
Effects of Allopolyploidization and Homoeologous Chromosomal Segment Exchange on Homoeolog Expression in a Synthetic Allotetraploid Wheat under Variable Environmental Conditions

Abstract
:1. Introduction
2. Results
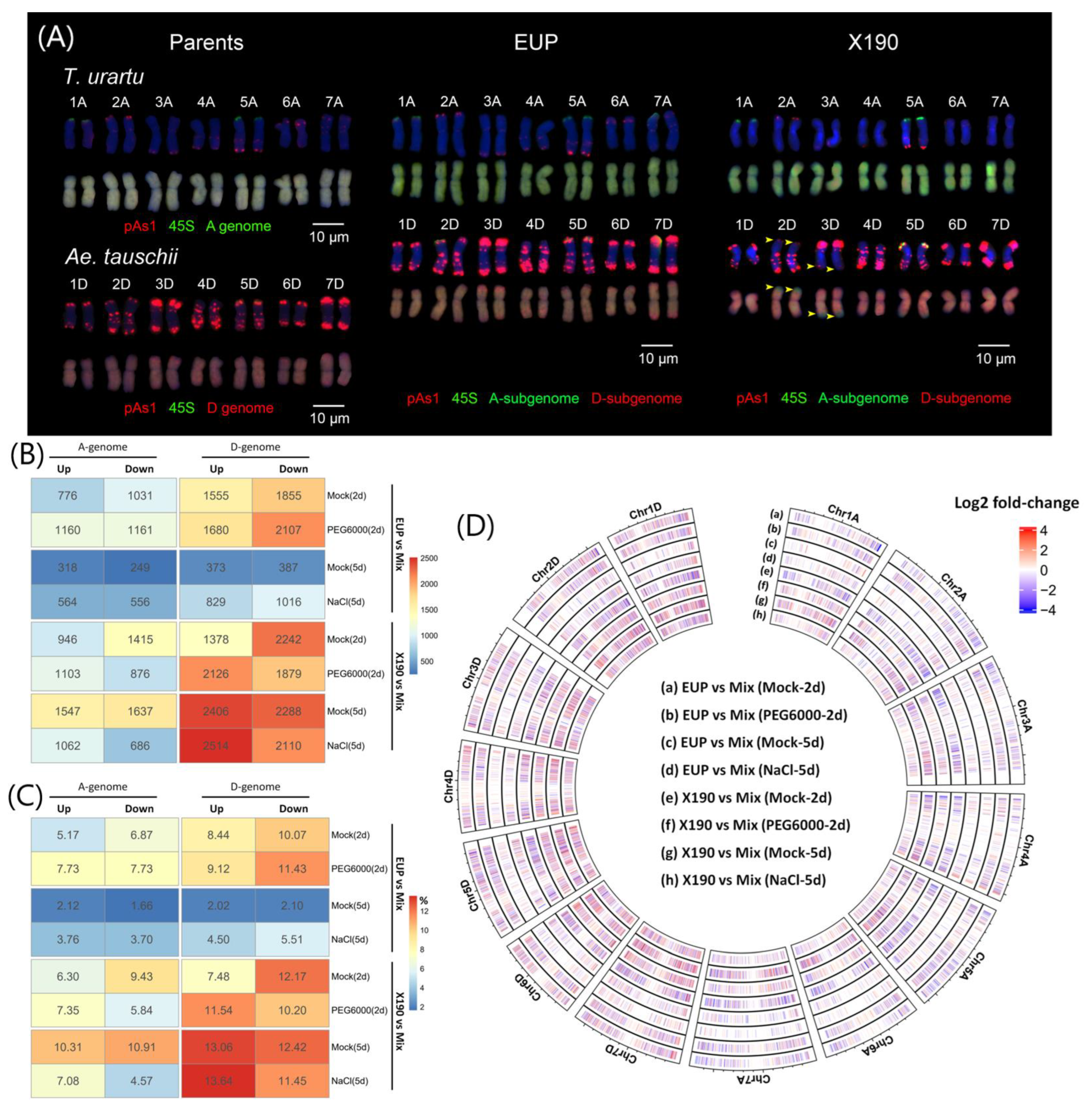
2.1. Subgenome Expression Changes following Allopolyploidization with and without Homoeologous Chromosomal Segment Exchanges under Different Environmental Conditions
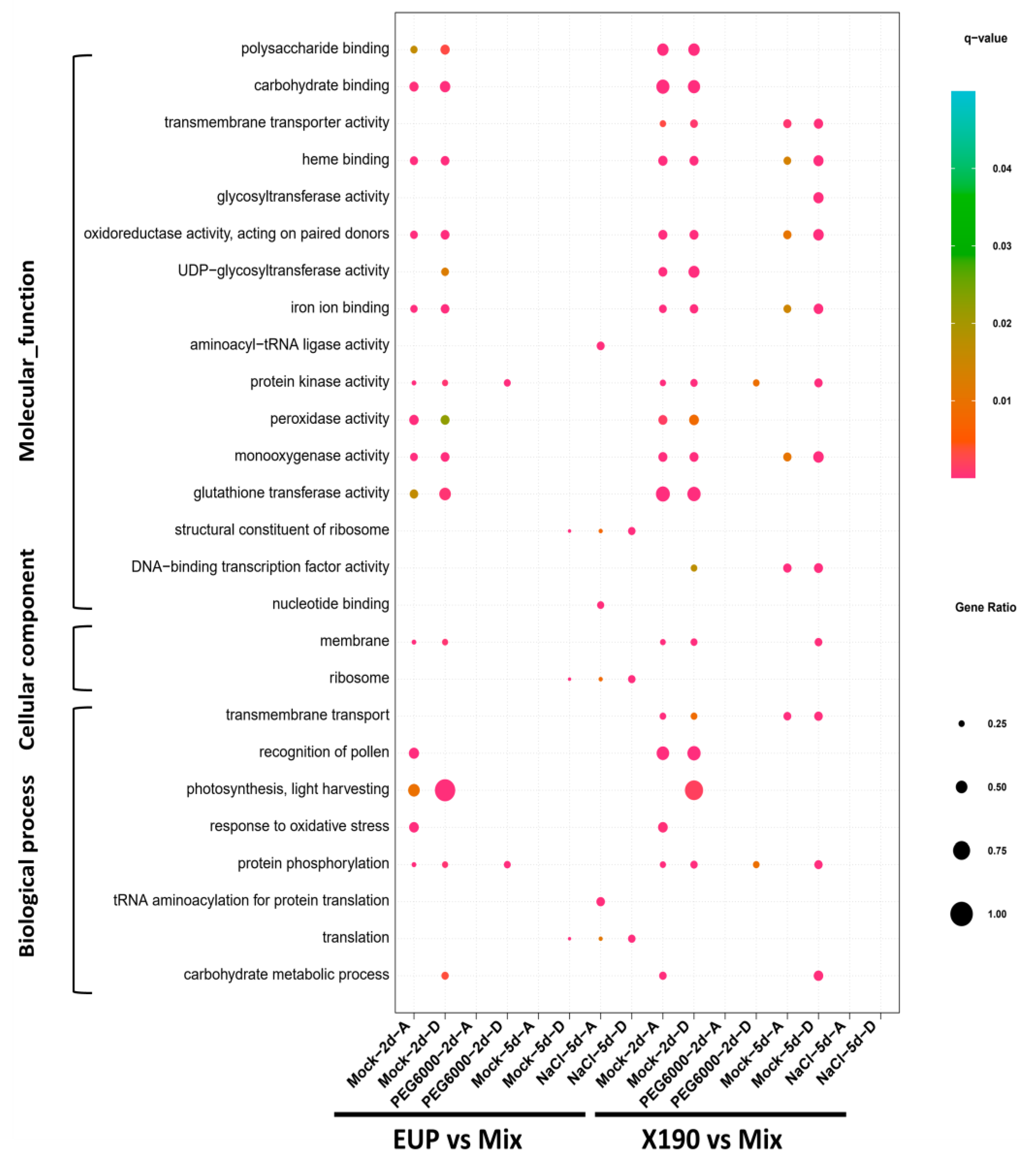
2.2. Functional Enrichment of Subgenome DEGs in the Synthetic Allotetraploid Wheat
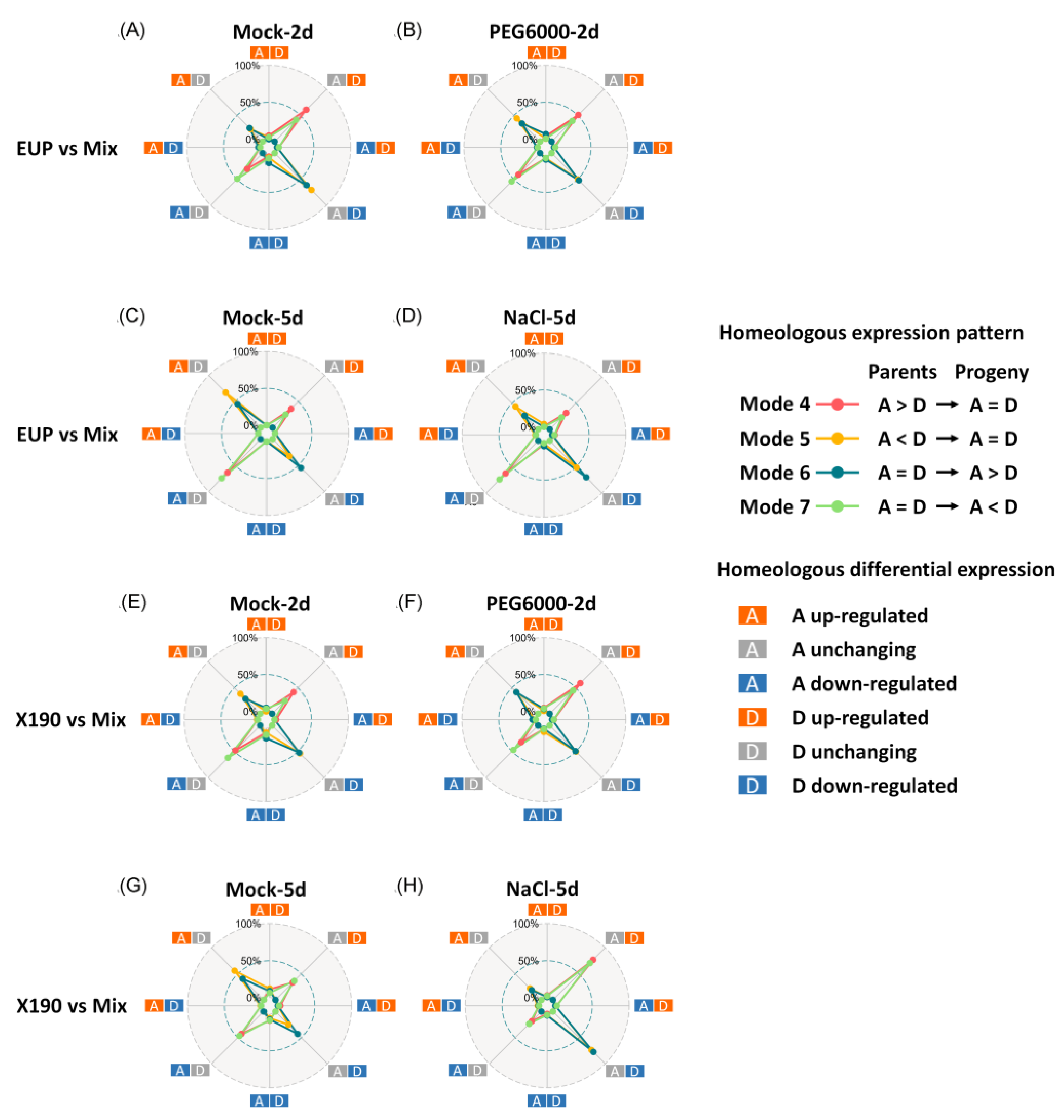
2.3. Modes of Homoeolog Expression in the Synthetic Allotetraploid Wheat
2.4. Homoeolog Expression Regulation in the Synthetic Allotetraploid Wheat
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Data Processing and Gene Expression Counts
4.3. Identification of Differentially Expressed Genes
4.4. Gene Ontology Enrichment Analysis
4.5. Identification of Collinear Homoeologous Gene Pairs
4.6. Comparison of Homoeologous Expression and Classification of Homoeologous Expression Modes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.A.; Maere, S.; Van de Peer, Y. Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc. Natl. Acad. Sci. USA 2009, 106, 5737–5742. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E. The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 2009, 60, 561–588. [Google Scholar] [CrossRef]
- Leitch, A.; Leitch, I. Genomic plasticity and the diversity of polyploid plants. Science 2008, 320, 481–483. [Google Scholar] [CrossRef]
- McClintock, B. The significance of responses of the genome to challenge. Science 1984, 226, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Comai, L.; Madlung, A.; Josefsson, C.; Tyagi, A. Do the different parental ‘heteromes’ cause genomic shock in newly formed allopolyploids? Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Ni, Z. Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. Bioessays 2006, 28, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Flagel, L.E.; Paterson, A.H.; Rapp, R.A.; Soltis, D.E.; Soltis, P.S.; Wendel, J.F. Evolutionary genetics of genome merger and doubling in plants. Annu. Rev. Genet. 2008, 42, 443–461. [Google Scholar] [CrossRef]
