
Multi-Omics Analysis of Vicia cracca Responses to Chronic Radiation Exposure in the Chernobyl Exclusion Zone
 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
2.1. Environmental Conditions in Experimental Plots
2.2. Analysis of Expression of Selected Genes in V. cracca Populations
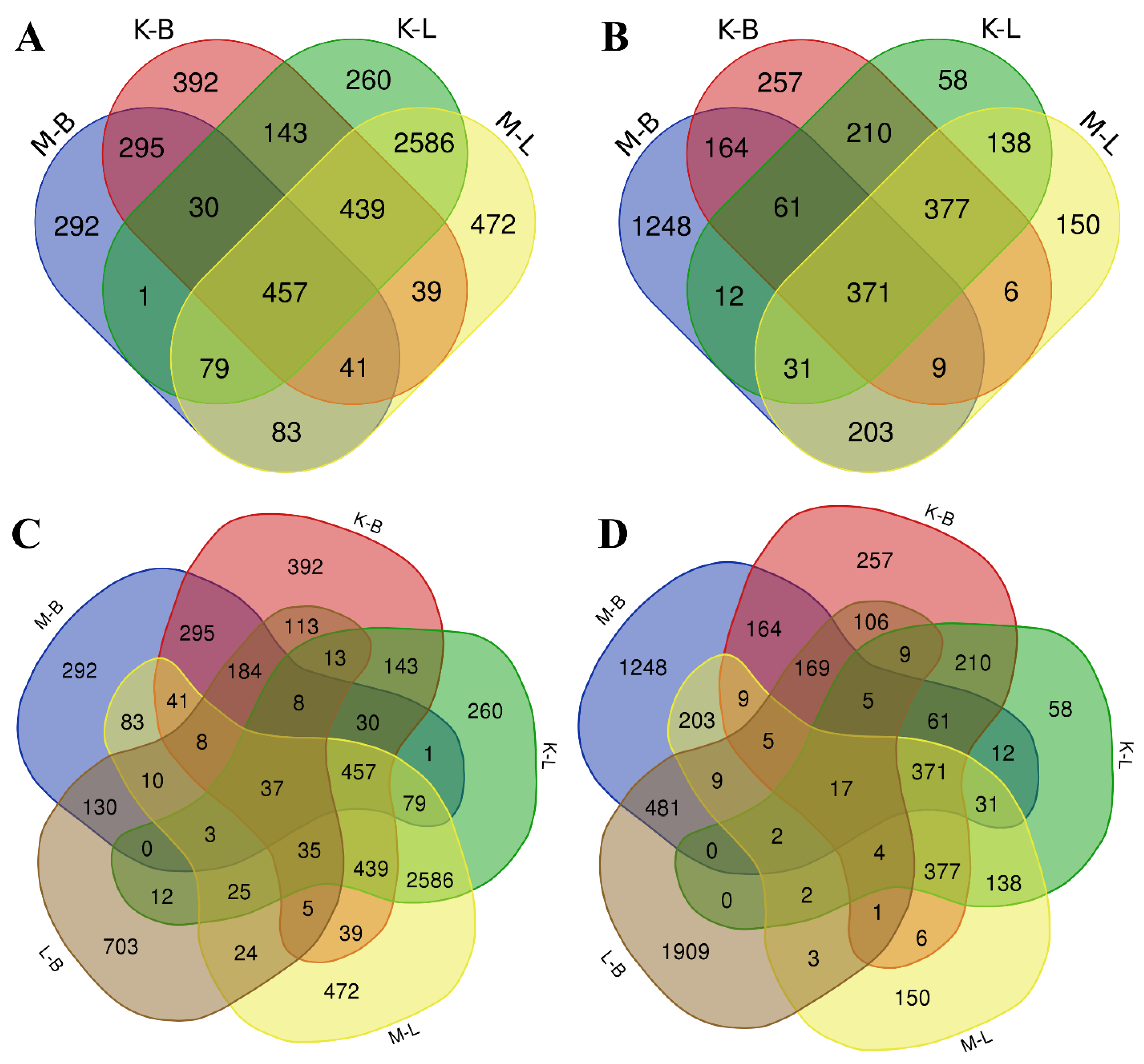
2.3. Whole-Transcriptome Analysis of V. cracca Populations
2.4. Non-Targeted Proteomic Analysis of V. cracca Populations
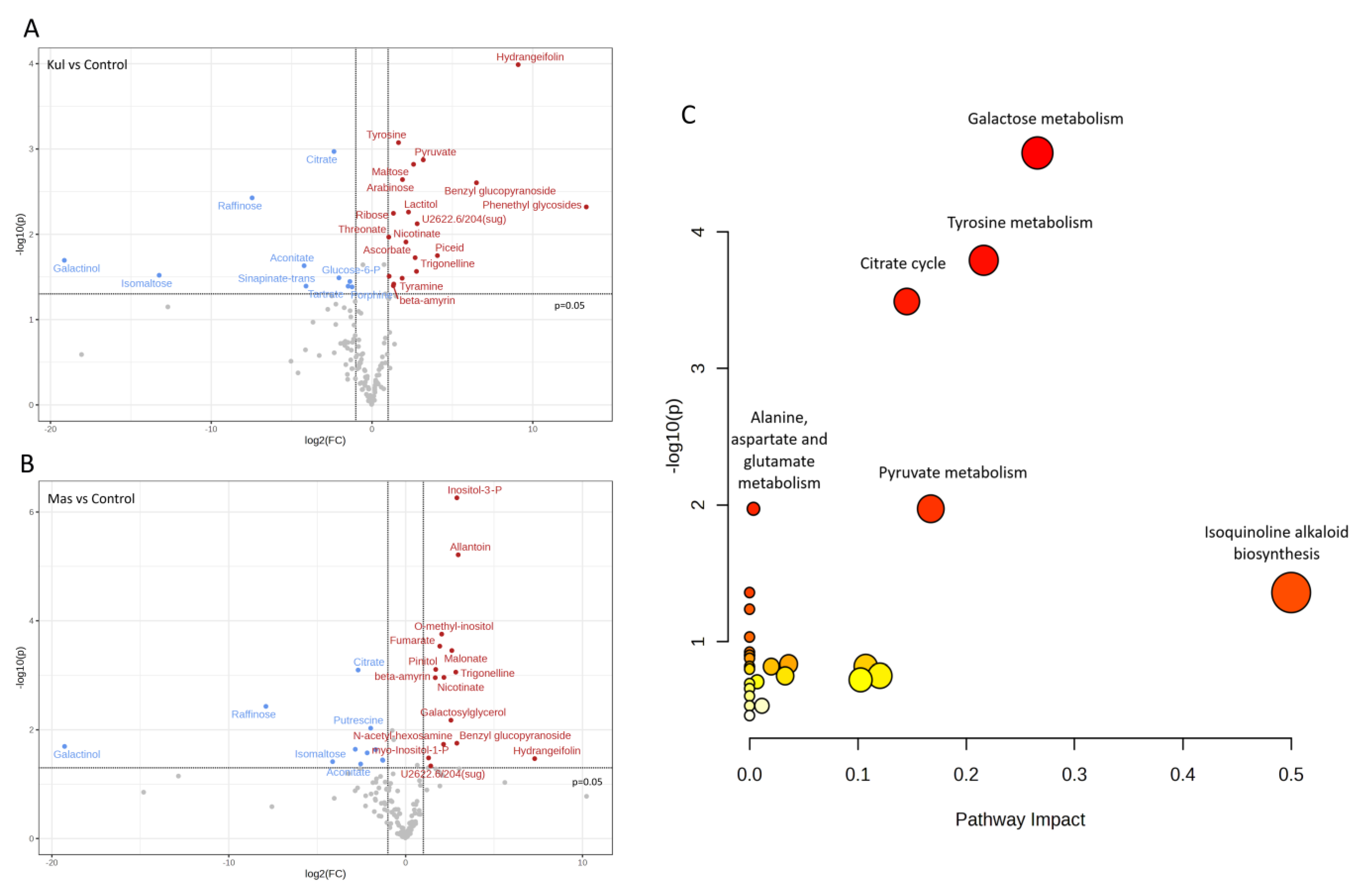
2.5. Non-Targeted Metabolomic Analysis of V. cracca Populations
2.6. Multi-Omics Data Integration
3. Discussion
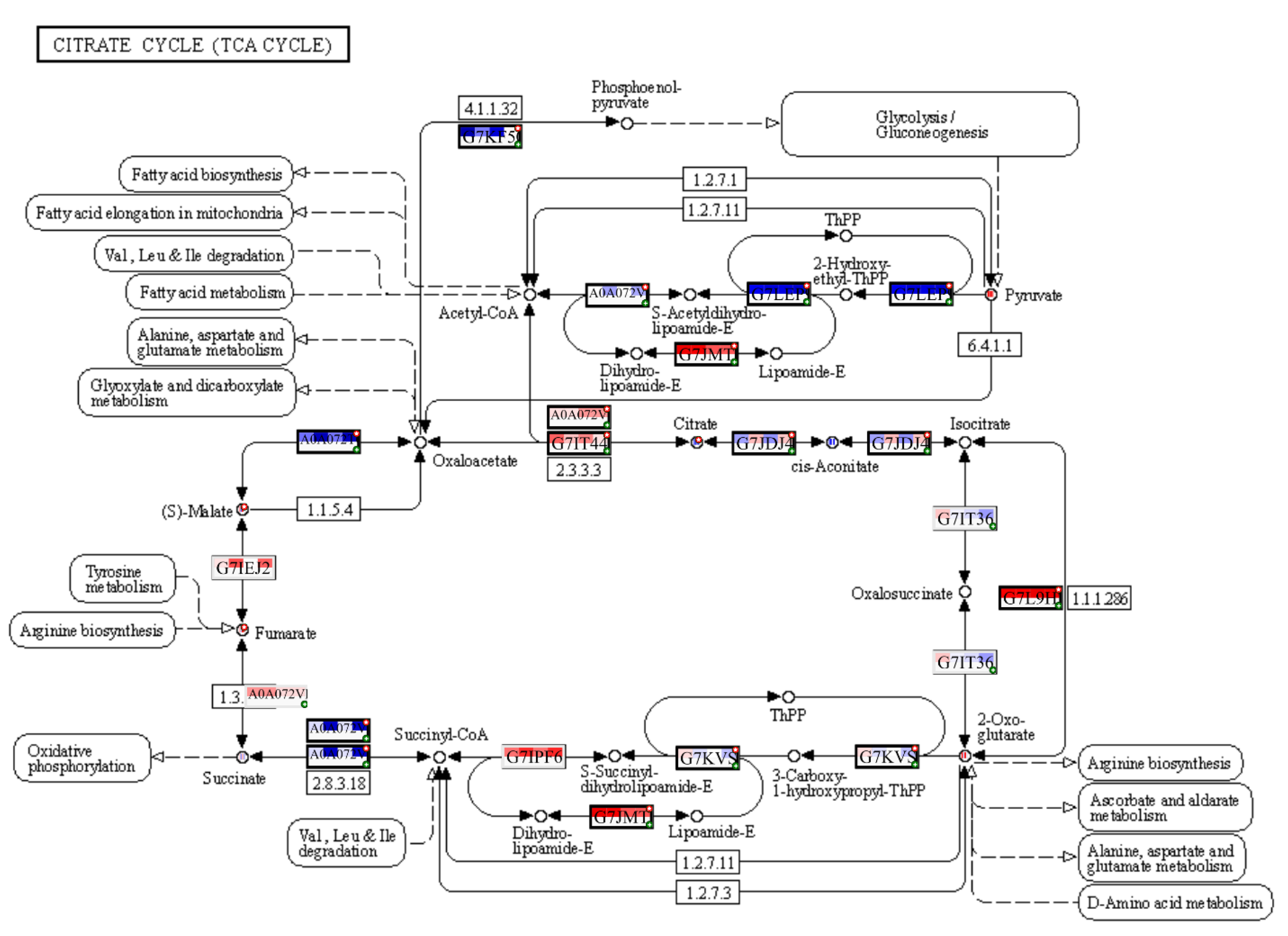
3.1. Carbon Metabolism and Photosynthesis
3.2. Chronic Radiation Stress Response: DNA Damage and Changes in the Redox Status
3.3. Secondary Metabolites
3.4. Protein Catabolism and RNA Processing
4. Materials and Methods
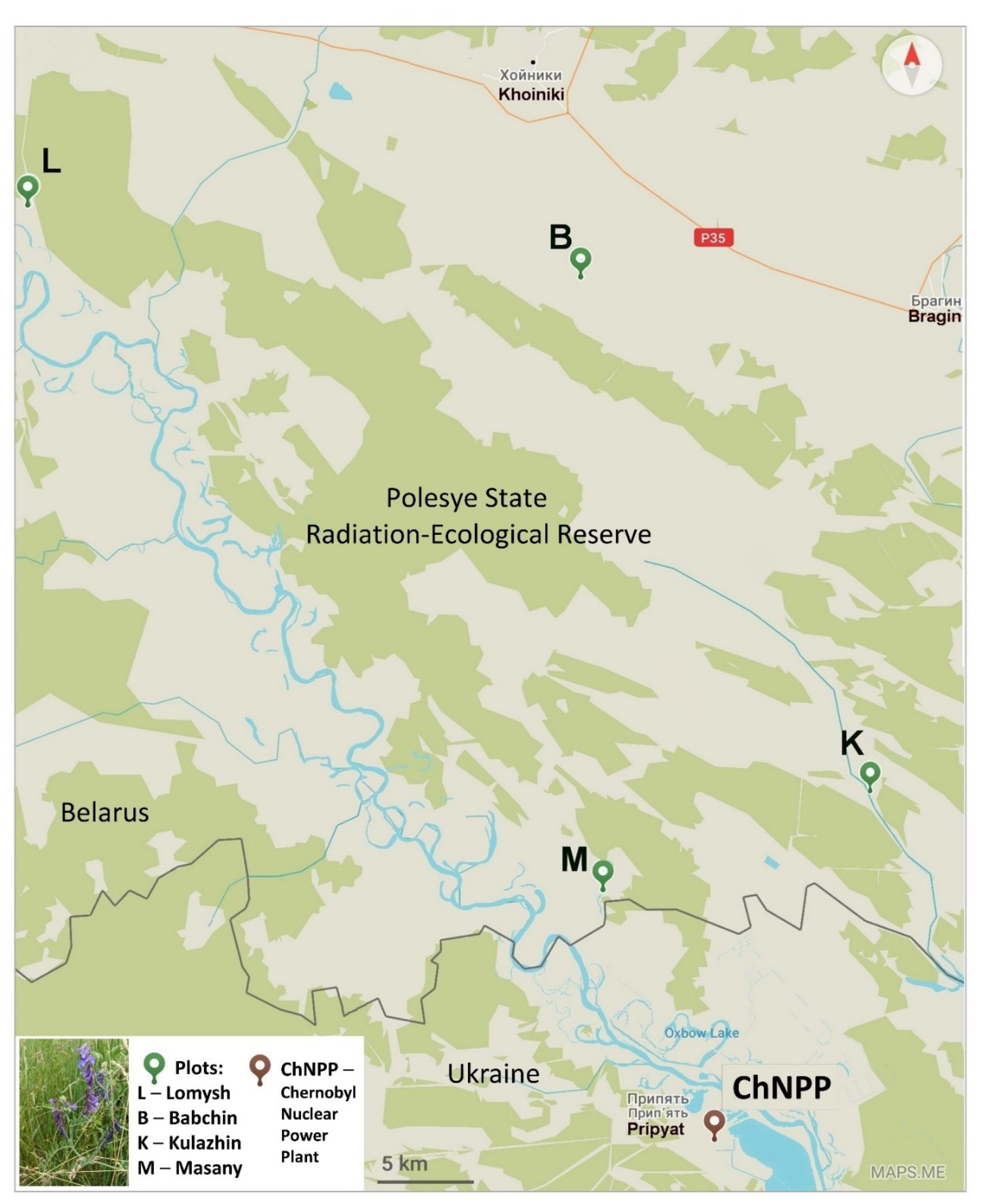
4.1. Experimental Plots and Soil Sampling
4.2. Plant Sampling
4.3. Gene Expression by qRT-PCR
4.4. RNA Sequencing and Transcriptomic Analysis
4.5. Protein Isolation and Proteomic Analysis
4.6. Metabolite Extraction and Metabolomic Analysis
4.7. Multi-Omics Data Integration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Volkova, P.Y.; Bondarenko, E.V.; Kazakova, E.A. Radiation Hormesis in Plants. Curr. Opin. Toxicol. 2022, 30, 100334. [Google Scholar] [CrossRef]
- Duarte, G.T.; Volkova, P.Y.; Fiengo Perez, F.; Horemans, N. Chronic Ionizing Radiation of Plants: An Evolutionary Factor from Direct Damage to Non-Target Effects. Plants 2023, 12, 1178. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, S.V.; Grinberg, M.A.; Sukhov, V.; Vodeneev, V. Effect of Ionizing Radiation on Physiological and Molecular Processes in Plants. J. Environ. Radioact. 2019, 202, 8–24. [Google Scholar] [CrossRef]
- De Micco, V.; Arena, C.; Pignalosa, D.; Durante, M. Effects of Sparsely and Densely Ionizing Radiation on Plants. Radiat. Environ. Biophys. 2011, 50, 1–19. [Google Scholar] [CrossRef]
- Lourenço, J.; Mendo, S.; Pereira, R. Radioactively Contaminated Areas: Bioindicator Species and Biomarkers of Effect in an Early Warning Scheme for a Preliminary Risk Assessment. J. Hazard. Mater. 2016, 317, 503–542. [Google Scholar] [CrossRef]
- Hoseini, P.S.; Poursafa, P.; Moattar, F.; Amin, M.M.; Rezaei, A.H. Ability of Phytoremediation for Absorption of Strontium and Cesium from Soils Using Cannabis sativa. Int. J. Environ. Health Eng. 2012, 1, 17. [Google Scholar] [CrossRef]
- Soudek, P.; Valenová, Š.; Vavříková, Z.; Vaněk, T. 137Cs and 90Sr Uptake by Sunflower Cultivated under Hydroponic Conditions. J. Environ. Radioact. 2006, 88, 236–250. [Google Scholar] [CrossRef]
