
Epigenetic Regulation of Subgenomic Gene Expression in Allotetraploid Brassica napus


Abstract
:1. Introduction
2. Results
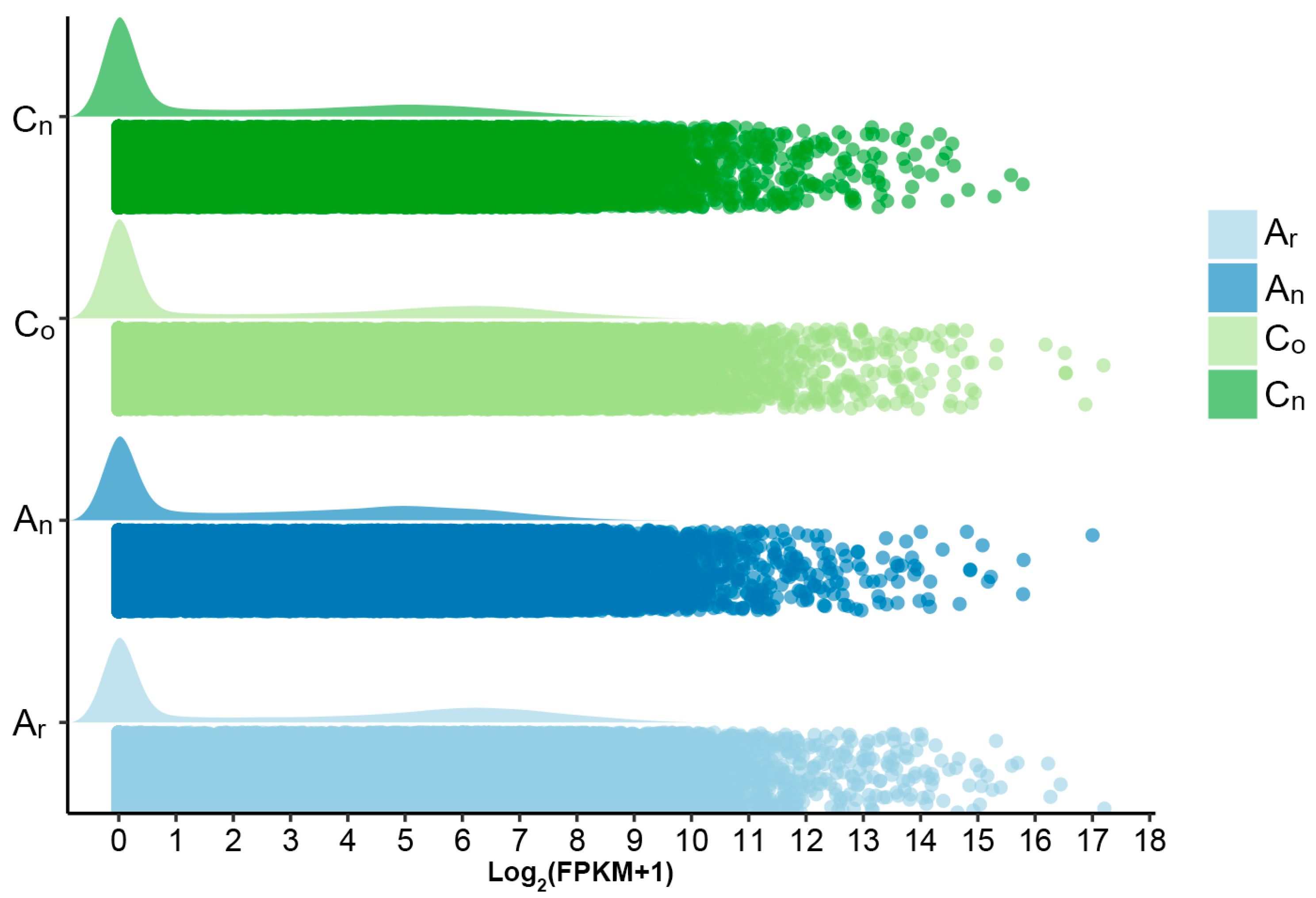
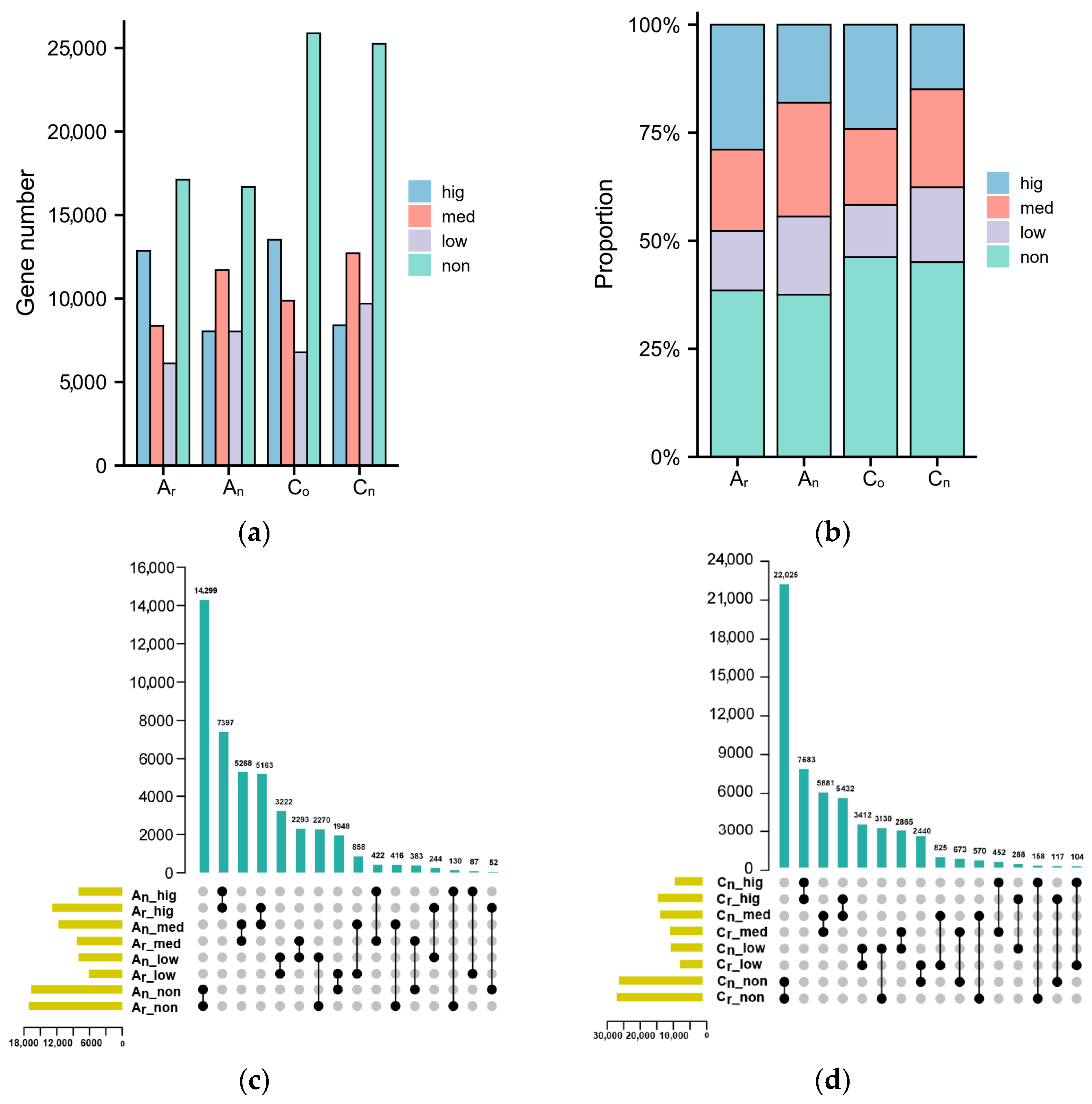
2.1. Subgenomic Gene Expression in B. napus
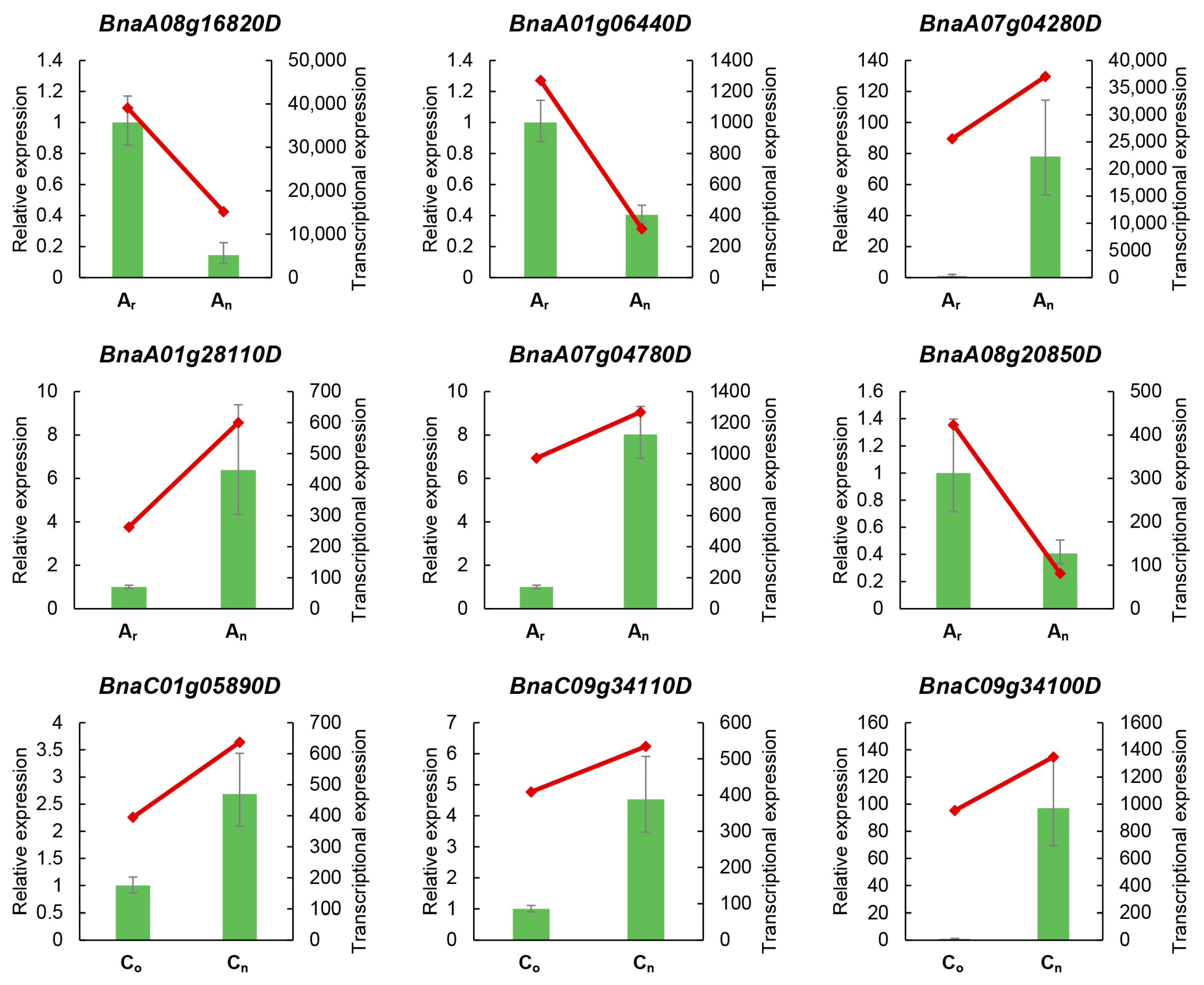
2.2. Gene Expression Levels Verified by Real-Time Quantitative PCR (RT-qPCR)
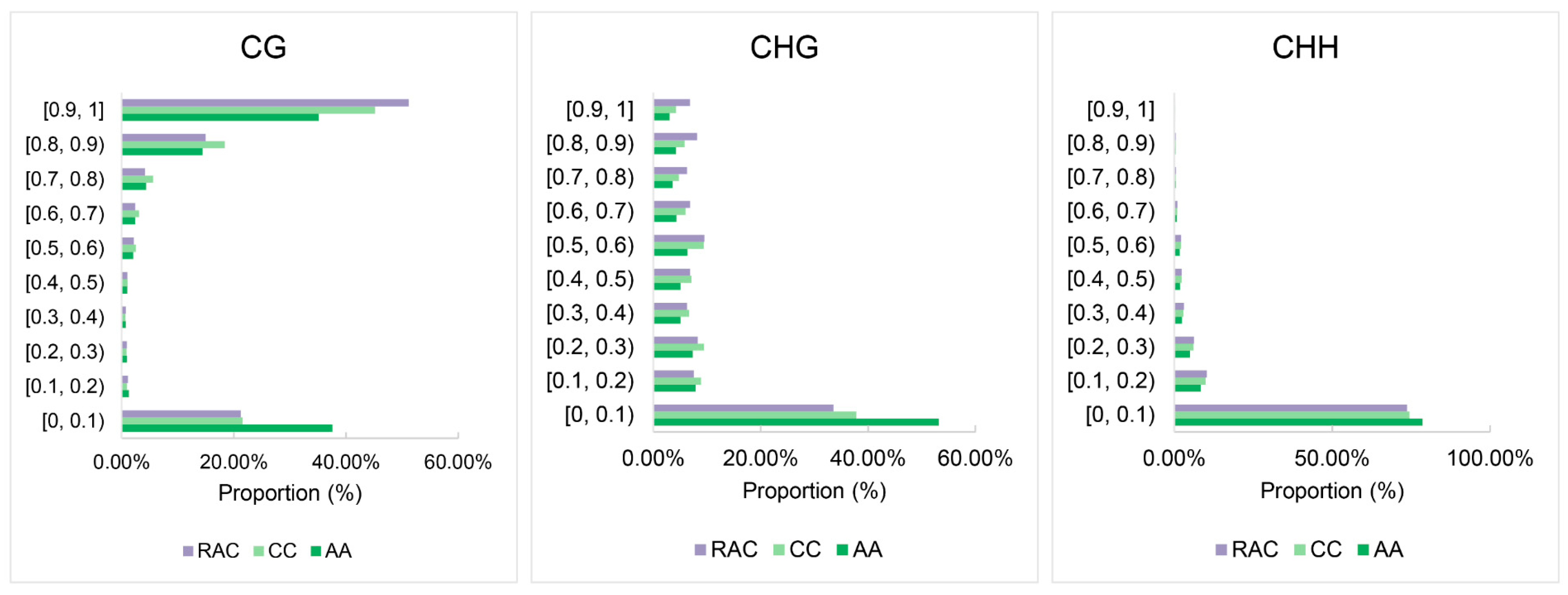
2.3. Detection and Analysis of Methylation Sites
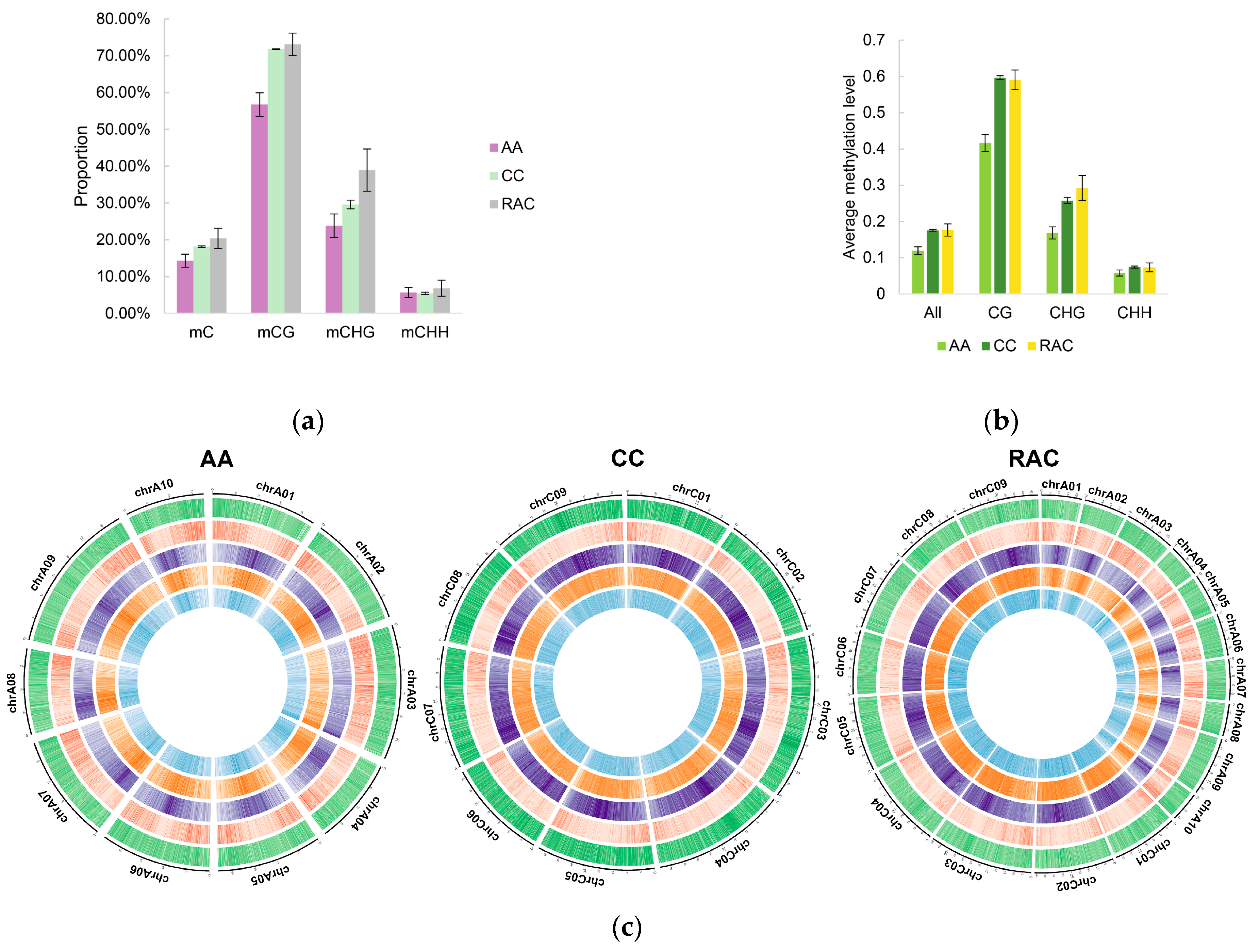
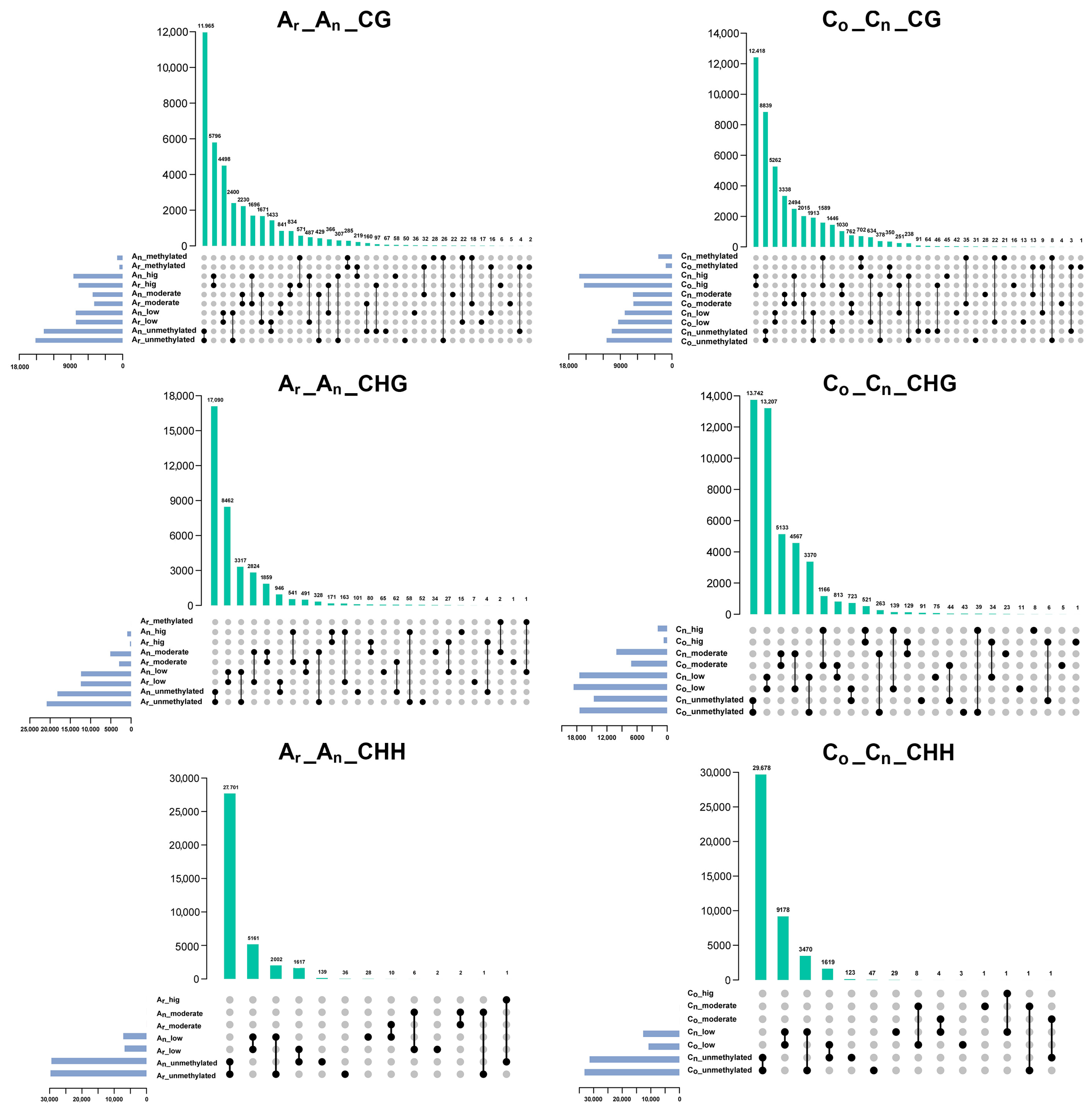
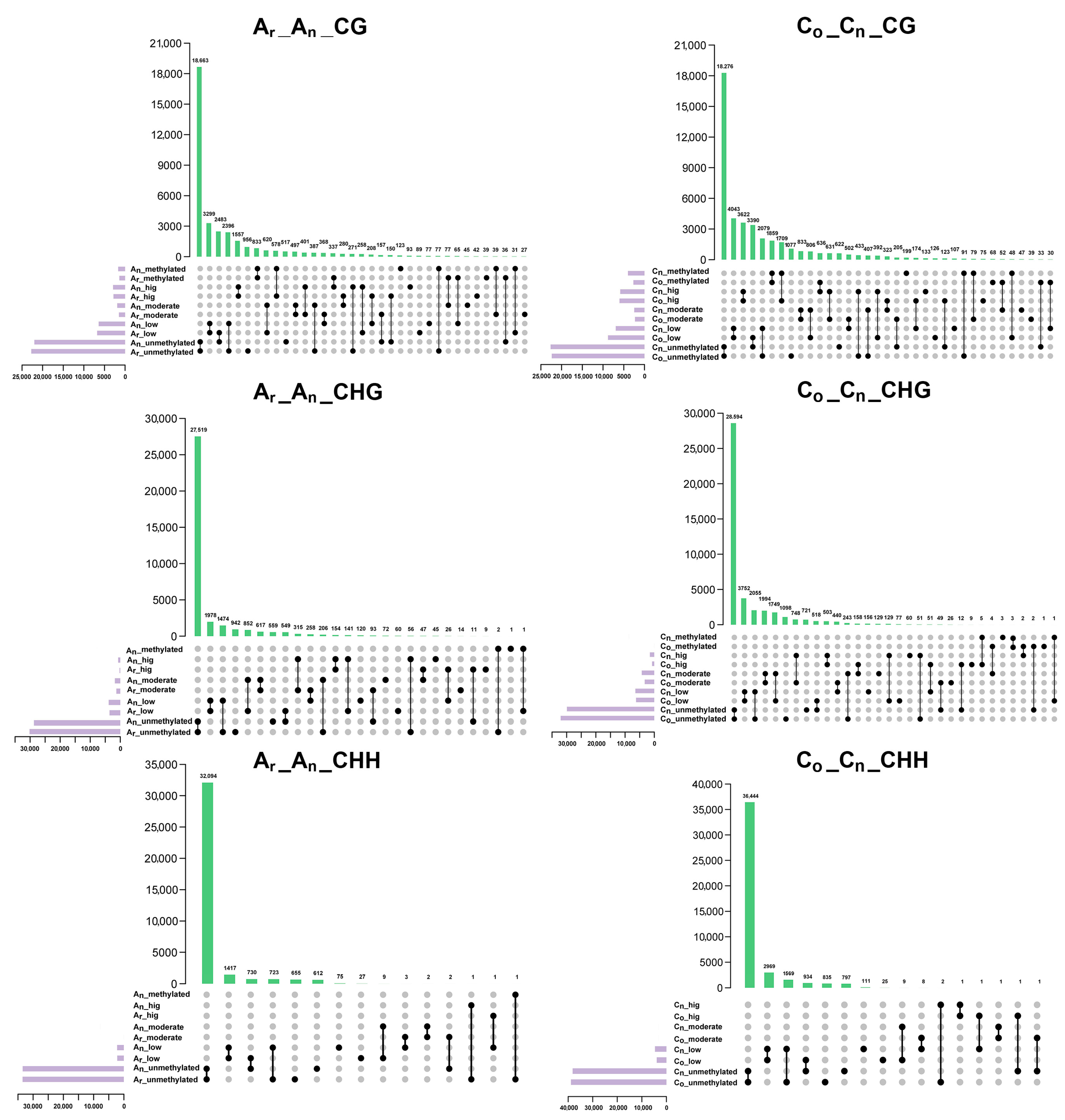
2.4. Changes of Subgenomic DNA Methylation in B. napus
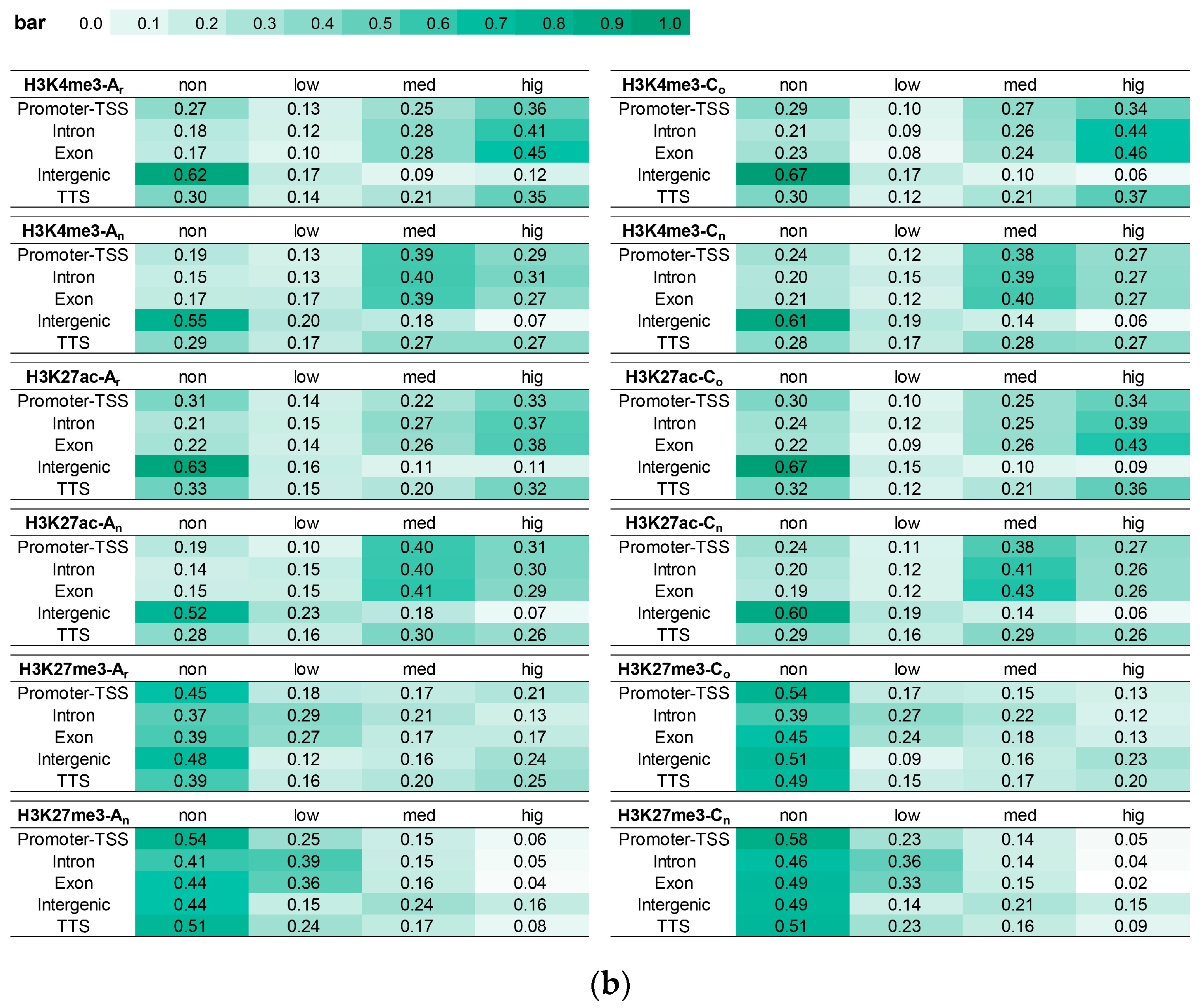
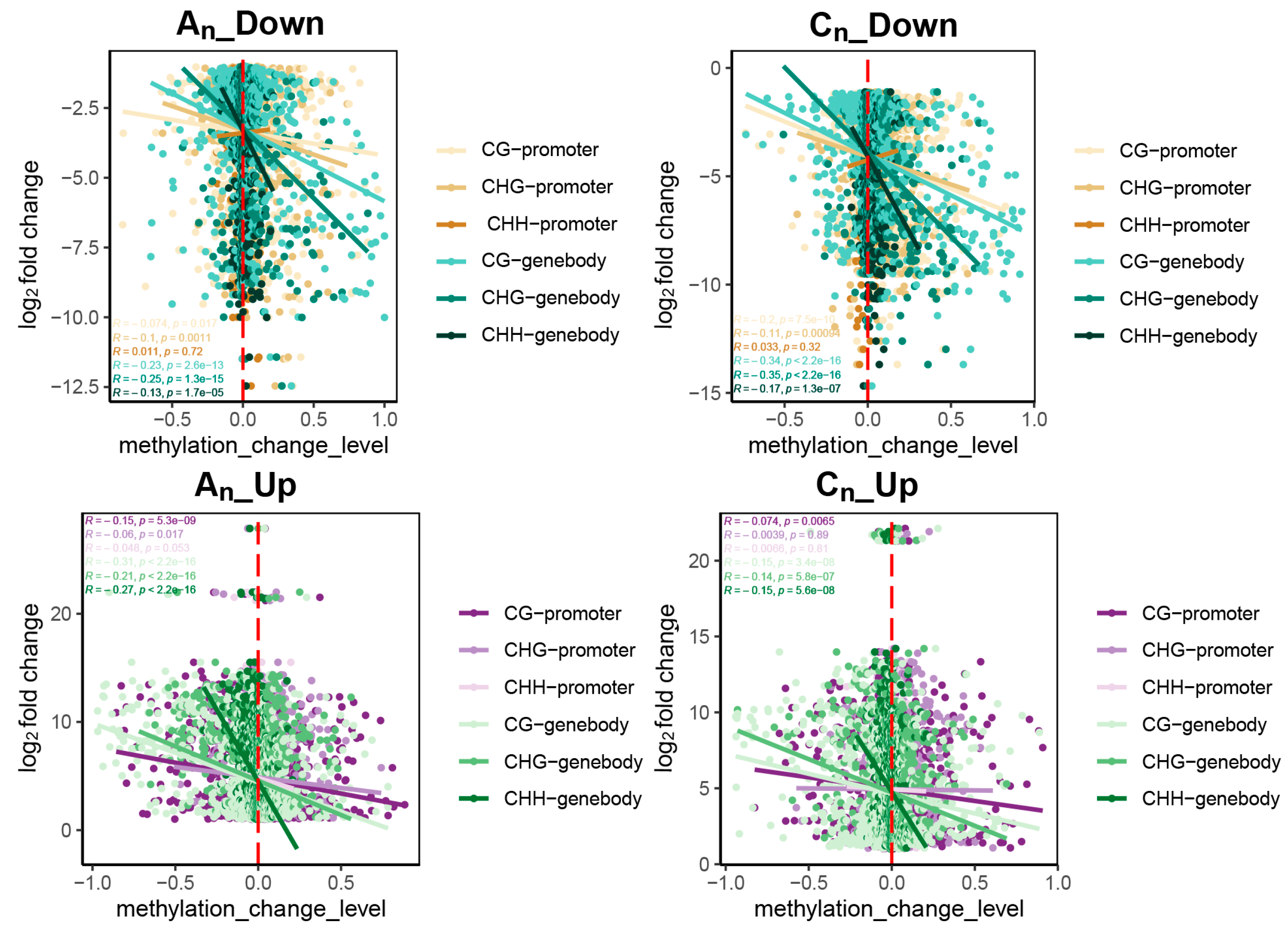
2.5. Effects of DNA Methylation on Gene Expression
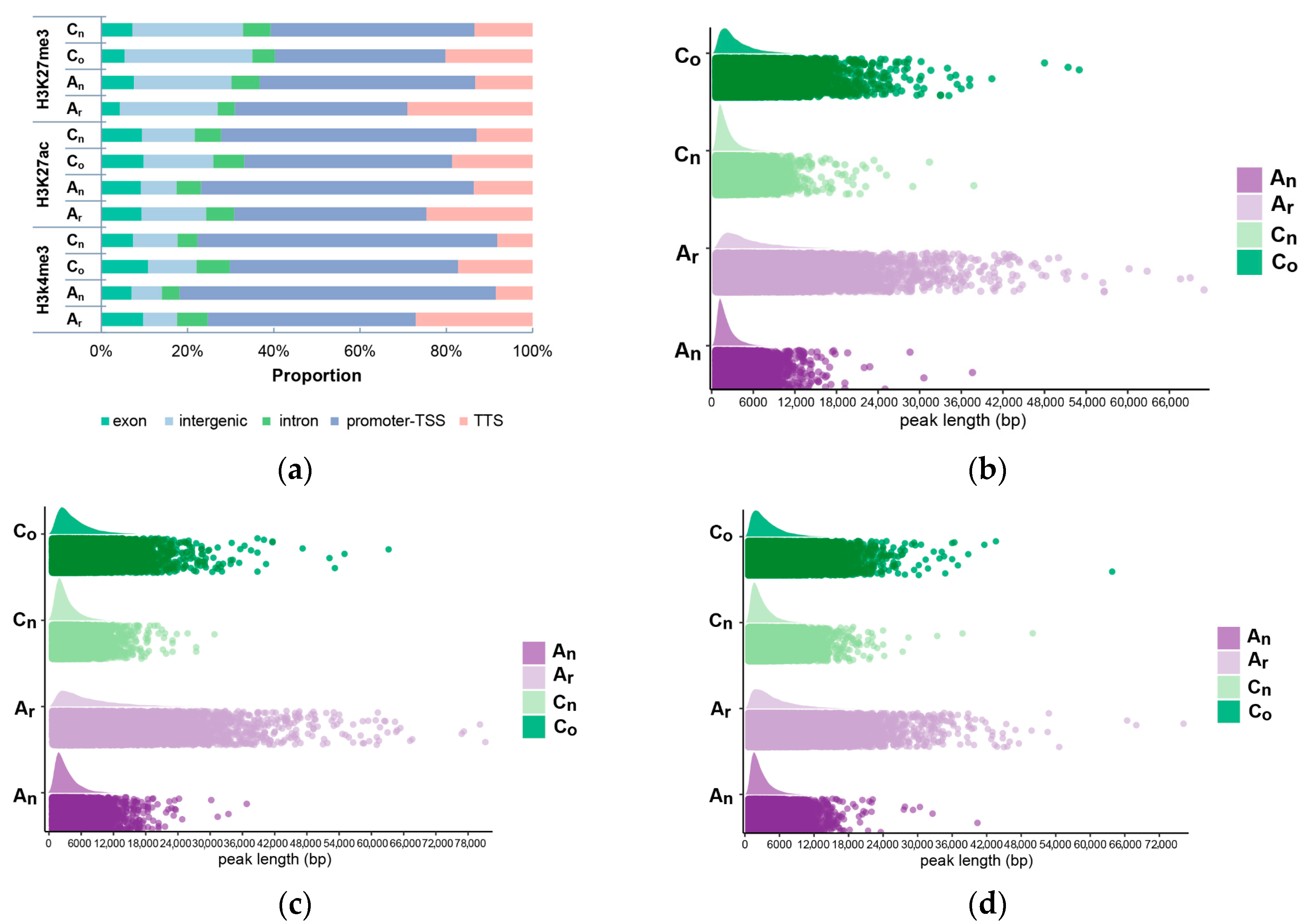
2.6. Subgenomic Histone Modification in B. napus
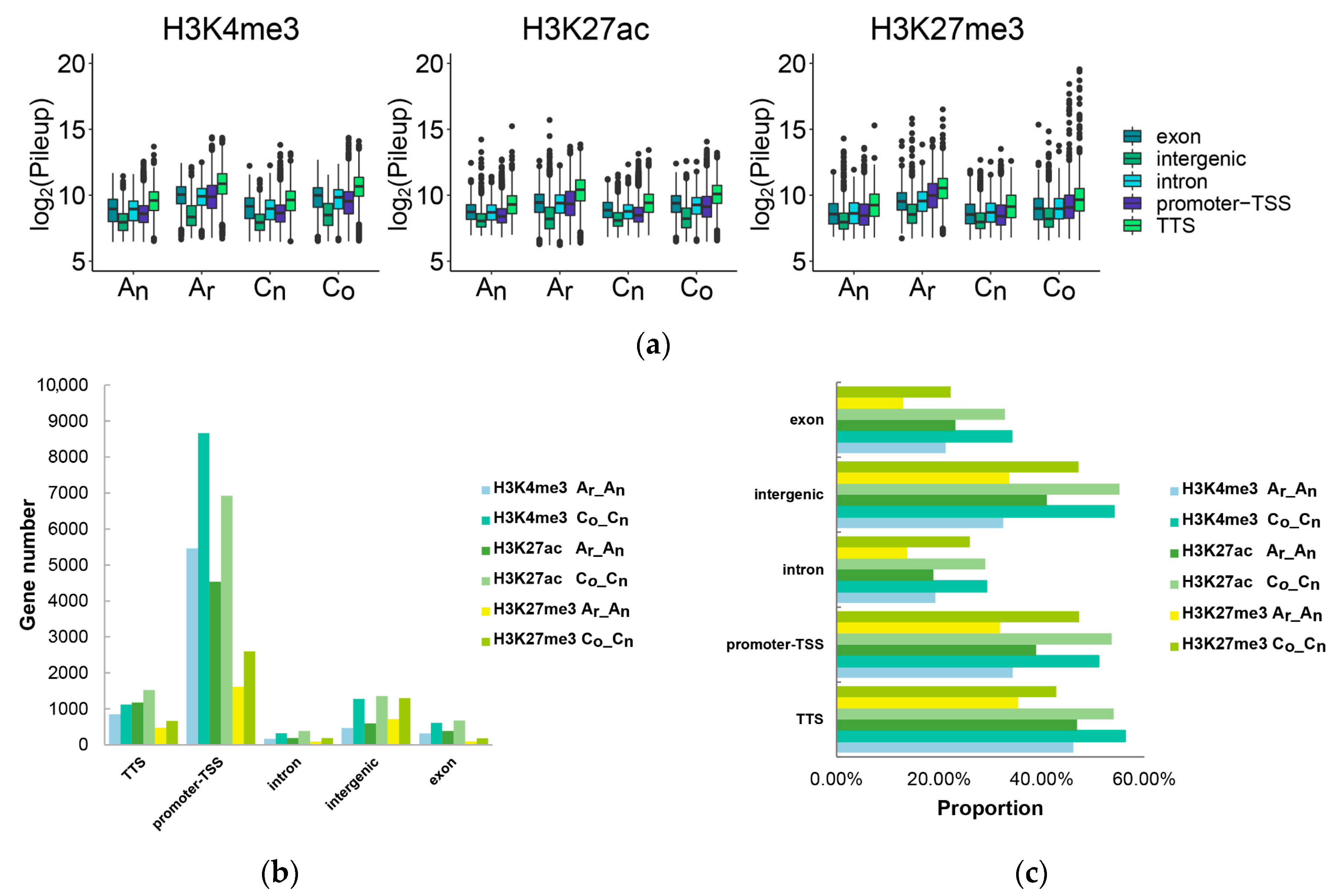
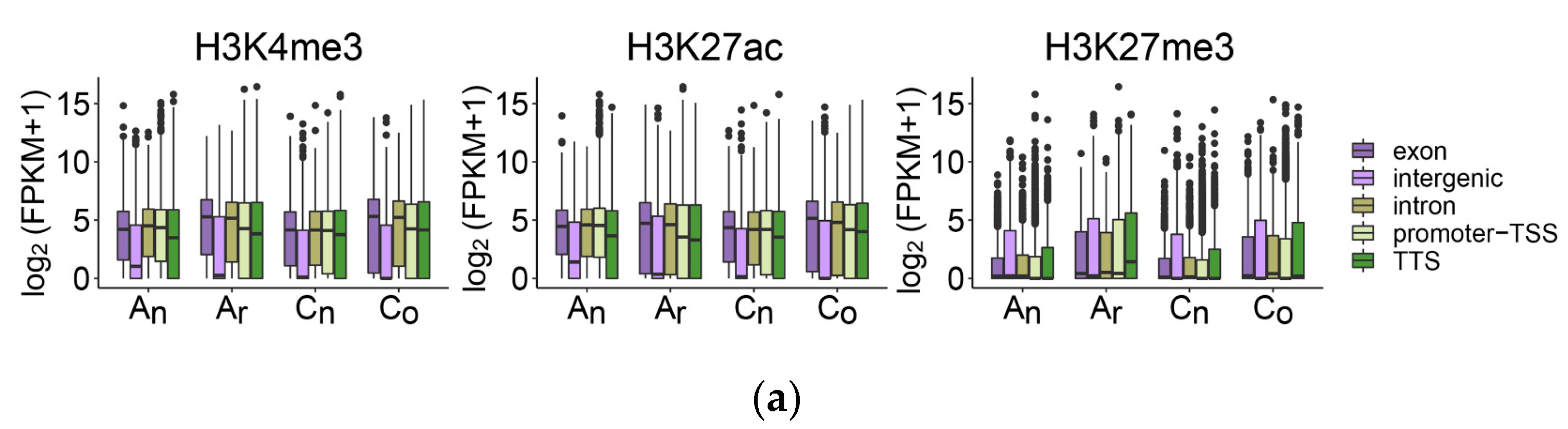
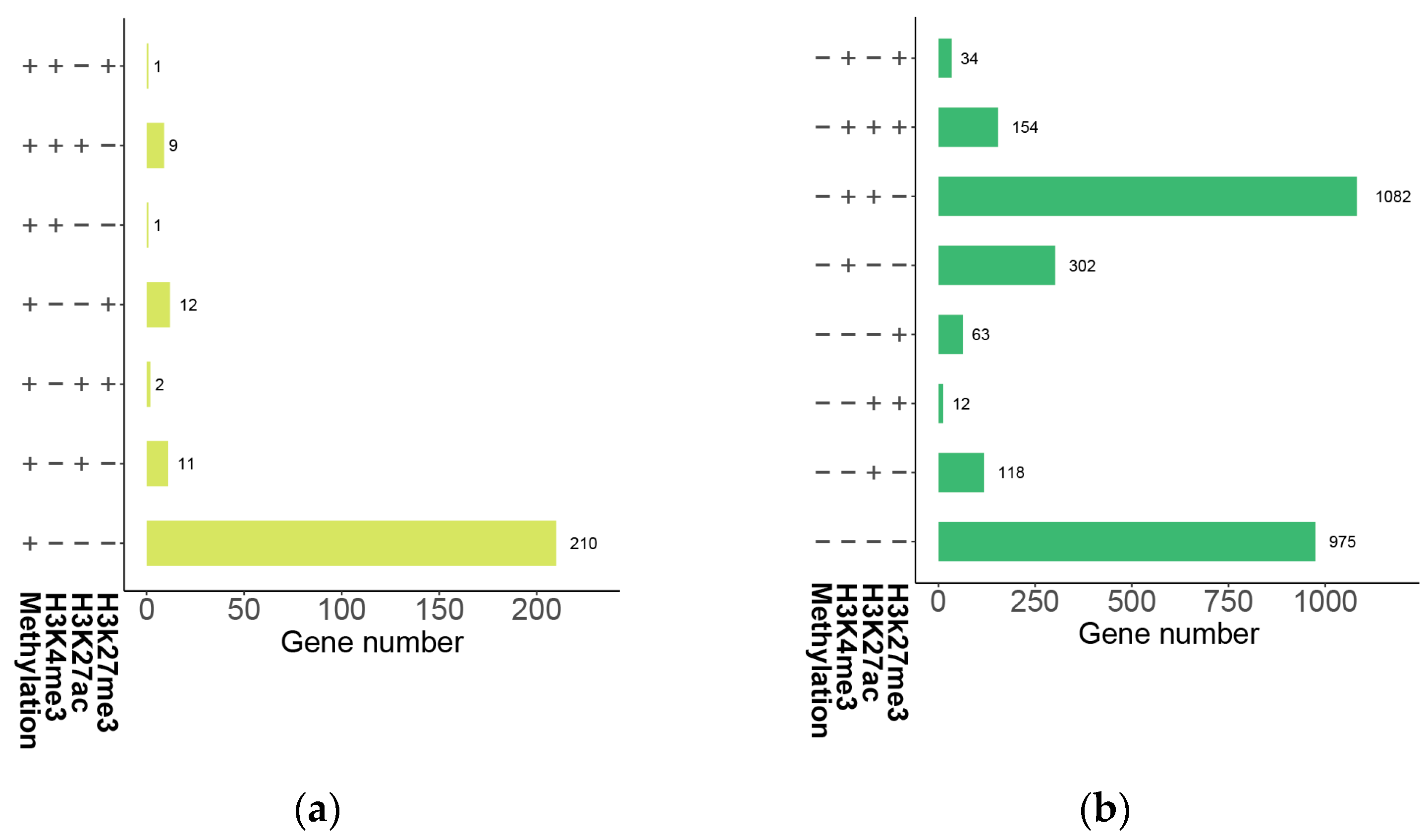
2.7. The Impact of Histone Modification on Gene Expression
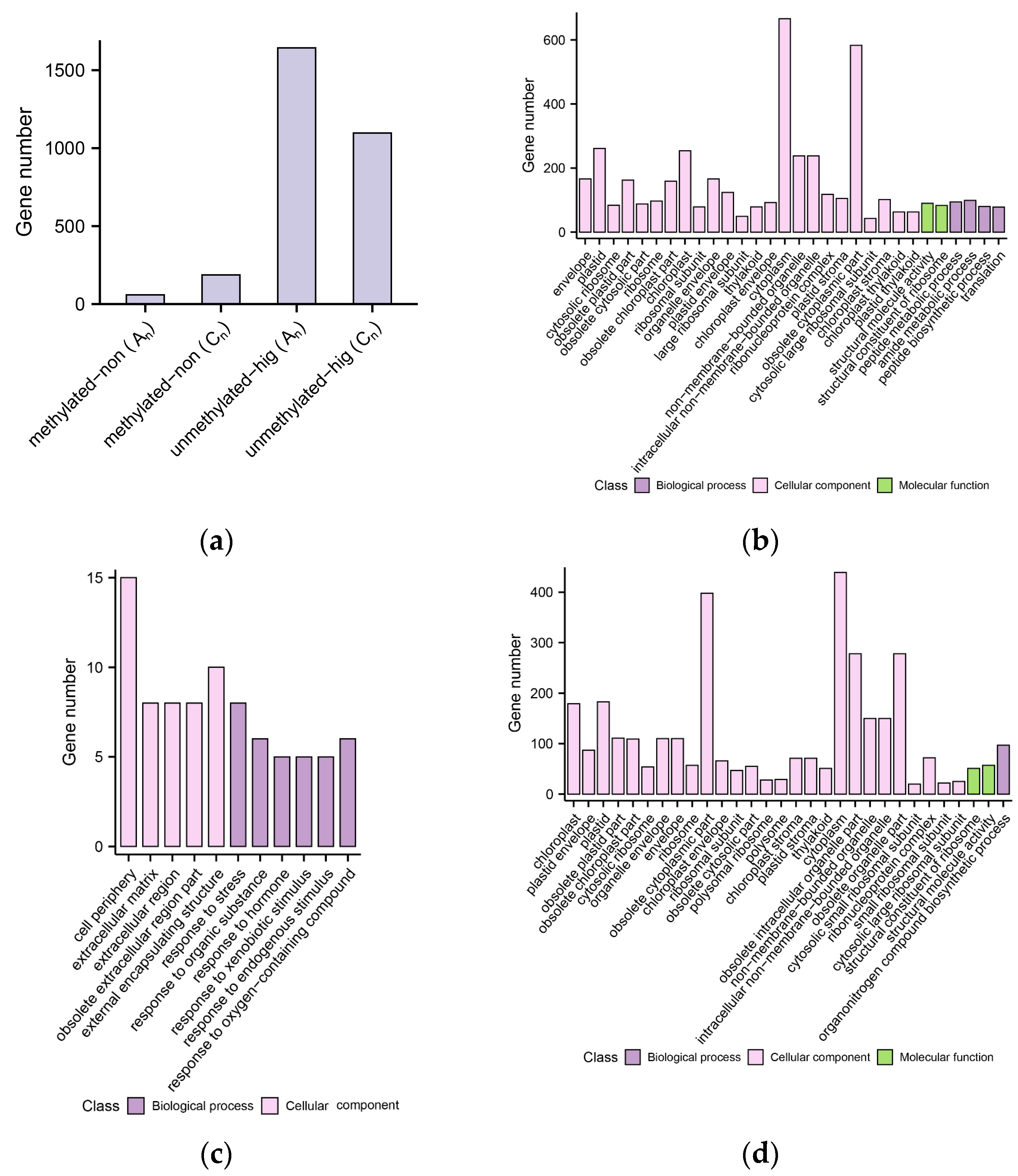
2.8. DNA Methylation Level Variations of DEGs in B. napus
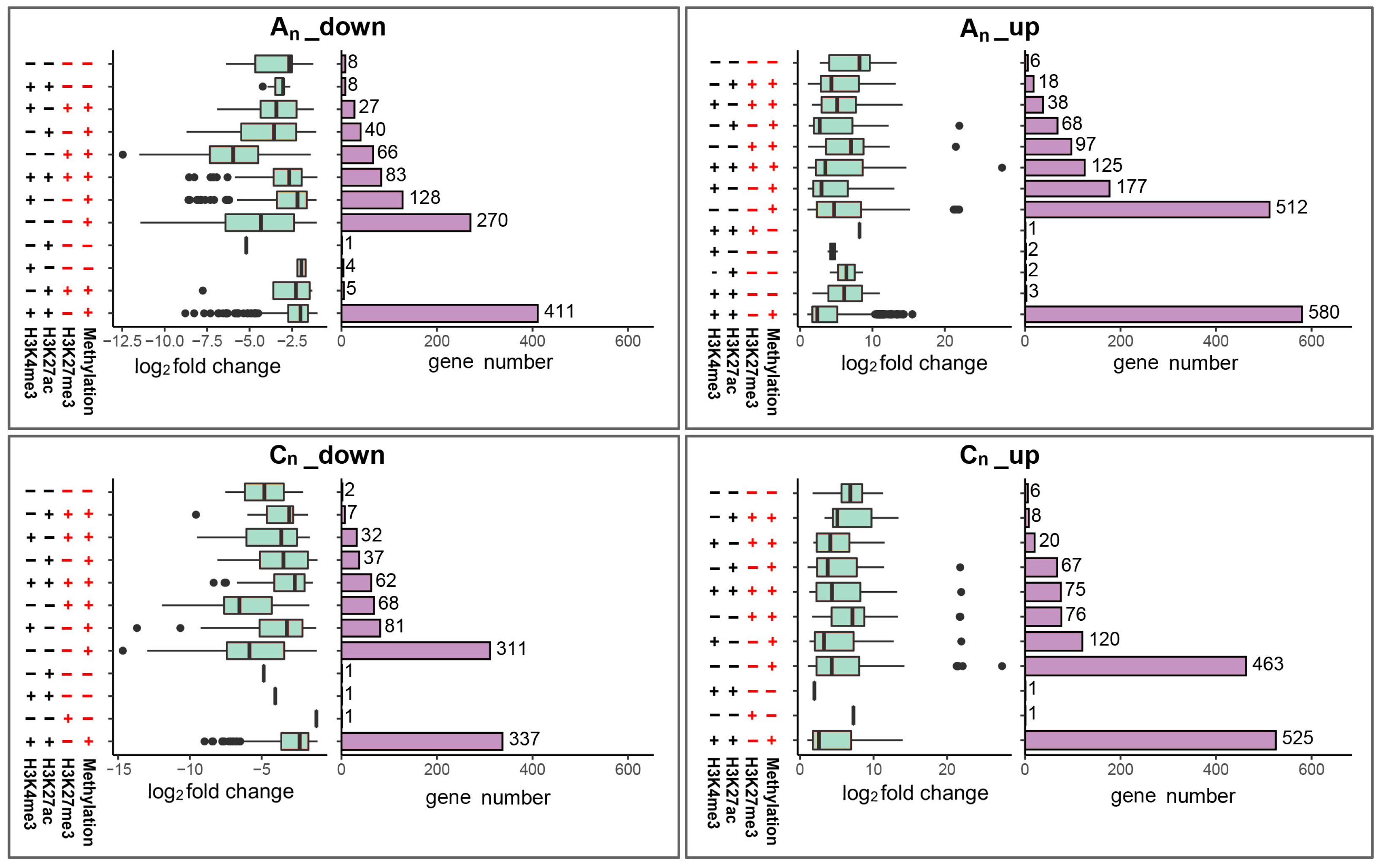
2.9. Combined Consequences of DNA Methylation and Histone Modification on Gene Expression
3. Discussion
3.1. Gene Expression Alteration and Asymmetric Subgenome Epigenetic Modification in B. napus
3.2. Various Epigenetic Markers Are Involved in Regulating Gene Expression in B. napus
4. Materials and Methods
4.1. Plant Materials
4.2. Transcriptome Sequencing and RT-qPCR
