Genome-Wide Association Analysis of Plant Height Using the Maize F1 Population

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Field Trials
2.2. Analysis of Phenotypic Data
2.3. Analysis of Genotypic Data
2.4. Genome-Wide Association Study
3. Result
3.1. Phenotypic Analysis and Heritability
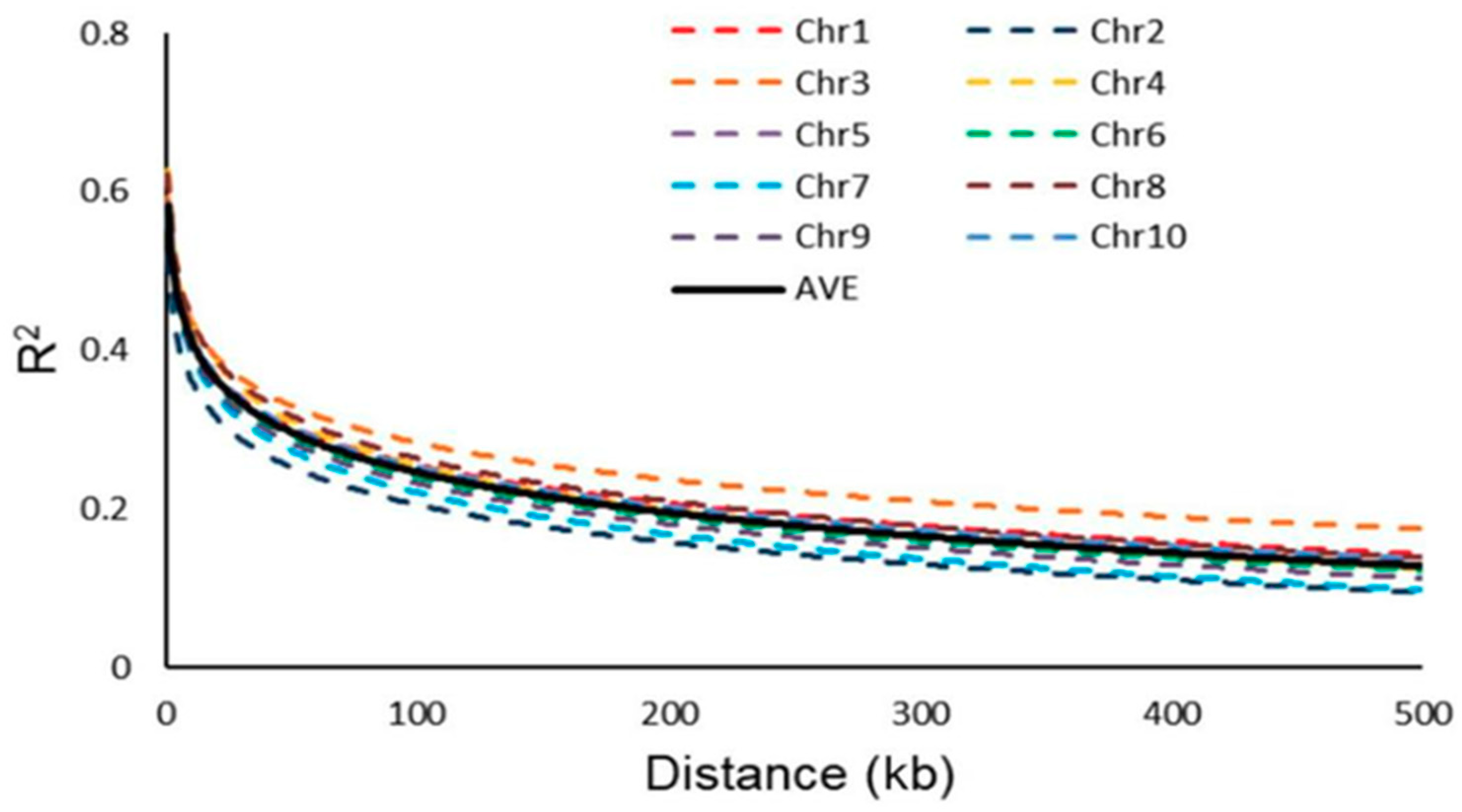
3.2. LD Analysis
3.3. Population Structure Analysis
3.4. Kinship Analysis
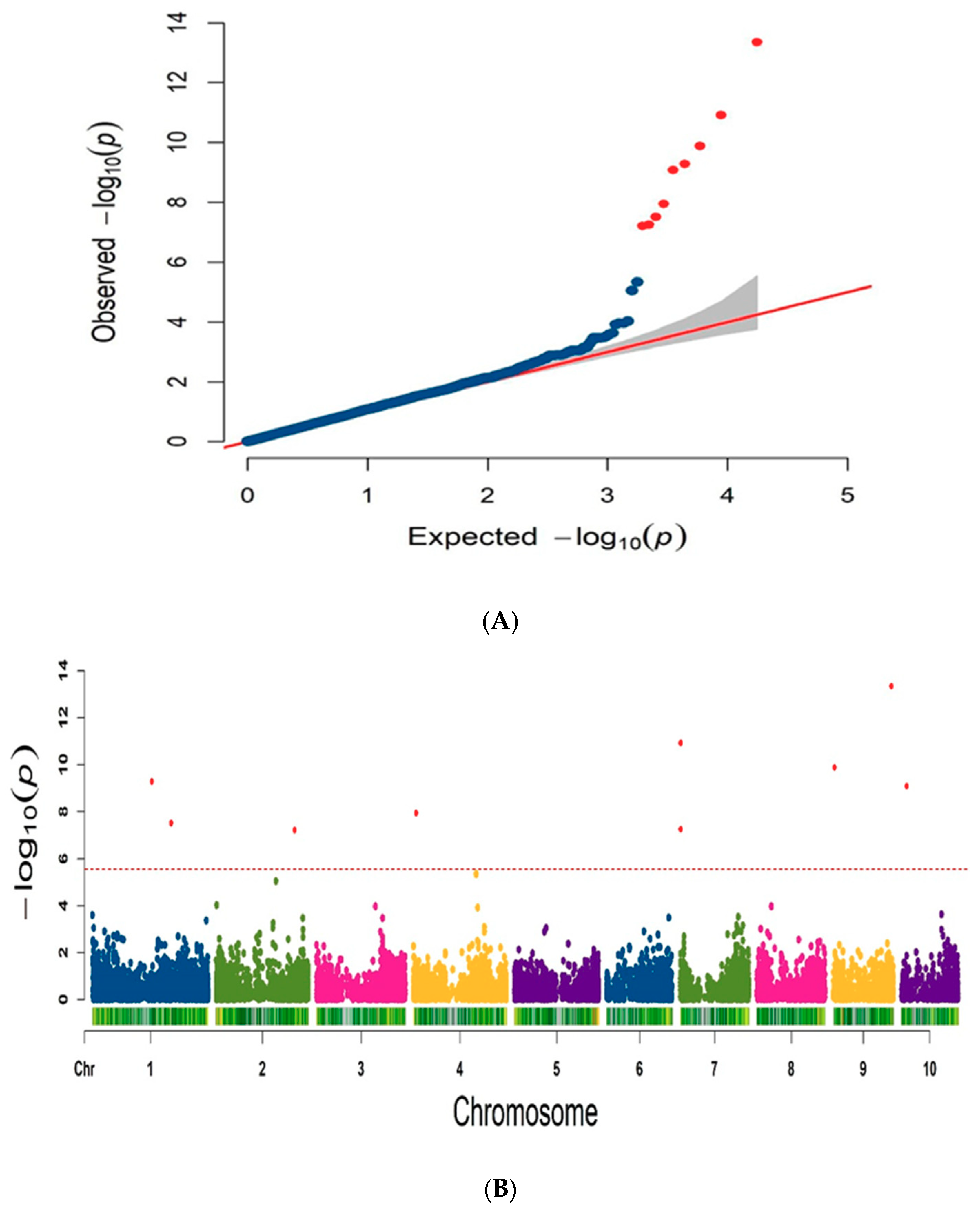
3.5. GWAS Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PH | Plant height |
| GWAS | Genome-wide association analysis |
| SNPs | single nucleotide polymorphisms |
| QTLs | quantitative trait loci |
| GBS | genotyping-by-sequencing |
| BLUP | the best linear unbiased prediction |
References
- Duvick, D. Genetic progress in yield of united states maize (zea mays l.). Maydica 2005, 50, 193. [Google Scholar]
- Cardwell, V. Fifty years of minnesota corn production: Sources of yield increase 1. Agron. J. 1982, 74, 984–990. [Google Scholar] [CrossRef]
- He, K.; Chang, L.; Cui, T.; Qu, J.; Guo, D.; Xu, S.; Zhang, X.; Zhang, R.; Xue, J.; Liu, J. Mapping qtl for plant height and ear height in maize under multi-environments. Sci. Agric. Sin. 2016, 49, 1443–1452. [Google Scholar]
- Lan, J.H.; Chu, D. Study on the genetic basis of plant height and ear height in maize (zea mays l.) by qtl dissection. Yi Chuan Hered. 2005, 27, 925–934. [Google Scholar]
- Li, H.; Chen, Q. QTL analysis of maize plant height and panicle height based on double haploid population. J. Henan Agric. Univ. 2016, 50, 161–166. [Google Scholar]
- Liu, K.; Zhang, X. Genome-Wide Association Studies of Plant Type Traits in Maize. Sci. Agric. Sin. 2018, 51, 821–834. [Google Scholar]
- Vanous, A.; Gardner, C.; Blanco, M.; Martin-Schwarze, A.; Lipka, A.E.; Flint-Garcia, S.; Bohn, M.; Edwards, J.; Lübberstedt, T. Association mapping of flowering and height traits in germplasm enhancement of maize doubled haploid (gem-dh) lines. Plant Genome 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Lawit, S.J.; Wych, H.M.; Xu, D.; Kundu, S.; Tomes, D.T. Maize della proteins dwarf plant8 and dwarf plant9 as modulators of plant development. Plant Cell Physiol. 2010, 51, 1854–1868. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, J.; Wang, M.; Wang, Z.; Li, G.; Qu, L.; Wang, G. Expression and functional analysis of zmdwf4, an ortholog of arabidopsis dwf4 from maize (zea mays L.). Plant Cell Rep. 2007, 26, 2091–2099. [Google Scholar] [CrossRef]