- Wang, G.; Wang, B.; Fan, W.; Deng, N. Enhanced Phytoremediation of Uranium-Contaminated Soils by Indian Mustard (Brassica juncea L.) Using Slow Release Citric Acid. Environ. Sci. Pollut. Res. 2021, 28, 61061–61071. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Le, Q.V.; Sonne, C.; Yang, Y.; Yang, H.; Gu, H.; Ma, N.L.; Lam, S.S.; Peng, W. Phytoremediation of Radionuclides in Soil, Sediments and Water. J. Hazard. Mater. 2021, 407, 124771. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.A.; Hughes, S.G. Vicia. In Wild Crop Relatives: Genomic and Breeding Resources: Legume Crops and Forages; Kole, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 273–289. ISBN 978-3-642-14387-8. [Google Scholar]
- Eckardt, N.A.; Ainsworth, E.A.; Bahuguna, R.N.; Broadley, M.R.; Busch, W.; Carpita, N.C.; Castrillo, G.; Chory, J.; DeHaan, L.R.; Duarte, C.M.; et al. Climate Change Challenges, Plant Science Solutions. Plant Cell 2023, 35, 24–66. [Google Scholar] [CrossRef]
- Ibañez, S.; Medina, M.I.; Agostini, E. Vicia: A Green Bridge to Clean up Polluted Environments. Appl. Microbiol. Biotechnol. 2020, 104, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, S.G.; Alderete, L.G.S.; Medina, M.I.; Agostini, E. Phytoremediation of Phenol Using Vicia sativa L. Plants and its Antioxidative Response. Environ. Sci. Pollut. Res. 2012, 19, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liu, J.; Wang, Y.; Zhang, X.; Shen, Z.; Hu, Z. Ectopic Expression of Vicia sativa Caffeoyl-CoA O-Methyltransferase (VsCCoAOMT) Increases the Uptake and Tolerance of Cadmium in Arabidopsis. Environ. Exp. Bot. 2018, 145, 47–53. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Chen, L.; Lu, Y.; An, Y. Ectopic Expression γ-Glutamylcysteine Synthetase of Vicia sativa Increased Cadmium Tolerance in Arabidopsis. Gene 2022, 823, 146358. [Google Scholar] [CrossRef]
- Halfadji, A.; Portet-Koltalo, F.; Touabet, A.; Le Derf, F.; Morin, C.; Merlet-Machour, N. Phytoremediation of PCB: Contaminated Algerian Soils Using Native Agronomics Plants. Environ. Geochem. Health 2022, 44, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Hamid, Y.; Zehra, A.; Sahito, Z.A.; He, Z.; Hussain, B.; Gurajala, H.K.; Yang, X. Characterization of Fava Bean (Vicia faba L.) Genotypes for Phytoremediation of Cadmium and Lead Co-Contaminated Soils Coupled with Agro-Production. Ecotoxicol. Environ. Saf. 2019, 171, 190–198. [Google Scholar] [CrossRef]
- Tang, L.; Hamid, Y.; Zehra, A.; Sahito, Z.A.; He, Z.; Beri, W.T.; Khan, M.B.; Yang, X. Fava Bean Intercropping with Sedum Alfredii Inoculated with Endophytes Enhances Phytoremediation of Cadmium and Lead Co-Contaminated Field. Environ. Pollut. 2020, 265, 114861. [Google Scholar] [CrossRef]