4.3. Analysis of Distribution Characteristics of Gene Expression
4.4. Screening of Differential Expression Genes (DEGs) and Annotation
4.5. Whole-Genome Bisulfite Sequencing (WGBS)
4.6. DNA Methylation Level Analysis
4.7. Histone Modification Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rapp, R.A.; Wendel, J.F. Epigenetics and plant evolution. New Phytol. 2005, 168, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.J.; Scheibe, M.; Wongpalee, S.P.; Liu, W.L.; Cornett, E.M.; Vaughan, R.M.; Li, X.Q.; Chen, W.; Xue, Y.; Zhong, Z.H.; et al. A DNA methylation reader complex that enhances gene transcription. Science 2018, 362, 1182–1186. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.L.; Zhang, J.Q.; Li, T.; Fu, X.; Satheesh, V.; Niu, Q.F.; Lang, Z.B.; Zhu, J.K.; Lei, M.G. A group of SUVH methyl-DNA binding proteins regulate expression of the DNA demethylase ROS1 in Arabidopsis. J. Integr. Plant Biol. 2019, 61, 110–119. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lu, F.; Cui, X.; Cao, X. Histone methylation in higher plants. Annu. Rev. Plant Biol. 2010, 61, 395–420. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.X.; Chen, Z.J. Epigenetic and developmental regulation in plant polyploids. Curr. Opin. Plant Biol. 2015, 24, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Soltis, D.E.; Visger, C.J.; Marchant, D.B.; Soltis, P.S. Polyploidy: Pitfalls and paths to a paradigm. Am. J. Bot. 2016, 103, 1146–1166. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Wendel, J.F. Epigenetic phenomena and the evolution of plant allopolyploids. Mol. Phylogenet. Evol. 2003, 29, 365–379. [Google Scholar] [CrossRef]