- Spray, C.R.; Kobayashi, M.; Suzuki, Y.; Phinney, B.O.; Gaskin, P.; MacMillan, J. The dwarf−1 (dt) mutant of zea mays blocks three steps in the gibberellin-biosynthetic pathway. Proc. Natl. Acad. Sci. USA 1996, 93, 10515–10518. [Google Scholar] [CrossRef]
- Fujioka, S.; Yamane, H.; Spray, C.R.; Gaskin, P.; Macmillan, J.; Phinney, B.O.; Takahashi, N. Qualitative and quantitative analyses of gibberellins in vegetative shoots of normal, dwarf−1, dwarf−2, dwarf−3, and dwarf−5 seedlings of zea mays l. Plant Physiol. 1988, 88, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.G.; Helentjaris, T. The maize dwarf3 gene encodes a cytochrome p450-mediated early step in gibberellin biosynthesis. Plant Cell 1995, 7, 1307–1317. [Google Scholar] [PubMed]
- Ogawa, M.; Kusano, T.; Koizumi, N.; Katsumi, M.; Sano, H. Gibberellin-responsive genes: High level of transcript accumulation in leaf sheath meristematic tissue from zea mays l. Plant Mol. Biol. 1999, 40, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Harberd, N.P.; Freeling, M. Genetics of dominant gibberellin-insensitive dwarfism in maize. Genetics 1989, 121, 827–838. [Google Scholar] [PubMed]
- Bensen, R.J.; Johal, G.S.; Crane, V.C.; Tossberg, J.T.; Schnable, P.S.; Meeley, R.B.; Briggs, S.P. Cloning and characterization of the maize an1 gene. Plant Cell 1995, 7, 75–84. [Google Scholar] [PubMed]
- Tao, Y.; Zheng, J.; Xu, Z.; Zhang, X.; Zhang, K.; Wang, G. Functional analysis of zmdwf1, a maize homolog of the arabidopsis brassinosteroids biosynthetic dwf1/dim gene. Plant Sci. 2004, 167, 743–751. [Google Scholar] [CrossRef]
- Teng, F.; Zhai, L.; Liu, R.; Bai, W.; Wang, L.; Huo, D.; Tao, Y.; Zheng, Y.; Zhang, Z. Zmga3ox2, a candidate gene for a major qtl, qph3. 1, for plant height in maize. Plant J. 2013, 73, 405–416. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (gbs) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Knapp, S.; Stroup, W.; Ross, W. Exact confidence intervals for heritability on a progeny mean basis 1. Crop Sci. 1985, 25, 192–194. [Google Scholar] [CrossRef]