- Radwan, S.S.; Dashti, N.; El-Nemr, I.M. Enhancing the Growth of Vicia faba Plants by Microbial Inoculation to Improve Their Phytoremediation Potential for Oily Desert Areas. Int. J. Phytoremediat. 2005, 7, 19–32. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, N.; Luo, Y.; Ni, Y.; Liu, Z.; Wu, G.; Zheng, T.; Dang, Y.; Chen, H.; Li, W. Effects of Strontium on the Morphological and Photosynthetic Physiological Characteristics of Vicia faba Seedlings. Int. J. Phytoremediat. 2022, 25, 811–821. [Google Scholar] [CrossRef]
- Ibañez, S.G.; Travaglia, C.N.; Medina, M.I.; Agostini, E. Vicia Villosa Roth: A Cover Crop to Phytoremediate Arsenic Polluted Environments. Environ. Sci. Pollut. Res. 2021, 28, 38604–38612. [Google Scholar] [CrossRef]
- Petukhov, A.S.; Kremleva, T.A.; Petukhova, G.A.; Khritokhin, N.A. Translocation of Heavy Metals in Herbs under Urban Anthropogenic Pollution Conditions. Environ. Process. 2020, 7, 1173–1196. [Google Scholar] [CrossRef]
- Evseeva, T.; Majstrenko, T.; Geras’kin, S.; Brown, J.E.; Belykh, E. Estimation of Ionizing Radiation Impact on Natural Vicia cracca Populations Inhabiting Areas Contaminated with Uranium Mill Tailings and Radium Production Wastes. Sci. Total Environ. 2009, 407, 5335–5343. [Google Scholar] [CrossRef]
- Hua, Y.; Chen, J.; Zhou, T.; Zhang, T.; Shen, D.; Feng, Y.; Guan, P.; Huang, S.; Zhou, Z.; Huang, J.; et al. Multiomics Reveals an Essential Role of Long-Distance Translocation in Regulating Plant Cadmium Resistance and Grain Accumulation in Allohexaploid Wheat (Triticum aestivum). J. Exp. Bot. 2022, 73, 7516–7537. [Google Scholar] [CrossRef] [PubMed]
- Duarte, G.T.; Volkova, P.Y.; Geras’kin, S.A. The Response Profile to Chronic Radiation Exposure Based on the Transcriptome Analysis of Scots Pine from Chernobyl Affected Zone. Environ. Pollut. 2019, 250, 618–626. [Google Scholar] [CrossRef]
- Volkova, P.Y.; Duarte, G.T.; Kazakova, E.A.; Makarenko, E.S.; Bitarishvili, S.V.; Bondarenko, V.S.; Perevolotskii, A.N.; Geras’kin, S.A.; Garbaruk, D.K.; Turchin, L.M. Radiosensitivity of Herbaceous Plants to Chronic Radiation Exposure: Field Study in the Chernobyl Exclusion Zone. Sci. Total Environ. 2021, 777, 146206. [Google Scholar] [CrossRef]
- Shesterikova, E.M.; Bondarenko, V.S.; Volkova, P.Y. Differential Gene Expression in Chronically Irradiated Herbaceous Species from the Chernobyl Exclusion Zone. Int. J. Radiat. Biol. 2023, 99, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Podlutskii, M.; Babina, D.; Podobed, M.; Bondarenko, E.; Bitarishvili, S.; Blinova, Y.; Shesterikova, E.; Prazyan, A.; Turchin, L.; Garbaruk, D.; et al. Arabidopsis thaliana Accessions from the Chernobyl Exclusion Zone Show Decreased Sensitivity to Additional Acute Irradiation. Plants 2022, 11, 3142. [Google Scholar] [CrossRef]
- STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/30476243/ (accessed on 12 April 2023).