- Jiang, X.; Song, Q.; Ye, W.; Chen, Z.J. Concerted genomic and epigenomic changes accompany stabilization of Arabidopsis allopolyploids. Nat. Ecol. Evol. 2021, 5, 1382–1393. [Google Scholar] [CrossRef]
- Yuan, J.; Jiao, W.; Liu, Y.; Ye, W.; Wang, X.; Liu, B.; Song, Q.; Chen, Z.J. Dynamic and reversible DNA methylation changes induced by genome separation and merger of polyploid wheat. BMC Biol. 2020, 18, 171. [Google Scholar] [CrossRef]
- Zheng, D.; Ye, W.; Song, Q.; Han, F.; Zhang, T.; Chen, Z.J. Histone modifications define expression bias of homoeologous genomes in allotetraploid cotton. Plant Physiol. 2016, 172, 1760–1771. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cao, S.; Wang, P.; Lu, K.; Song, Q.; Zhao, F.J.; Chen, Z.J. DNA hypomethylation in tetraploid rice potentiates stress-responsive gene expression for salt tolerance. Proc. Natl. Acad. Sci. USA 2021, 118, e2023981118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, Q.; He, L.; Deng, L.; Lozano-Duran, R.; Li, G.; Zhu, J.K. DNA methylation underpins the epigenomic landscape regulating genome transcription in Arabidopsis. Genome Biol. 2022, 23, 197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yuan, J.; Jia, G.; Ye, W.; Jeffrey Chen, Z.; Song, Q. Histone H3K27 dimethylation landscapes contribute to genome stability and genetic recombination during wheat polyploidization. Plant J. 2021, 105, 678–690. [Google Scholar] [CrossRef]