- Goldberger, A.S. Best linear unbiased prediction in the generalized linear regression model. J. Am. Stat. Assoc. 1962, 57, 369–375. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, H.; Bo, C.; Dai, W.; Zhang, X.; Cai, R.; Gu, L.; Ma, Q.; Jiang, H.; Zhu, J. Genome-wide association study of maize plant architecture using f 1 populations. Plant Mol. Biol. 2019, 99, 1–15. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Liu, M.; Tan, X.; Yang, Y.; Liu, P.; Zhang, X.; Zhang, Y.; Wang, L.; Hu, Y.; Ma, L.; Li, Z. Analysis of the genetic architecture of maize kernel size traits by combined linkage and association mapping. Plant Biotechnol. J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Zhao, X.; Luo, L.; Cao, Y.; Liu, Y.; Li, Y.; Wu, W.; Lan, Y.; Jiang, Y.; Gao, S.; Zhang, Z. Genome-wide association analysis and qtl mapping reveal the genetic control of cadmium accumulation in maize leaf. BMC Genom. 2018, 19, 91. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A. The b73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xue, Y.; Guo, Z.; Li, W.; Tang, J. Genome-wide association study identifies candidate genes for starch content regulation in maize kernels. Front. Plant Sci. 2016, 7, 1046. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Z.; Ding, J.; Wu, Y.; Zhou, B.; Wang, R.; Ma, J.; Wang, S.; Zhang, X.; Xia, Z. Combined linkage and association mapping reveals qtl and candidate genes for plant and ear height in maize. Front. Plant Sci. 2016, 7, 833. [Google Scholar] [CrossRef]
- Monna, L.; Kitazawa, N.; Yoshino, R.; Suzuki, J.; Masuda, H.; Maehara, Y.; Tanji, M.; Sato, M.; Nasu, S.; Minobe, Y. Positional cloning of rice semidwarfing gene, sd−1: Rice “green revolution gene” encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 2002, 9, 11–17. [Google Scholar] [CrossRef]
- Peng, J.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F. ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 1999, 400, 256. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Xie, C.; Hao, Z.; Wang, J.; Liu, C.; Li, M.; Zhang, D.; Bai, L.; Zhang, S.; Li, X. Genome-wide association study identifies candidate genes that affect plant height in chinese elite maize (zea mays L.) inbred lines. PLoS ONE 2011, 6, e29229. [Google Scholar] [CrossRef] [PubMed]