- Gaufichon, L.; Masclaux-Daubresse, C.; Tcherkez, G.; Reisdorf-Cren, M.; Sakakibara, Y.; Hase, T.; Clément, G.; Avice, J.-C.; Grandjean, O.; Marmagne, A.; et al. Arabidopsis thaliana ASN2 Encoding Asparagine Synthetase Is Involved in the Control of Nitrogen Assimilation and Export during Vegetative Growth. Plant Cell Environ. 2013, 36, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Igamberdiev, A.U.; Eprintsev, A.T. Organic Acids: The Pools of Fixed Carbon Involved in Redox Regulation and Energy Balance in Higher Plants. Front. Plant Sci. 2016, 7, 1042. [Google Scholar] [CrossRef] [Green Version]
- Igamberdiev, A.U. Citrate Valve Integrates Mitochondria into Photosynthetic Metabolism. Mitochondrion 2020, 52, 218–230. [Google Scholar] [CrossRef]
- Lucas, K.A.; Filley, J.R.; Erb, J.M.; Graybill, E.R.; Hawes, J.W. Peroxisomal Metabolism of Propionic Acid and Isobutyric Acid in Plants. J. Biol. Chem. 2007, 282, 24980–24989. [Google Scholar] [CrossRef] [Green Version]
- Elango, D.; Rajendran, K.; Van der Laan, L.; Sebastiar, S.; Raigne, J.; Thaiparambil, N.A.; El Haddad, N.; Raja, B.; Wang, W.; Ferela, A.; et al. Raffinose Family Oligosaccharides: Friend or Foe for Human and Plant Health? Front. Plant Sci. 2022, 13, 829118. [Google Scholar] [CrossRef]
- Sanyal, R.; Pradhan, B.; Jawed, D.M.; Tribhuvan, K.U.; Dahuja, A.; Kumar, M.; Kumar, N.; Mishra, G.P.; Ram, C.; Mahatma, M.K.; et al. Spatio-Temporal Expression Pattern of Raffinose Synthase Genes Determine the Levels of Raffinose Family Oligosaccharides in Peanut (Arachis hypogaea L.). Seed Sci. Rep. 2023, 13, 795. [Google Scholar] [CrossRef]
- Strasser, R. Recent Developments in Deciphering the Biological Role of Plant Complex N-Glycans. Front. Plant Sci. 2022, 13, 897549. [Google Scholar] [CrossRef]
- Van Holle, S.; Van Damme, E.J.M. Signaling through Plant Lectins: Modulation of Plant Immunity and Beyond. Biochem. Soc. Trans. 2018, 46, 217–233. [Google Scholar] [CrossRef]
- Hong, M.J.; Kim, J.; Yoon, Y.H.; Kim, S.H.; Ahn, J.W.; Jeong, I.Y.; Kang, S.Y.; Seo, Y.W.; Kim, D.S. The Effects of Chronic Gamma Irradiation on Oxidative Stress Response and the Expression of Anthocyanin Biosynthesis-Related Genes in Wheat (Triticum aestivum). Int. J. Radiat. Biol. 2014, 90, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Ahuja, S.; Singhal, R.K.; Venu Babu, P. Effect of Gamma Radiation on Wheat Plant Growth Due to Impact on Gas Exchange Characteristics and Mineral Nutrient Uptake and Utilization. J. Radioanal. Nucl. Chem. 2013, 298, 249–257. [Google Scholar] [CrossRef]
- Vanhoudt, N.; Vandenhove, H.; Horemans, N.; Wannijn, J.; Van Hees, M.; Vangronsveld, J.; Cuypers, A. The Combined Effect of Uranium and Gamma Radiation on Biological Responses and Oxidative Stress Induced in Arabidopsis thaliana. J. Environ. Radioact. 2010, 101, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-H.; Chang, C.-Y.; Hsu, S.-J.; Chen, J.-J. Chloroplast Localization of Methylerythritol 4-Phosphate Pathway Enzymes and Regulation of Mitochondrial Genes in IspD and IspE Albino Mutants in Arabidopsis. Plant Mol. Biol. 2008, 66, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, C.; Mo, R.; Zhu, Z.; Dong, Z.; Hu, X.; Deng, W.; Zhuang, C. Screening and Verification of Photosynthesis and Chloroplast-Related Genes in Mulberry by Comparative RNA-Seq and Virus-Induced Gene Silencing. Int. J. Mol. Sci. 2022, 23, 8620. [Google Scholar] [CrossRef] [PubMed]
- Mishra, L.S.; Funk, C. The FtsHi Enzymes of Arabidopsis thaliana: Pseudo-Proteases with an Important Function. Int. J. Mol. Sci. 2021, 22, 5917. [Google Scholar] [CrossRef]
- He, X.; Long, F.; Li, Y.; Xu, Y.; Hu, L.; Yao, T.; Huang, Y.; Hu, D.; Yang, Y.; Fei, Y. Comparative Transcriptome Analysis Revealing the Potential Mechanism of Low-Temperature Stress in Machilus microcarpa. Front. Plant Sci. 2022, 13, 900870. [Google Scholar] [CrossRef] [PubMed]
- Pontier, D.; Albrieux, C.; Joyard, J.; Lagrange, T.; Block, M. Knock-out of the Magnesium Protoporphyrin IX Methyltransferase Gene in Arabidopsis: Effects on Chloroplast Development and on Chloroplast-to-Nucleus Signaling. J. Biol. Chem. 2007, 282, 2297–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, L.; Fan, Z.; Zhang, Q.; Wang, C.; Gao, Y.; Deng, Y.; Zhu, B.; Zhu, H.; Chen, J.; Shan, W.; et al. BEL1-LIKE HOMEODOMAIN 11 Regulates Chloroplast Development and Chlorophyll Synthesis in Tomato Fruit. Plant J. 2018, 94, 1126–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangappa, S.N.; Botto, J.F. The Multifaceted Roles of HY5 in Plant Growth and Development. Mol. Plant 2016, 9, 1353–1365. [Google Scholar] [CrossRef] [Green Version]
- Laanen, P.; Cuypers, A.; Saenen, E.; Horemans, N. Flowering under Enhanced Ionising Radiation Conditions and Its Regulation through Epigenetic Mechanisms. Plant Physiol. Biochem. 2023, 196, 246–259. [Google Scholar] [CrossRef]
- Kim, J.-H.; Moon, Y.R.; Kim, J.-S.; Oh, M.-H.; Lee, J.-W.; Chung, B.Y. Transcriptomic Profile of Arabidopsis Rosette Leaves during the Reproductive Stage after Exposure to Ionizing Radiation. Radiat. Res. 2007, 168, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Szurman-Zubrzycka, M.; Jędrzejek, P.; Szarejko, I. How Do Plants Cope with DNA Damage? A Concise Review on the DDR Pathway in Plants. Int. J. Mol. Sci. 2023, 24, 2404. [Google Scholar] [CrossRef]
- Han, J.; Li, H.; Yin, B.; Zhang, Y.; Liu, Y.; Cheng, Z.; Liu, D.; Lu, H. The Papain-like Cysteine Protease CEP1 Is Involved in Programmed Cell Death and Secondary Wall Thickening during Xylem Development in Arabidopsis. J. Exp. Bot. 2019, 70, 205–215. [Google Scholar] [CrossRef]