- He, L.; Huang, H.; Bradai, M.; Zhao, C.; You, Y.; Ma, J.; Zhao, L.; Lozano-Duran, R.; Zhu, J.K. DNA methylation-free Arabidopsis reveals crucial roles of DNA methylation in regulating gene expression and development. Nat. Commun. 2022, 13, 1335. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Wang, H.Z.; Wang, J.; Sun, R.F.; Wu, J.; Liu, S.Y.; Bai, Y.Q.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Wu, J.; Wang, X.W. Genome triplication drove the diversification of Brassica plants. Hortic. Res. 2014, 1, 14024. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.Y.; Gaeta, R.T.; Pires, J.C. Homoeologous shuffling and chromosome compensation maintain genome balance in resynthesized allopolyploid Brassica napus. Proc. Natl. Acad. Sci. USA 2011, 108, 7908–7913. [Google Scholar] [CrossRef]
- Liu, S.Y.; Liu, Y.M.; Yang, X.H.; Tong, C.B.; Edwards, D.; Parkin, I.A.P.; Zhao, M.X.; Ma, J.X.; Yu, J.Y.; Huang, S.M.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef] [Green Version]
- Bottani, S.; Zabet, N.R.; Wendel, J.F.; Veitia, R.A. Gene expression dominance in allopolyploids: Hypotheses and models. Trends Plant Sci. 2018, 23, 393–402. [Google Scholar] [CrossRef]
- Wendel, J.F.; Lisch, D.; Hu, G.J.; Mason, A.S. The long and short of doubling down: Polyploidy, epigenetics, and the temporal dynamics of genome fractionation. Curr. Opin. Genet. Dev. 2018, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liang, J.L.; Lin, R.M.; Cai, X.; Zhang, L.; Guo, X.L.; Wang, T.P.; Chen, H.X.; Wang, X.W. Investigation of Brassica and its relative genomes in the post-genomics era. Hortic. Res. 2022, 9, uhac182. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.; Yuan, J.Y.; Jiang, S.; Liu, Y.F.; Wang, L.L.; Liu, M.M.; Zheng, D.W.; Ye, W.X.; Wang, X.E.; Chen, Z.J. Asymmetrical changes of gene expression, small RNAs and chromatin in two resynthesized wheat allotetraploids. Plant J. 2018, 93, 828–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Z.L.; Li, Z.J.; Wang, M.Y.; Zhao, F.; Zhang, W.J.; Li, C.P.; Gong, L.; Zhang, Y.; Mason, A.S.; Liu, B. Conservation and trans-regulation of histone modification in the A and B subgenomes of polyploid wheat during domestication and ploidy transition. BMC Biol. 2021, 19, 42. [Google Scholar] [CrossRef]
- de Carvalho, J.F.; Stoeckel, S.; Eber, F.; Lode-Taburel, M.; Gilet, M.M.; Trotoux, G.; Morice, J.; Falentin, C.; Chevre, A.M.; Rousseau-Gueutin, M. Untangling structural factors driving genome stabilization in nascent Brassica napus allopolyploids. New Phytol. 2021, 230, 2072–2084. [Google Scholar] [CrossRef]
- Zhang, Q.; Guan, P.; Zhao, L.; Ma, M.; Xie, L.; Li, Y.; Zheng, R.; Ouyang, W.; Wang, S.; Li, H.; et al. Asymmetric epigenome maps of subgenomes reveal imbalanced transcription and distinct evolutionary trends in Brassica napus. Mol. Plant 2021, 14, 604–619. [Google Scholar] [CrossRef]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef]
- Karan, R.; DeLeon, T.; Biradar, H.; Subudhi, P.K. Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE 2012, 7, e40203. [Google Scholar] [CrossRef]