- Multani, D.S.; Briggs, S.P.; Chamberlin, M.A.; Blakeslee, J.J.; Murphy, A.S.; Johal, G.S. Loss of an mdr transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science 2003, 302, 81–84. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean | Minimum | Maximum | SD | Skewness | Kurtosis | CV (%) | H2 (%) |
|---|---|---|---|---|---|---|---|---|
| PH | 290.67 | 239.15 | 324.50 | 16.34 | −0.56 | 0.26 | 5.62 | 83.32 |
| Trait | Source | SS | DF | MS | F Value | Sig |
|---|---|---|---|---|---|---|
| PH | G | 954,834.54 | 299 | 3193.43 | 32.98 | <0.01 ** |
| E | 1,122,082.30 | 4 | 280,520.57 | 2897.20 | <0.01 ** | |
| G×E | 363,150.96 | 1194 | 304.15 | 3.14 | <0.01 ** | |
| Error | 142,235.55 | 1469 | 96.83 |
| Trait | Chr | SNP ID | SNP Physical Position | p Value |
|---|---|---|---|---|
| PH | 1 | SNP−41465 | 157,566,180 | 5.19 × 10−10 |
| 1 | SNP−54654 | 208,218,658 | 3.05 × 10−8 | |
| 2 | SNP−137325 | 207,979,043 | 6.08 × 10−8 | |
| 4 | SNP−214914 | 7,407,530 | 1.14 × 10−8 | |
| 7 | SNP−382338 | 1,148,863 | 5.57 × 10−8 | |
| 7 | SNP−382339 | 1,148,876 | 1.20× 10−11 | |
| 9 | SNP−480424 | 3,878,215 | 1.31 × 10−10 | |
| 9 | SNP−516792 | 154,433,535 | 4.49 × 10−14 | |
| 10 | SNP−524015 | 13,705,944 | 8.83 × 10−10 |
| Trait | Chr | SNP ID | Gen ID | Encoding |
|---|---|---|---|---|
| PH | 7 | SNP−382338 | Zm00001d018617 | gibberellin 2–oxidase 12 |
| 10 | SNP−524015 | Zm00001d023659 | auxin response factor 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wan, J.; He, L.; Lan, H.; Li, L. Genome-Wide Association Analysis of Plant Height Using the Maize F1 Population. Plants 2019, 8, 432. https://doi.org/10.3390/plants8100432
Zhang Y, Wan J, He L, Lan H, Li L. Genome-Wide Association Analysis of Plant Height Using the Maize F1 Population. Plants. 2019; 8(10):432. https://doi.org/10.3390/plants8100432
Chicago/Turabian StyleZhang, Yong, Jiyu Wan, Lian He, Hai Lan, and Lujiang Li. 2019. "Genome-Wide Association Analysis of Plant Height Using the Maize F1 Population" Plants 8, no. 10: 432. https://doi.org/10.3390/plants8100432