- Buggs, R.J.; Zhang, L.; Miles, N.; Tate, J.A.; Gao, L.; Wei, W.; Schnable, P.S.; Barbazuk, W.B.; Soltis, P.S.; Soltis, D.E. Transcriptomic shock generates evolutionary novelty in a newly formed, natural allopolyploid plant. Curr. Biol. 2011, 21, 551–556. [Google Scholar] [CrossRef]
- Yoo, M.-J.; Liu, X.; Pires, J.C.; Soltis, P.S.; Soltis, D.E. Nonadditive gene expression in polyploids. Annu. Rev. Genet. 2014, 48, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Edger, P.P.; Smith, R.; McKain, M.R.; Cooley, A.M.; Vallejo-Marin, M.; Yuan, Y.; Bewick, A.J.; Ji, L.; Platts, A.E.; Bowman, M.J. Subgenome dominance in an interspecific hybrid, synthetic allopolyploid, and a 140-year-old naturally established neo-allopolyploid monkeyflower. Plant Cell 2017, 29, 2150–2167. [Google Scholar] [CrossRef] [PubMed]
- Bird, K.A.; VanBuren, R.; Puzey, J.R.; Edger, P.P. The causes and consequences of subgenome dominance in hybrids and recent polyploids. New Phytol. 2018, 220, 87–93. [Google Scholar] [CrossRef]
- Jiang, X.; Song, Q.; Ye, W.; Chen, Z.J. Concerted genomic and epigenomic changes accompany stabilization of Arabidopsis allopolyploids. Nat. Ecol. Evol. 2021, 5, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Chen, Z.J. Genomic and expression plasticity of polyploidy. Curr. Opin. Plant Biol. 2010, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Rapp, R.A.; Udall, J.A.; Wendel, J.F. Genomic expression dominance in allopolyploids. BMC Biol. 2009, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tian, L.; Lee, H.S.; Wei, N.E.; Jiang, H.; Watson, B.; Madlung, A.; Osborn, T.C.; Doerge, R.W.; Comai, L.; et al. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 2006, 172, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.J.; Szadkowski, E.; Wendel, J.F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 2013, 110, 171–180. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Li, Y.; Zhang, Z.; Li, L.; Liu, B. Transcriptome asymmetry in synthetic and natural allotetraploid wheats, revealed by RNA-sequencing. New Phytol. 2016, 209, 1264–1277. [Google Scholar] [CrossRef]