- Kumar, S.; Mohapatra, T. Dynamics of DNA methylation and its functions in plant growth and development. Front Plant Sci. 2021, 12, 596236. [Google Scholar] [CrossRef]
- Bird, K.A.; VanBuren, R.; Puzey, J.R.; Edger, P.P. The causes and consequences of subgenome dominance in hybrids and recent polyploids. New Phytol. 2018, 220, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.J.; Szadkowski, E.; Wendel, J.F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 2013, 110, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Chalhoub, B.; Denoeud, F.; Liu, S.Y.; Parkin, I.A.P.; Tang, H.B.; Wang, X.Y.; Chiquet, J.; Belcram, H.; Tong, C.B.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichten, S.R.; Briskine, R.; Song, J.; Li, Q.; Swanson-Wagner, R.; Hermanson, P.J.; Waters, A.J.; Starr, E.; West, P.T.; Tiffin, P.; et al. Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell 2013, 25, 2783–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.K. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef]
- Li, S.F.; Liu, L.; Li, S.B.; Gao, L.; Zhao, Y.Y.; Kim, Y.J.; Chen, X.M. SUVH1, a Su(var)3-9 family member, promotes the expression of genes targeted by DNA methylation. Nucleic Acids Res. 2016, 44, 608–620. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.C.; Howard, M.; Dean, C. Antagonistic roles for H3K36me3 and H3K27me3 in the cold-induced epigenetic switch at Arabidopsis FLC. Curr. Biol. 2014, 24, 1793–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.Y.; Wang, L.; Chen, L.F.; Pan, X.C.; Lin, K.D.; Fang, Y.; Wang, X.E.; Zhang, W.L. Salt-responsive genes are differentially regulated at the chromatin levels between seedlings and roots in rice. Plant Cell Physiol. 2019, 60, 1790–1803. [Google Scholar] [CrossRef]