- Song, Y.; Chen, Q.; Ci, D.; Shao, X.; Zhang, D. Effects of High Temperature on Photosynthesis and Related Gene Expression in Poplar. BMC Plant Biol. 2014, 14, 111. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Wang, Q.; Song, Z.; Wu, Y.; Liang, Q.; Wang, Q.; Yang, J.; Bi, Y.; Zhou, W.; Fan, L.-M. O-Fucosylation of CPN20 by SPINDLY Derepresses Abscisic Acid Signaling During Seed Germination and Seedling Development. Front. Plant Sci. 2021, 12, 724144. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Jiang, T.; Wu, Z.; Du, S.-Y.; Yu, Y.-T.; Jiang, S.-C.; Lu, K.; Feng, X.-J.; Wang, X.-F.; Zhang, D.-P. Cochaperonin CPN20 Negatively Regulates Abscisic Acid Signaling in Arabidopsis. Plant Mol. Biol. 2013, 83, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Brader, G.; Helenius, E.; Kariola, T.; Palva, E.T. Biotin Deficiency Causes Spontaneous Cell Death and Activation of Defense Signaling. Plant J. 2012, 70, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Alban, C. Chapter 2—Biotin (Vitamin B8) Synthesis in Plants. In Advances in Botanical Research; Rébeillé, F., Douce, R., Eds.; Biosynthesis of Vitamins in Plants Part B; Academic Press: Cambridge, MA, USA, 2011; Volume 59, pp. 39–66. [Google Scholar]
- Jones, D.H. Phenylalanine Ammonia-Lyase: Regulation of Its Induction, and Its Role in Plant Development. Phytochemistry 1984, 23, 1349–1359. [Google Scholar] [CrossRef]
- Benoît, M.A.; D’Aprano, G.; Lacroix, M. Effect of γ-Irradiation on Phenylalanine Ammonia-Lyase Activity, Total Phenolic Content, and Respiration of Mushrooms (Agaricus bisporus). J. Agric. Food Chem. 2000, 48, 6312–6316. [Google Scholar] [CrossRef]
- Oufedjikh, H.; Mahrouz, M.; Amiot, M.J.; Lacroix, M. Effect of γ-Irradiation on Phenolic Compounds and Phenylalanine Ammonia-Lyase Activity during Storage in Relation to Peel Injury from Peel of Citrus Clementina Hort. Ex. Tanaka. J. Agric. Food Chem. 2000, 48, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Hussain, P.R.; Wani, A.M.; Meena, R.S.; Dar, M.A. Gamma Irradiation Induced Enhancement of Phenylalanine Ammonia-Lyase (PAL) and Antioxidant Activity in Peach (Prunus Persica Bausch, Cv. Elberta). Radiat. Phys. Chem. 2010, 79, 982–989. [Google Scholar] [CrossRef]
- Xu, J.-J.; Fang, X.; Li, C.-Y.; Yang, L.; Chen, X.-Y. General and Specialized Tyrosine Metabolism Pathways in Plants. aBIOTECH 2020, 1, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K. Explorations of Plant’s Chemodiversity: Role of Nitrogen-Containing Secondary Metabolites in Plant Defense. In Molecular Aspects of Plant-Pathogen Interaction; Singh, A., Singh, I.K., Eds.; Springer: Singapore, 2018; pp. 309–332. [Google Scholar] [CrossRef]
- Baral, B.; Teixeira da Silva, J.A.; Izaguirre-Mayoral, M.L. Early Signaling, Synthesis, Transport and Metabolism of Ureides. J. Plant Physiol. 2016, 193, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Chowrasia, S.; Gaur, V.S.; Monda, T.K. Allantoin: Emerging Role in Plant Abiotic Stress Tolerance. Plant Mol. Biol. Rep. 2021, 39, 648–661. [Google Scholar] [CrossRef]
- Berglund, T.; Wallström, A.; Nguyen, T.-V.; Laurell, C.; Ohlsson, A.B. Nicotinamide; Antioxidative and DNA Hypomethylation Effects in Plant Cells. Plant Physiol. Biochem. PPB 2017, 118, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Hidalgo, M.; León-González, A.J.; Gálvez-Peralta, M.; González-Mauraza, N.H.; Martin-Cordero, C. D-Pinitol: A Cyclitol with Versatile Biological and Pharmacological Activities. Phytochem. Rev. 2021, 20, 211–224. [Google Scholar] [CrossRef]
- Ashihara, H. Trigonelline (N-Methylnicotinic Acid) Biosynthesis and Its Biological Role in Plants. Nat. Prod. Commun. 2008, 3, 1423–1428. [Google Scholar] [CrossRef] [Green Version]
- Sunil, C.; Irudayaraj, S.S.; Duraipandiyan, V.; Al-Dhabi, N.A.; Agastian, P.; Ignacimuthu, S. Antioxidant and Free Radical Scavenging Effects of β-Amyrin Isolated from S. Cochinchinensis Moore. Leaves. Ind. Crops Prod. 2014, 61, 510–516. [Google Scholar] [CrossRef]
- Hamerski, L.; Bomm, M.D.; Silva, D.H.S.; Young, M.C.M.; Furlan, M.; Eberlin, M.N.; Castro-Gamboa, I.; Cavalheiro, A.J.; da Silva Bolzani, V. Phenylpropanoid Glucosides from Leaves of Coussarea hydrangeifolia (Rubiaceae). Phytochemistry 2005, 66, 1927–1932. [Google Scholar] [CrossRef]
- Fabris, S.; Momo, F.; Ravagnan, G.; Stevanato, R. Antioxidant Properties of Resveratrol and Piceid on Lipid Peroxidation in Micelles and Monolamellar Liposomes. Biophys. Chem. 2008, 135, 76–83. [Google Scholar] [CrossRef]
- Whittle, C.A.; Krochko, J.E. Transcript Profiling Provides Evidence of Functional Divergence and Expression Networks among Ribosomal Protein Gene Paralogs in Brassica napus. Plant Cell 2009, 21, 2203–2219. [Google Scholar] [CrossRef] [Green Version]
- Moin, M.; Bakshi, A.; Saha, A.; Dutta, M.; Madhav, S.M.; Kirti, P.B. Rice Ribosomal Protein Large Subunit Genes and Their Spatio-Temporal and Stress Regulation. Front. Plant Sci. 2016, 7, 1284. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lan, P.; Gao, H.; Zheng, L.; Li, W.; Schmidt, W. Expression Changes of Ribosomal Proteins in Phosphate- and Iron-Deficient Arabidopsis Roots Predict Stress-Specific Alterations in Ribosome Composition. BMC Genom. 2013, 14, 783. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Lan, T.; Mo, B. Extraribosomal Functions of Cytosolic Ribosomal Proteins in Plants. Front. Plant Sci. 2021, 12, 607157. [Google Scholar] [CrossRef] [PubMed]
- Haag, J.R.; Pikaard, C.S. Multisubunit RNA Polymerases IV and V: Purveyors of Non-Coding RNA for Plant Gene Silencing. Nat. Rev. Mol. Cell Biol. 2011, 12, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Vásquez, J.; Delseny, M. Ribosome Biogenesis in Plants: From Functional 45S Ribosomal DNA Organization to Ribosome Assembly Factors. Plant Cell 2019, 31, 1945–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ISO/TC 190—Soil Quality. Available online: https://www.iso.org/committee/54328.html (accessed on 7 April 2023).