- Zhao, N.; Dong, Q.; Nadon, B.D.; Ding, X.; Wang, X.; Dong, Y.; Liu, B.; Jackson, S.A.; Xu, C. Evolution of Homeologous Gene Expression in Polyploid Wheat. Genes 2020, 11, 1401. [Google Scholar] [CrossRef]
- Liu, Z.; Adams, K.L. Expression partitioning between genes duplicated by polyploidy under abiotic stress and during organ development. Curr. Biol. 2007, 17, 1669–1674. [Google Scholar] [CrossRef]
- Dong, S.; Adams, K.L. Differential contributions to the transcriptome of duplicated genes in response to abiotic stresses in natural and synthetic polyploids. New Phytol. 2011, 190, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Bardil, A.; de Almeida, J.D.; Combes, M.C.; Lashermes, P.; Bertrand, B. Genomic expression dominance in the natural allopolyploid Coffea arabica is massively affected by growth temperature. New Phytol. 2011, 192, 760–774. [Google Scholar] [CrossRef]
- Combes, M.C.; Cenci, A.; Baraille, H.; Bertrand, B.; Lashermes, P. Homeologous gene expression in response to growing temperature in a recent Allopolyploid (Coffea arabica L.). J. Hered. 2012, 103, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Combes, M.C.; Dereeper, A.; Severac, D.; Bertrand, B.; Lashermes, P. Contribution of subgenomes to the transcriptome and their intertwined regulation in the allopolyploid Coffea arabica grown at contrasted temperatures. New Phytol. 2013, 200, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Hu, G.; Grover, C.E.; Miller, E.R.; Zhu, S.; Wendel, J.F. Parental legacy versus regulatory innovation in salt stress responsiveness of allopolyploid cotton (Gossypium) species. Plant J. 2022, 111, 872–887. [Google Scholar] [CrossRef]
- Consortium, I.W.G.S.; Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar]
- Levy, A.A.; Feldman, M. Evolution and origin of bread wheat. Plant Cell 2022, 34, 2549–2567. [Google Scholar] [CrossRef]
