole genome resequencing | F2 and F 2:3 | Illumina HiSeqPE150 machine. | 10× | [138] | |
| 45 | QTL-seq | Chr 4 (QtlPC-C04), 11 QtlPC-C11) and 14 (QtlPC-C14) | Resistance to Phytophthora crown rot in squash | Squash (Cucurbita moschata) | QTL-seq bulk segregant analysis | F2 population | Illumina HiSeq X Sequencer | 45× | [139] |
| 46 | RNA-seq (BSR-Seq) | hoxb1bb1219, nhsl1bfh131, vangl2m209, egr2bfh227 | - | Zebrafish Xenopustropicalis | RNA sequencing based bulked segregant analysis | BC1F2 | Illumina HiSeq 2000 machine | - | [17] |
| 47 | RNA-seq (BSR-Seq) | QTL detected for races TTTTF and TTKSK | Stem resistance locus in Aegilops umbellulata | Asiatic grass (Aegilops umbellulata) | RNA sequencing based bulked segregant analysis | F2, bi- parental mapping populations | Illumina HiSeq 2500 | - | [140] |
| 48 | RNA-seq based BSA (BSR-Seq) | Net2 gene | Synthetic wheat | Wheat (Triticum aestivum L.) | RNA sequencing-based bulked segregant analysis | bi-parental mapping population | Illumina MiSeq sequencer | - | [141] |
| 49 | SHOREmap | AT4G35090 | Slow growth light green leaves | Arabidopsis (Arabidopsis thaliana) | Mapping by sequencing through BSA | BC1F2 | Illumina Whole-genome sequencing | 22× | [11] |
| 50 | deep CAndidateREsequencing (dCARE) | Heterochromatin protein1 (lhp1) | Chromatin-mediated gene repression | Arabidopsis (Arabidopsis thaliana) | Mapping by sequencing through BSA | BC1F2 | Illumina Whole-genome sequencing | 41× | [14] |
| 51 | Simultaneous Identification of Multiple Causal Mutations (SIMM) | LOC_Os03g43670 (H-224 mutant) LOC_Os03g58600 (H-190 mutant) | Open hull and brownish palea/lemma Male sterility | Rice (Oryza sativa L.) | SIMM simultaneous analyze the multiple mutants derived from the same parental plants, with no parental reference genome. It follows Mapping by sequencing through BSA approach. | BC1F2 | Whole-genome sequencing at Illumina Hiseq 2000 platform | >20× | [24] |
| 52 | AgRenSeq | Sr33, Sr45, Sr46 and SrTA1662 | Stem rust resistance | Wheat (Triticum aestivum L.) | AgRenSeq exploits entire gene set of all strains of a species to isolate the uncharacterized R-genes | Germplasm lines | Illumina short-read sequencing | - | [26] |
| S. No. | NGS Based Technique/Approach | Principle | Population Required | Reference Genome Required (Yes/No) | Applicability/Scope (All Species or Any Specific) | Firstly Demonstrated by |
|---|---|---|---|---|---|---|
| 1 | MutMap | Mapping by Whole Genome Sequencing through BSA | BC1F2 | Yes | Applicable in all where crossing is possible and reference genome is available | [13] |
| 2 | MutMap Gap | Mapping by sequencing through BSA | BC1F2 | Yes | Applicable in all where crossing is possible and reference genome is available | [16] |
| 3 | MutMap+ | Mapping by Sequencing through BSA | M3 | Yes | Applicable for the mutants where crossing is difficult or the traits which appear early | [15] |
| 4 | MutChromSeq | Gene cloning by chromosome flow sorting and sequencing and mutational genomics | M3 population | No | Applicable to wheat, barley, rye and other crop species where mutagenesis is possible | [22] |
| 5 | MutRenSeq | Exome sequencing of R-gene complements (NB-LRR sequence) and mutational genomics | M2 /M3/M4/M5 | No | Applicable to plant species with large genome size (wheat, barley, rye) where mutagenesis is possible | [23] |
| 6 | NIKS (needle in the k-stack) | Estimation of the frequencies of k-mers (short subsequences) on the WGS data of two highly related genomes | M3 | No | Applicable to all organisms. However, especially useful for non-model organism where genome has not been sequenced and where mutagenesis is feasible. | [21] |
| 7 | LNISKS (longer needle in a scanter k-stack) | Estimation of the frequencies of k-mers (short subsequences) on the WGS data of two highly related genomes with custom k-filters. | BC1F2 or F2 | No | Applicable to all organisms. However, especially useful for complex genomes and large and repetitive crop genomes like wheat (17 Gbp). | [27] |
| 8 | TACCA (Targeted chromosome based via Long-range assembly) | Generation of a long range scaffold of chromosome with help of either chromosome contact map method or proximity ligation of in-vitroreconstituted chromatin (Chicago). | BC1F2 or M2 lines | No | Applicable to all crop species. | [25] |
| 9 | NGM (Next Generation Mapping) | Identification of causal mutation using sequencing of F2 bulks and computational short downing to SNP present in genomic region of low recombination. | BC1F2 | Yes | Applicable to all species with good quality reference genome. | [12] |
| 10 | Exome capture | Mapping of traits only in the expressed portion of genome, to avoid complexities due to size, repetitive elements etc. present in genome. | BC1F2 bulk or M2 lines. | No | Applicable to all species, but more powerful in sequenced genomes. | [19] |
| 11 | QTL-Seq | QTL-seq combines bulked-segregant analysis and whole-genome resequencing | BC1F2, RIL and DH | Yes | Applicable to all species where whole genome sequence and mapping population is available | [18] |