- ISO 11047:1998. Soil Quality—Determination of Cadmium, Chromium, Cobalt, Copper, Lead, Manganese, Nickel and Zinc—Flame and Electrothermal Atomic Absorption Spectrometric Methods. Available online: https://www.iso.org/standard/24010.html (accessed on 7 April 2023).
- Laanen, P.; Saenen, E.; Mysara, M.; Van de Walle, J.; Van Hees, M.; Nauts, R.; Van Nieuwerburgh, F.; Voorspoels, S.; Jacobs, G.; Cuypers, A.; et al. Changes in DNA Methylation in Arabidopsis thaliana Plants Exposed Over Multiple Generations to Gamma Radiation. Front. Plant Sci. 2021, 12, 611783. [Google Scholar] [CrossRef]
- Grinberg, M.A.; Gudkov, S.V.; Balalaeva, I.V.; Gromova, E.; Sinitsyna, Y.; Sukhov, V.; Vodeneev, V. Effect of Chronic β-Radiation on Long-Distance Electrical Signals in Wheat and Their Role in Adaptation to Heat Stress. Environ. Exp. Bot. 2021, 184, 104378. [Google Scholar] [CrossRef]
- Gautam, A.; Pandey, A.K. Aquaporins Responses under Challenging Environmental Conditions and Abiotic Stress Tolerance in Plants. Bot. Rev. 2021, 87, 467–495. [Google Scholar] [CrossRef]
- Gutierrez, N.; Giménez, M.J.; Palomino, C.; Avila, C.M. Assessment of Candidate Reference Genes for Expression Studies in Vicia faba L. by Real-Time Quantitative PCR. Mol. Breed. 2010, 28, 13–24. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Duarte, G.T.; Volkova, P.Y.; Geras’kin, S.A. A Pipeline for Non-Model Organisms for de Novo Transcriptome Assembly, Annotation, and Gene Ontology Analysis Using Open Tools: Case Study with Scots Pine. Biol. Protoc. 2021, 11, e3912. [Google Scholar] [CrossRef]
- The Galaxy Community. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2022 Update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef] [PubMed]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction; Humana: New York, NY, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar] [CrossRef]
- Eliášová, A.; Trávníček, P.; Mandák, B.; Münzbergová, Z. Autotetraploids of Vicia cracca Show a Higher Allelic Richness in Natural Populations and a Higher Seed Set after Artificial Selfing than Diploids. Ann. Bot. 2014, 113, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliášová, A.; Münzbergová, Z. Factors Influencing Distribution and Local Coexistence of Diploids and Tetraploids of Vicia cracca: Inferences from a Common Garden Experiment. J. Plant Res. 2017, 130, 4. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER Web Server: 2018 Update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating Signal Peptides from Transmembrane Regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and Rapid Annotation of Ribosomal RNA Genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.-H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolite Profiling in Arabidopsis. In Arabidopsis Protocols; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2006; Volume 323, pp. 439–447. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.E. An Integrated Method for Spectrum Extraction and Compound Identification from Gas Chromatography/Mass Spectrometry Data. J. Am. Soc. Mass Spectrom. 1999, 10, 770–781. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Chong, J.; Zhou, G.; De Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Liu, T.; Salguero, P.; Petek, M.; Martinez-Mira, C.; Balzano-Nogueira, L.; Ramšak, Ž.; McIntyre, L.; Gruden, K.; Tarazona, S.; Conesa, A. PaintOmics 4: New Tools for the Integrative Analysis of Multi-Omics Datasets Supported by Multiple Pathway Databases. Nucleic Acids Res. 2022, 50, W551–W559. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
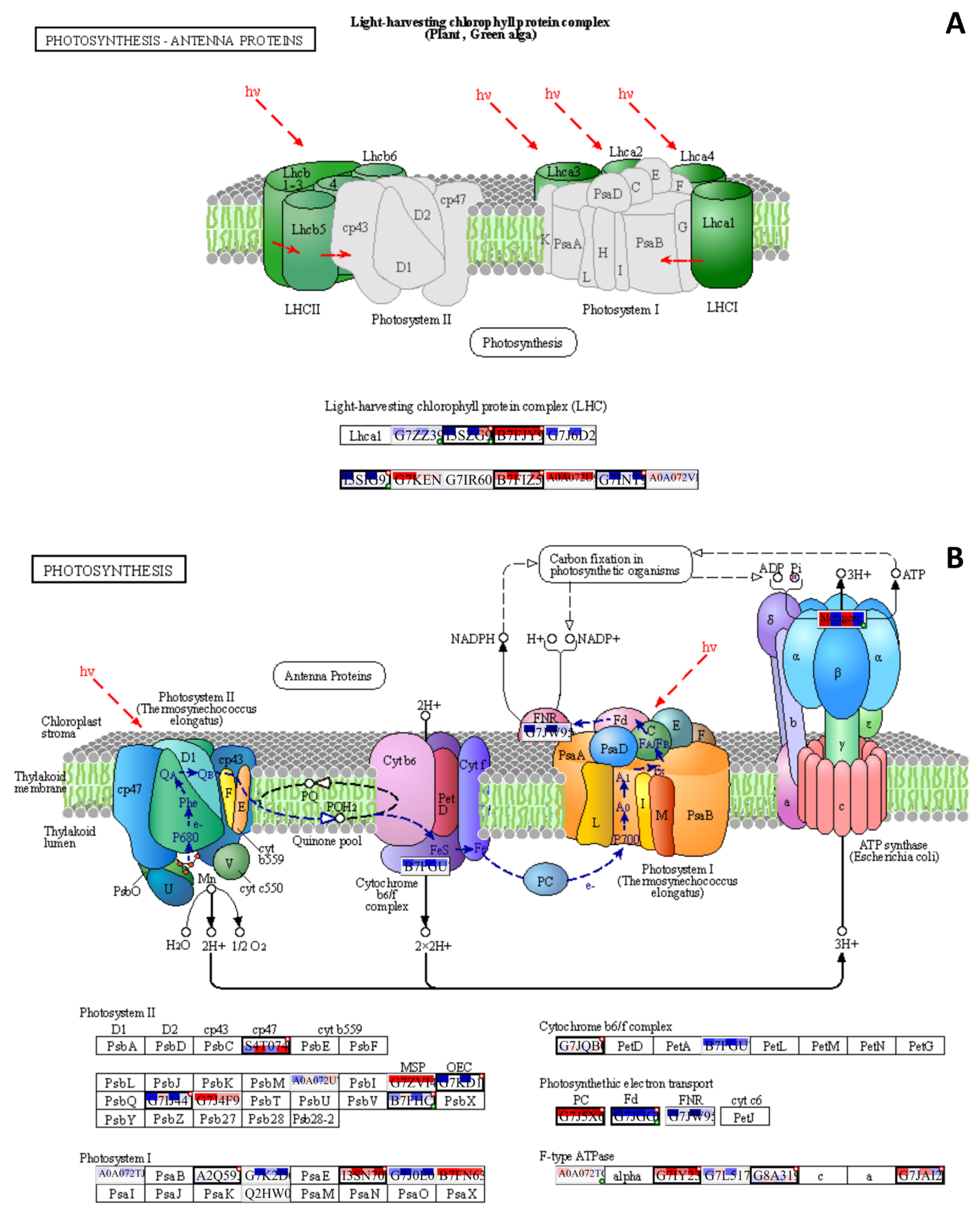{kind=link}
| Experimental Plots | Plot Type | GPS Coordinates | Ambient Dose Rate (γ) μSv × h−1 | α-Particles Flux Density min−1 × sm−2 | β-Particles Flux Density min−1 × sm−2 | Specific Activity of 137Cs in Soil Bq × kg−1 | Specific Activity of 90Sr in Soil Bq × kg−1 |
|---|---|---|---|---|---|---|---|
| Babchin (Bab, B) | Reference | N 51°47′29.43″ E 30°00′03.86″ | 0.29 | 0.4 | 4.7 | 126 ± 4.5 | 9.86 ± 1.6 |
| Lomysh (Lom, L) | Reference | N 51°49′50.56″ E 29°34′37.03″ | 0.35 | 2.5 | 2.4 | 1027 ± 30 | 16.09 ± 2.6 |
| Kulazhin (Kul, K) | Polluted | N 51°33′07.6″ E 30°13′10.5″ | 6.84 | 42.0 | 31.0 | 36,990 ± 935 | 1396.6 ± 209.8 |
| Masany (Mas, M) | Polluted | N 51°30′22.36″ E 30°00′56.86″ | 3.20 | 38.7 | 47.2 | 9587 ± 246 | 1598.5 ± 240.1 |
| Experimental Plots | pH | HA | TEB | Humus | Av. P2O5 | Av. K2O | Av. Ca | Av. Mg | Av. Na |
|---|---|---|---|---|---|---|---|---|---|
| mg-eqv Per 100 | % | mg/kg | mg-eqv Per 100 g | ||||||
| Babchin (Bab, B) | 6.4 | 0.90 ± 0.01 | 6.1 ± 0.7 | 1.17 ± 0.01 | 210.7 ± 5.2 | 91.0 ± 1.0 | 3.41 ± 0.20 | 0.85 ± 0.02 | 0.61 ± 0.04 |