- Xiao, J.; Liu, B.; Yao, Y.; Guo, Z.; Jia, H.; Kong, L.; Zhang, A.; Ma, W.; Ni, Z.; Xu, S. Wheat genomic study for genetic improvement of traits in China. Sci. China Life Sci. 2022, 65, 1718–1775. [Google Scholar] [CrossRef]
- Sha, Y.; Li, Y.; Zhang, D.; Lv, R.; Wang, H.; Wang, R.; Ji, H.; Li, S.; Gong, L.; Li, N. Genome shock in a synthetic allotetraploid wheat invokes subgenome-partitioned gene regulation, meiotic instability, and karyotype variation. J. Exp. Bot. 2023, erad247. [Google Scholar] [CrossRef]
- Wang, B.; Lv, R.; Zhang, Z.; Yang, C.; Xun, H.; Liu, B.; Gong, L. Homoeologous exchange enables rapid evolution of tolerance to salinity and hyper-osmotic stresses in a synthetic allotetraploid wheat. J. Exp. Bot. 2022, 73, 7488–7502. [Google Scholar] [CrossRef]
- Hu, G.; Wendel, J.F. Cis–trans controls and regulatory novelty accompanying allopolyploidization. New Phytol. 2019, 221, 1691–1700. [Google Scholar] [CrossRef]
- Alger, E.I.; Edger, P.P. One subgenome to rule them all: Underlying mechanisms of subgenome dominance. Curr. Opin. Plant Biol. 2020, 54, 108–113. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Gou, X.; Bian, Y.; Zhang, A.; Zhang, H.; Wang, B.; Lv, R.; Li, J.; Zhu, B.; Gong, L.; Liu, B. Transgenerationally precipitated meiotic chromosome instability fuels rapid karyotypic evolution and phenotypic diversity in an artificially constructed allotetraploid wheat (AADD). Mol. Biol. Evol. 2018, 35, 1078–1091. [Google Scholar] [CrossRef]
- Lv, R.; Wang, C.; Wang, R.; Wang, X.; Zhao, J.; Wang, B.; Aslam, T.; Han, F.; Liu, B. Chromosomal instability and phenotypic variation in a specific lineage derived from a synthetic allotetraploid wheat. Front. Plant Sci. 2022, 13, 981234. [Google Scholar] [CrossRef]
- Zhao, J.; Li, J.; Lv, R.; Wang, B.; Zhang, Z.; Yu, T.; Liu, S.; Xun, H.; Xu, C.; Wendel, J.F. Meiotic pairing irregularity and homoeologous chromosome compensation cause rapid karyotype variation in synthetic allotetraploid wheat. New Phytol. 2023, 239, 606–623. [Google Scholar] [CrossRef]