| 12 | RNA seq based mapping | RNA sequencing based bulked segregant analysis | Mutant and sibling pools | Yes | Applicable to all species where whole genome sequence and mapping population is available | [17] |
| 13 | SHOREmap | Mapping by Sequencing | BC1F2 | Yes | Applicable in all where crossing is possible and reference genome is available | [11] |
| 14 | deep CAndidateREsequencing (dCARE) | Mapping by Sequencing through BSA | BC1F2 | Yes | Applicable in all where crossing is possible and reference genome is available | [14] |
| 15 | Simultaneous Identification of Multiple Causal Mutations (SIMM) | Simultaneous identification of multiple causal mutations in the lines derived from the same parental plant, without requiring a wild- type reference genome. It follows Mapping by sequencing through BSA approach. | BC1F2 | Yes | Identification of causal mutations in multiple mutations at a time by analyzing simultaneously their sequence data. It is Applicable to all. | [24] |
| 16 | AgRenSeq | AgRenSeq exploits entire gene set of all strains of a species to isolate the uncharacterized R-genes | Germplasm lines | No | Discovery and cloning of broad range of resistance genes from diverse germplasm | [26] |
| S. No. | Name of Pipelines/Softwares/Tools | Data/File Requirements | Used in the Genome of Organism | Web Browser Interface or Standalone Software | Applicability/Usefulness | Source Site/URL | Firstly Designed/Developed by |
|---|---|---|---|---|---|---|---|
| 1 | MAQGene | WGS reads in Fastq format | Caenorhabditis elegans | Web browser interface | To detect the causative mutations to further classify the mutations based on associated exon annotations | http://maqweb.sourceforge.net | [29] |
| 2 | CandiSNP | Whole genome high-throughput sequencing data | Arabidopsis thaliana | Web-application | To identify the causal mutations | http://candisnp.tsl.ac.uk | [34] |
| 3 | Next-Generation Mapping (NGM) | SNP data from output of either Maq or SAMtools. | Arabidopsis thaliana | Web browser interface | To identify causal mutation from F2 bulk sequence data. | http://www.bar.utoronto.ca/NGM/index.html | [12] |
| 4 | The SNPtrack tool | Paired files having sequencing reads in fastq format | Zebrafish and Mouse | Web browser interface | Mutation mapping in all model systems | http://genetics.bwh.harvard.edu/snptrack | [32] |
| 5 | artMAP | Data in BAM or FASTQ formats | Arabidopsis thaliana | Standalone software | Identification of EMS-induced mutations in Arabidopsis | https://github.com/RihaLab/artMAP | [28] |
| 6 | CloudMap | Sequencing data. | Caenorhabditis elegans Applicable to other organism also | Web, or cloud or local installation | To detect causal mutations, check for candidate genes, complementation tests. | http://www.usegalaxy.org/cloudmap http://mimodd.readthedocs.io/en/latest/ | [33] |
| 7 | MASS (Mapping and Assembly with Short Sequences) | Paired end reads obtained from direct sequencing | Arabidopsis thaliana | Individual software | Simultaneous mapping and sequencing at a genome-wide level. Identification of a small number of candidate genes/causal mutations within a relatively small interval of 1–2 Mb | http://jcclab.science.oregonstate.edu/MASS | [31] |
| 8 | SHORE and SHOREmap | WGS reads in Fastq format, SHOREmap‘interval’ plot and ‘annotate’ | Arabidopsis thaliana | Web browser interface | To detect the causal mutation site from large pool of recombinant lines | http://1001genomes.org/downloads/shore.html | [11] |
| 9 | GenomeMapper | SBS sequencing reads | Arabidopsis thaliana | Standalone software | Simultaneous alignments of short reads against multiple genomes | http://1001genomes.org | [30] |
| 10 | SIMPLE pipeline | NGS reads in Fastq format | Arabidopsisthaliana, Oryza sativa L. | Individual software | Implemented for mapping causal mutations in any diploid organism with a sequenced genome | https://github.com/wacguy/Simple | [35] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahu, P.K.; Sao, R.; Mondal, S.; Vishwakarma, G.; Gupta, S.K.; Kumar, V.; Singh, S.; Sharma, D.; Das, B.K. Next Generation Sequencing Based Forward Genetic Approaches for Identification and Mapping of Causal Mutations in Crop Plants: A Comprehensive Review. Plants 2020, 9, 1355. https://doi.org/10.3390/plants9101355
Sahu PK, Sao R, Mondal S, Vishwakarma G, Gupta SK, Kumar V, Singh S, Sharma D, Das BK. Next Generation Sequencing Based Forward Genetic Approaches for Identification and Mapping of Causal Mutations in Crop Plants: A Comprehensive Review. Plants. 2020; 9(10):1355. https://doi.org/10.3390/plants9101355
Chicago/Turabian StyleSahu, Parmeshwar K., Richa Sao, Suvendu Mondal, Gautam Vishwakarma, Sudhir Kumar Gupta, Vinay Kumar, Sudhir Singh, Deepak Sharma, and Bikram K. Das. 2020. "Next Generation Sequencing Based Forward Genetic Approaches for Identification and Mapping of Causal Mutations in Crop Plants: A Comprehensive Review" Plants 9, no. 10: 1355. https://doi.org/10.3390/plants9101355