- Zhang, A.C.; Wei, Y.Y.; Shi, Y.N.; Deng, X.J.; Gao, J.J.; Feng, Y.L.; Zheng, D.Y.; Cheng, X.J.; Li, Z.G.; Wang, T.; et al. Profiling of H3K4me3 and H3K27me3 and their roles in gene subfunctionalization in allotetraploid Cotton. Front Plant Sci. 2021, 12, 761059. [Google Scholar] [CrossRef]
- Dasgupta, P.; Prasad, P.; Bag, S.K.; Chaudhuri, S. Dynamicity of histone H3K27ac and H3K27me3 modifications regulate the cold-responsive gene expression in Oryza sativa L. ssp. indica. Genomics 2022, 114, 110433. [Google Scholar] [CrossRef]
- Liu, J.; Chang, C. Concerto on chromatin: Interplays of different epigenetic mechanisms in plant development and environmental adaptation. Plants 2021, 10, 2766. [Google Scholar] [CrossRef]
- Chen, Z.J.; Tian, L. Roles of dynamic and reversible histone acetylation in plant development and polyploidy. Biochim. Biophys. Acta 2007, 1769, 295–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.M.; Bostick, M.; Zhang, X.Y.; Kraft, E.; Henderson, I.; Callis, J.; Jacobsen, S.E. The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr. Biol. 2007, 17, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.Y.; Bernatavichute, Y.V.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009, 10, R62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.L.; He, N.; Niu, L.J.; Huang, Z.; Shen, W.; Zhang, Y.D.; Li, L.; Hou, C.H. Global quantitative mapping of enhancers in rice by STARR-seq. Genom. Proteom. Bioinform. 