- Boideau, F.; Richard, G.; Coriton, O.; Huteau, V.; Belser, C.; Deniot, G.; Eber, F.; Falentin, C.; Ferreira de Carvalho, J.; Gilet, M. Epigenomic and structural events preclude recombination in Brassica napus. New Phytol. 2022, 234, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Rönspies, M.; Schmidt, C.; Schindele, P.; Lieberman-Lazarovich, M.; Houben, A.; Puchta, H. Massive crossover suppression by CRISPR–Cas-mediated plant chromosome engineering. Nat. Plants 2022, 8, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Maherali, H.; Walden, A.E.; Husband, B.C. Genome duplication and the evolution of physiological responses to water stress. New Phytol. 2009, 184, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Hu, G.; Yu, J.; Thu, S.W.; Grover, C.E.; Zhu, S.; Wendel, J.F. Salt-tolerance diversity in diploid and polyploid cotton (Gossypium) species. Plant J. 2020, 101, 1135–1151. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Guo, Z.; Feng, X.; Shao, S.; Yang, Y.; Li, J.; Zhong, C.; He, Z.; Shi, S. Where whole-genome duplication is most beneficial: Adaptation of mangroves to a wide salinity range between land and sea. Mol. Ecol. 2023, 32, 460–475. [Google Scholar] [CrossRef]
- Lloyd, A.; Blary, A.; Charif, D.; Charpentier, C.; Tran, J.; Balzergue, S.; Delannoy, E.; Rigaill, G.; Jenczewski, E. Homoeologous exchanges cause extensive dosage-dependent gene expression changes in an allopolyploid crop. New Phytol. 2018, 217, 367–377. [Google Scholar] [CrossRef]
- Zhang, Z.; Xun, H.; Lv, R.; Gou, X.; Ma, X.; Li, J.; Zhao, J.; Li, N.; Gong, L.; Liu, B. Effects of homoeologous exchange on gene expression and alternative splicing in a newly formed allotetraploid wheat. Plant J. 2022, 111, 1267–1282. [Google Scholar] [CrossRef]
- Powell, J.J.; Fitzgerald, T.L.; Stiller, J.; Berkman, P.J.; Gardiner, D.M.; Manners, J.M.; Henry, R.J.; Kazan, K. The defence-associated transcriptome of hexaploid wheat displays homoeolog expression and induction bias. Plant Biotechnol. J. 2017, 15, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Adams, K.L. Global insights into duplicated gene expression and alternative splicing in polyploid Brassica napus under heat, cold, and drought stress. Plant Genome 2020, 13, e20057. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Ashman, T.-L.; Soltis, P.S.; Soltis, D.E. Polyploidy: An evolutionary and ecological force in stressful times. Plant Cell 2021, 33, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, W.; Wang, F.; Wu, X.; Wang, J. Asymmetric epigenetic modification and homoeolog expression bias in the establishment and evolution of allopolyploid Brassica napus. New Phytol. 2021, 232, 898–913. [Google Scholar] [CrossRef]
- Zhang, Q.; Guan, P.; Zhao, L.; Ma, M.; Xie, L.; Li, Y.; Zheng, R.; Ouyang, W.; Wang, S.; Li, H. Asymmetric epigenome maps of subgenomes reveal imbalanced transcription and distinct evolutionary trends in Brassica napus. Mol. Plant 2021, 14, 604–619. [Google Scholar] [CrossRef]
- Rayburn, A.L.; Gill, B. Molecular identification of the D-genome chromosomes of wheat. J. Hered. 1986, 77, 253–255. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Mode | Progeny | Mix vs. EUP | Mix vs. X190 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parents | Mock 2d | PEG6000 2d | Mock 5d | NaCl 5d | Mock 2d | PEG6000 2d | Mock 5d | NaCl 5d | ||
| Conserved regulation | ||||||||||
| Mode 1 | A > D | A > D | 360 (8.14%) | 762 (17.24%) | 452 (10.23%) | 702 (15.88%) | 351 (7.94%) | 776 (17.56%) | 341 (7.71%) | 779 (17.62%) |
| Mode 2 | A < D | A < D | 357 (8.08%) | 790 (17.87%) | 420 (9.50%) | 758 (17.15%) | 341 (7.71%) | 785 (17.76%) | 352 (7.96%) | 791 (17.90%) |
| Mode 3 | A = D | A = D | 2645 (59.84%) | 1633 (36.95%) | 2648 (59.91%) | 1679 (37.99%) | 2648 (59.91%) | 1552 (35.11%) | 2640 (59.73%) | 1524 (34.48%) |
| Subtotal | 3362 (76.06%) | 3185 (72.06%) | 3520 (79.64%) | 3139 (71.02%) | 3340 (75.57%) | 3113 (70.43%) | 3333 (75.41%) | 3094 (70.00%) | ||
| Convergent regulation | ||||||||||
| Mode 4 | A > D | A = D | 425 (9.62%) | 401 (9.07%) | 305 (6.90%) | 436 (9.86%) | 435 (9.84%) | 381 (8.62%) | 413 (9.34%) | 358 (8.10%) |