| Lomysh (Lom, L) | 5.5 | 1.58 ± 0.02 | 3.1 ± 0.1 | 1.78 ± 0.04 | 209.1 ± 5.2 | 38.7 ± 0.1 | 2.49 ± 0.08 | 0.53 ± 0.01 | 0.57 ± 0.02 |
| Kulazhin (Kul, K) | 4.6 | 7.04 ± 0.07 | 6.2 ± 0.4 | 5.28 ± 0.07 | 337.1 ± 6.4 | 47.5 ± 0.7 | 5.13 ± 0.01 | 2.09 ± 0.02 | 1.18 ± 0.14 |
| Masany (Mas, M) | 5.5 | 3.71 ± 0.08 | 7.6 ± 0.2 | 2.67 ± 0.02 | 943.2 ± 19.9 | 153.5 ± 2.6 | 5.06 ± 0.19 | 0.79 ± 0.01 | 0.64 ± 0.05 |
| Upregulated | Downregulated | ||
|---|---|---|---|
| GO:0000786 | nucleosome | GO:0009793 | embryo development ending in seed dormancy |
| GO:0003677 | DNA binding | GO:0009534 | chloroplast thylakoid |
| GO:0046982 | protein heterodimerisation activity | GO:0009506 | plasmodesma |
| GO:0009507 | chloroplast | ||
| GO:0009941 | chloroplast envelope | ||
| GO:0009570 | chloroplast stroma | ||
| GO:0009536 | plastid | ||
| GO:0009706 | chloroplast inner membrane | ||
| GO:0000325 | plant-type vacuole | ||
| GO:0003729 | mRNA binding | ||
| DOWNREGULATED GENES | ||||||
| Uniprot ID | Physiological Process | Description | Log2FC | |||
| B | L | |||||
| M | K | M | K | |||
| TKTC_CRAPL | Photosynthesis, the Calvin cycle | Transketolase, chloroplastic | −11.6 | −9.8 | −10.4 | −10.4 |
| RBS_MEDSA | Ribulose bisphosphate carboxylase small subunit, chloroplastic | −11.5 | −11.7 | −9.8 | −9.7 | |
| KPPR_MESCR | Phosphoribulokinase, chloroplastic | −10.6 | −9.8 | −9.1 | −9.9 | |
| RCA1_LARTR | Ribulose bisphosphate carboxylase/oxygenase activase 1, chloroplastic | −10.1 | −9.2 | −10.1 | −9.0 | |
| GLGS2_VICFA | Glucose-1-phosphate adenylyltransferase small subunit 2, chloroplastic | −10.0 | −10.6 | −6.7 | −6.3 | |
| PGL1B_ARATH | Photosynthesis, light-dependent reactions | Ferredoxin-plastoquinone reductase | −10.6 | −10.7 | −9.1 | −9.2 |
| FTSI1_ARATH | Chlorophyll biosynthesis | FtsH extracellular protease family | −10.1 | −10.1 | −6.9 | −7.0 |
| CHLM_ARATH | Magnesium-protoporphyrin IX methyltransferase | −10.1 | −10.1 | −6.9 | −7.0 | |
| CHLP_ARATH | Pyridine nucleotide-disulfide oxidoreductase family protein | −10.8 | −10.8 | −7.4 | −7.6 | |
| THI4_CITSI | Thiamine biosynthesis | Thiamine thiazole synthase, chloroplastic-like | −11.0 | −10.9 | −8.1 | −8.3 |
| PUR5_VIGUN | Phosphoribosylformylglycinamidine cyclo-ligase, chloroplastic/mitochondrial | −10.7 | −10.6 | −7.0 | −7.1 | |
| SYWM_ARATH | Translation | Nucleotidylyl transferase superfamily protein | −10.6 | −10.5 | −7.3 | −7.4 |
| RS6_ASPOF | 40S ribosomal protein S6 | −10.1 | −10.3 | −8.4 | −8.6 | |
| IF5A_SENVE | Eukaryotic translation initiation factor 5A | −10.2 | −10.2 | −8.1 | −8.3 | |
| HSP11_PEA | Stress response | 18.1 kDa class I heat shock protein | −10.9 | −10.9 | −8.8 | −9.0 |
| STEP1_ARATH | Stress-enhanced protein 1 | −10.5 | −11.0 | −6.5 | −7.1 | |
| ROC5_NICSY | RNA processing | 33 kDa ribonucleoprotein, chloroplastic | −11.2 | −11.1 | −8.4 | −8.9 |
| STR9_ARATH | Sulphur metabolism | Rhodanese-like domain-containing protein 9, chloroplastic | −11.2 | −11.1 | −6.9 | −7.1 |
| ISPE_SOLLC | Secondary metabolism | 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase, chloroplastic/chromoplastic | −10.9 | −8.0 | −9.0 | −6.5 |
| LRX4_ARATH | Cell wall | Leucine-rich repeat (LRR) family protein | −10.9 | −10.8 | −7.0 | −7.2 |
| SNAT2_ORYSJ | Melatonin biosynthesis | Serotonin N-acetyltransferase 2, chloroplastic-like | −10.7 | −8.6 | −7.1 | −6.9 |
| CP31A_ARATH | Telomere elongation | 31-kDa RNA-binding protein | −10.7 | −11.6 | −9.3 | −9.6 |
| PGKH_WHEAT | Glycolysis | Phosphoglycerate kinase, chloroplastic-like | −10.3 | −10.2 | −8.6 | −8.8 |
| FT_ARATH | Flowering | Protein FLOWERING LOCUS T | −10.1 | −9.2 | −9.1 | −8.9 |
| CNIH4_ARATH | Vesicle-mediated transport | Cornichon family protein | −10.0 | −9.9 | −7.8 | −7.9 |
| UPREGULATED GENES | ||||||
| Uniprot ID | Physiological process | Description | Log2FC | |||
| B | L | |||||
| M | K | M | K | |||
| CEP1_ARATH | Programmed cell death | Cysteine proteinases superfamily protein | 10.3 | 7.1 | 9.3 | 7.1 |
| PER72_ARATH | Antioxidant system | Peroxidase superfamily protein | 9.6 | 6.0 | 7.4 | 6.0 |
| PERE5_ARMRU | Peroxidase E5 | 9.1 | 6.4 | 8.2 | 6.5 | |
| LECS_VATGU | Defence response | Seed lectin | 9.4 | 7.7 | 8.7 | 7.7 |
| GUN4C_ARATH | Chlorophyll synthesis | Tetrapyrrole-binding protein, chloroplastic | 9.2 | 8.2 | 7.6 | 8.2 |
| ASPG_LUPAL | Protein catabolism | Isoaspartyl peptidase/L-asparaginase | 9.4 | 5.8 | 7.3 | 5.8 |
| UBP21_ARATH | Ubiquitin-specific protease 21 | 9.4 | 6.0 | 8.5 | 6.0 | |
| RS31_CANAL | Ubiquitin-ribosomal 40S subunit protein S31 fusion protein | 8.8 | 6.9 | 8.7 | 6.9 | |