2019, 17, 140–153. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant graphics for data analysis. Meas-Interdiscip. Res. 2019, 17, 160–167. [Google Scholar]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.H.; Yin, H.; Qi, K.J.; Li, L.T.; Wang, R.Z.; Zhang, S.L.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.X.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar]
- Li, M.D.; Sun, W.Q.; Wang, F.; Wu, X.M.; Wang, J.B. Asymmetric epigenetic modification and homoeolog expression bias in the establishment and evolution of allopolyploid Brassica napus. New Phytol. 2021, 232, 898–913. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Ar | An | Co | Cn |
|---|---|---|---|---|
| H3K4me3 | 16,902 | 21,674 | 24,070 | 24,396 |
| H3K27ac | 21,249 | 18,417 | 25,959 | 21,808 |
| H3K27me3 | 11,393 | 10,118 | 15,303 | 11,656 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, M.; Xi, Z.; Wang, J. Epigenetic Regulation of Subgenomic Gene Expression in Allotetraploid Brassica napus. Plants 2023, 12, 2608. https://doi.org/10.3390/plants12142608
Hu M, Xi Z, Wang J. Epigenetic Regulation of Subgenomic Gene Expression in Allotetraploid Brassica napus. Plants. 2023; 12(14):2608. https://doi.org/10.3390/plants12142608
Chicago/Turabian StyleHu, Meimei, Zengde Xi, and Jianbo Wang. 2023. "Epigenetic Regulation of Subgenomic Gene Expression in Allotetraploid Brassica napus" Plants 12, no. 14: 2608. https://doi.org/10.3390/plants12142608