| Mode 5 | A < D | A = D | 279 (6.31%) | 353 (7.99%) | 232 (5.25%) | 403 (9.12%) | 297 (6.72%) | 355 (8.03%) | 298 (6.74%) | 362 (8.19%) |
| Subtotal | 704 (15.93%) | 754 (17.06%) | 537 (12.15%) | 839 (18.98%) | 732 (16.56%) | 736 (16.65%) | 711 (16.09%) | 720 (16.29%) | ||
| Divergent regulation | ||||||||||
| Mode 6 | A = D | A > D | 136 (3.08%) | 210 (4.75%) | 162 (3.67%) | 210 (4.75%) | 142 (3.21%) | 235 (5.32%) | 146 (3.30%) | 289 (6.54%) |
| Mode 7 | A = D | A < D | 204 (4.62%) | 260 (5.88%) | 197 (4.46%) | 220 (4.98%) | 195 (4.41%) | 316 (7.15%) | 221 (5.00%) | 296 (6.70%) |
| Subtotal | 340 (7.69%) | 470 (10.63%) | 359 (8.12%) | 430 (9.73%) | 337 (7.62%) | 551 (12.47%) | 367 (8.30%) | 585 (13.24%) | ||
| Reversed regulation | ||||||||||
| Mode 8 | A > D | A < D | 11 (0.25%) | 6 (0.14%) | 3 (0.07%) | 7 (0.16%) | 10 (0.23%) | 12 (0.27%) | 6 (0.14%) | 8 (0.18%) |
| Mode 9 | A < D | A > D | 3 (0.07%) | 5 (0.11%) | 1 (0.02%) | 5 (0.11%) | 1 (0.02%) | 8 (0.18%) | 3 (0.07%) | 13 (0.29%) |
| Subtotal | 14 (0.32%) | 11 (0.25%) | 4 (0.09%) | 12 (0.27%) | 11 (0.25%) | 20 (0.45%) | 9 (0.20%) | 21 (0.48%) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Lv, R.; Wang, B.; Xun, H.; Liu, B.; Xu, C. Effects of Allopolyploidization and Homoeologous Chromosomal Segment Exchange on Homoeolog Expression in a Synthetic Allotetraploid Wheat under Variable Environmental Conditions. Plants 2023, 12, 3111. https://doi.org/10.3390/plants12173111
Zhang Z, Lv R, Wang B, Xun H, Liu B, Xu C. Effects of Allopolyploidization and Homoeologous Chromosomal Segment Exchange on Homoeolog Expression in a Synthetic Allotetraploid Wheat under Variable Environmental Conditions. Plants. 2023; 12(17):3111. https://doi.org/10.3390/plants12173111
Chicago/Turabian StyleZhang, Zhibin, Ruili Lv, Bin Wang, Hongwei Xun, Bao Liu, and Chunming Xu. 2023. "Effects of Allopolyploidization and Homoeologous Chromosomal Segment Exchange on Homoeolog Expression in a Synthetic Allotetraploid Wheat under Variable Environmental Conditions" Plants 12, no. 17: 3111. https://doi.org/10.3390/plants12173111