| UFSP_ORYSJ | Probable Ufm1-specific protease | 8.7 | 5.2 | 5.4 | 5.2 | |
| FB330_ARATH | F-box protein | 10.1 | 8.6 | 9.1 | 8.6 | |
| TI10A_ORYSJ | Phytohormones | Protein TIFY 10a-like (jasmonate response) | 8.8 | 6.6 | 5.7 | 6.7 |
| AHP2_ARATH | Histidine-containing phosphotransmitter 2 (cytokinin response) | 8.8 | 5.9 | 6.2 | 5.9 | |
| ILL1_ORYSI | IAA-amino acid hydrolase ILR1-like 1 (auxin response) | 8.7 | 6.1 | 5.6 | 6.1 | |
| XRN2_ARATH | RNA processing | Exoribonuclease 2 | 9.6 | 6.8 | 7.2 | 6.8 |
| C3H54_ORYSJ | Zinc finger CCCH domain-containing protein 54-like | 8.7 | 7.8 | 8.7 | 7.8 | |
| PP182_ARATH | Pentatricopeptide repeat (PPR) superfamily protein | 8.6 | 6.6 | 6.4 | 6.6 | |
| GLSN_MEDSA | Nitrogen assimilation | Glutamate synthase (NADH), amyloplastic | 9.0 | 6.6 | 6.0 | 6.6 |
| LHCA2_ARATH | Chloroplast biogenesis | Photosystem I light-harvesting complex protein | 9.0 | 6.5 | 6.6 | 6.5 |
| IQD32_ARATH | Transport | IQ-domain 32 | 8.9 | 5.9 | 6.5 | 5.9 |
| PITC_DICDI | Phosphatidylinositol transfer protein 3 | 8.8 | 5.9 | 8.0 | 5.9 | |
| MED7A_ARATH | Transcription | Mediator complex, subunit Med7(AT5G03220) | 8.9 | 6.7 | 7.1 | 6.7 |
| H4_SOYBN | Histone | Histone H4 | 8.8 | 6.8 | 5.9 | 6.8 |
| PALY_MEDSA | Secondary metabolism | Phenylalanine ammonia-lyase | 8.8 | 7.2 | 9.6 | 7.3 |
| TOP3A_ARATH | Cell cycle | Topoisomerase 3-alpha | 9.5 | 6.6 | 7.2 | 6.6 |
| CCNB1_MEDSA | G2/mitotic-specific cyclin-1 | 8.6 | 8.2 | 8.8 | 8.2 | |
| Metabolite | F Value (ANOVA) | FDR | Fisher’s LSD |
|---|---|---|---|
| Allantoin | 55.705 | 5.84 × 10−4 | Mas > Control; Mas > Kul |
| O-methyl-inositol | 21.045 | 4.05 × 10−2 | Mas > Control; Mas > Kul |
| Citrate | 20.873 | 4.05 × 10−2 | Control > Kul; Control > Mas |
| Inositol-3-phosphate | 18.018 | 5.72 × 10−2 | Mas > Control; Mas > Kul |
| Fumarate | 17.492 | 5.72 × 10−2 | Mas > Control; Mas > Kul |
| Hydrangeifolin | 16.433 | 6.39 × 10−2 | Kul > Control; Kul > Mas |
| Tyrosine | 14.835 | 8.76 × 10−2 | Kul > Control; Kul > Mas |
| Raffinose | 13.806 | 9.28 × 10−2 | Control > Kul; Control > Mas |
| Maltose | 13.706 | 9.28 × 10−2 | Kul > Control; Kul > Mas |
| Pinitol | 13.519 | 9.28 × 10−2 | Kul > Control; Mas > Control; Mas > Kul |
| Arabinose | 12.781 | 1.07 × 10−1 | Kul > Control; Kul > Mas |
| Malonate | 12.547 | 1.07 × 10−1 | Mas > Control; Mas > Kul |
| Benzyl glucopyranoside | 11.172 | 0.016 | Kul > Control; Kul > Mas |
| Pyruvate | 9.4194 | 0.029 | Kul > Control; Kul > Mas |
| Nicotinate | 8.5022 | 0.039 | Kul > Control; Mas > Control |
| Lactitol | 8.4345 | 0.039 | Kul > Control; Mas > Control |
| Phenethyl glycosides | 8.2561 | 0.040 | Kul > Control; Kul > Mas |
| Ribose | 7.9636 | 0.043 | Kul > Control; Kul > Mas |
| Galactosylglycerol | 7.4886 | 0.049 | Mas > Control; Mas > Kul |
| Galactinol | 7.4159 | 0.049 | Control > Kul; Control > Mas |
| Gene (Arabidopsis thaliana) | AGI | Protein | Function |
|---|---|---|---|
| APX1 | AT1G07890 | Cytosolic ascorbate peroxidase APX1 | Antioxidant |
| CAB1 | AT1G29930 | Light-harvesting complex subunit II (LHCII) | Photosynthesis |
| RBOH-F | AT1G64060 | Respiratory burst oxidase homologue F | Response to ABA and ROS |
| Hy-5 | AT5G11260 | Transcription factor HY5 | |
| PIP1 | AT3G61430 | Plasma membrane intrinsic protein 1 | Aquaporin |
| TIP1 | AT2G36830 | Tonoplast intrinsic protein 1 | |
| CPN60A | AT2G28000 | Chaperonin 60-α | Protein folding |
| CPN20 | AT5G20720 | Chloroplast chaperonin 20 | |
| H2B | AT5G22880 | Histone 2B | Nucleosome assembly |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voronezhskaya, V.; Volkova, P.; Bitarishvili, S.; Shesterikova, E.; Podlutskii, M.; Clement, G.; Meyer, C.; Duarte, G.T.; Kudin, M.; Garbaruk, D.; et al. Multi-Omics Analysis of Vicia cracca Responses to Chronic Radiation Exposure in the Chernobyl Exclusion Zone. Plants 2023, 12, 2318. https://doi.org/10.3390/plants12122318
Voronezhskaya V, Volkova P, Bitarishvili S, Shesterikova E, Podlutskii M, Clement G, Meyer C, Duarte GT, Kudin M, Garbaruk D, et al. Multi-Omics Analysis of Vicia cracca Responses to Chronic Radiation Exposure in the Chernobyl Exclusion Zone. Plants. 2023; 12(12):2318. https://doi.org/10.3390/plants12122318
Chicago/Turabian StyleVoronezhskaya, Viktoria, Polina Volkova, Sofia Bitarishvili, Ekaterina Shesterikova, Mikhail Podlutskii, Gilles Clement, Christian Meyer, Gustavo Turqueto Duarte, Maksim Kudin, Dmitrii Garbaruk, and et al. 2023. "Multi-Omics Analysis of Vicia cracca Responses to Chronic Radiation Exposure in the Chernobyl Exclusion Zone" Plants 12, no. 12: 2318. https://doi.org/10.3390/plants12122318