
Population Genetic Analysis in Persimmons (Diospyros kaki Thunb.) Based on Genome-Wide Single-Nucleotide Polymorphisms

Abstract
:1. Introduction
2. Results
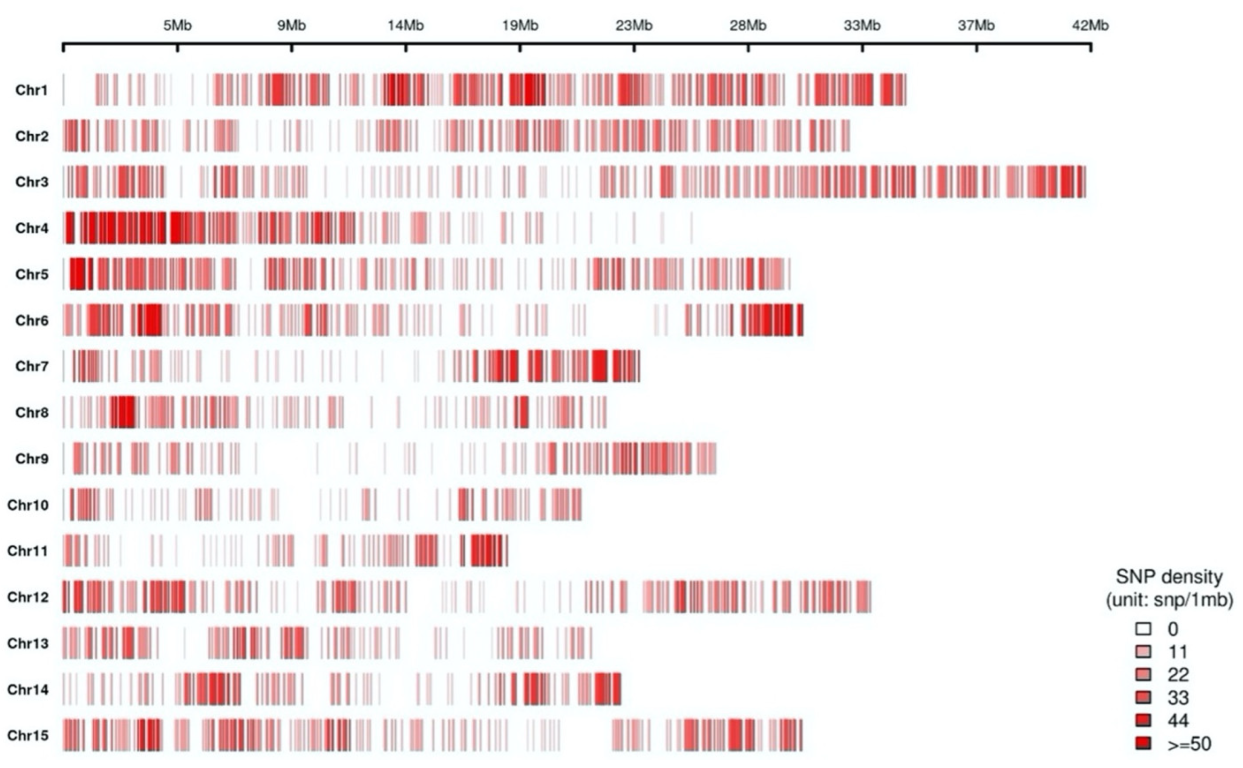
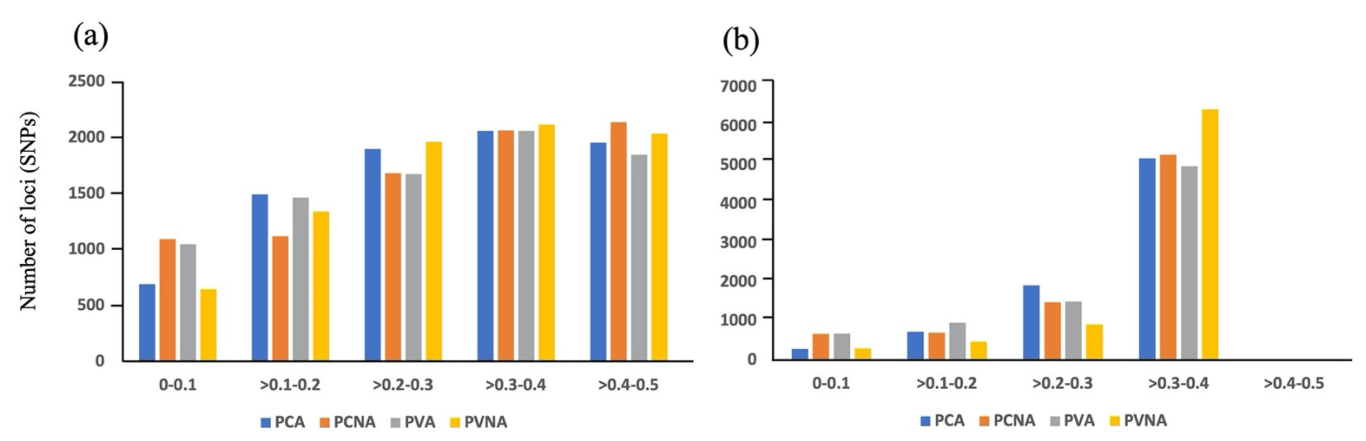
2.1. GBS and SNP Discovery
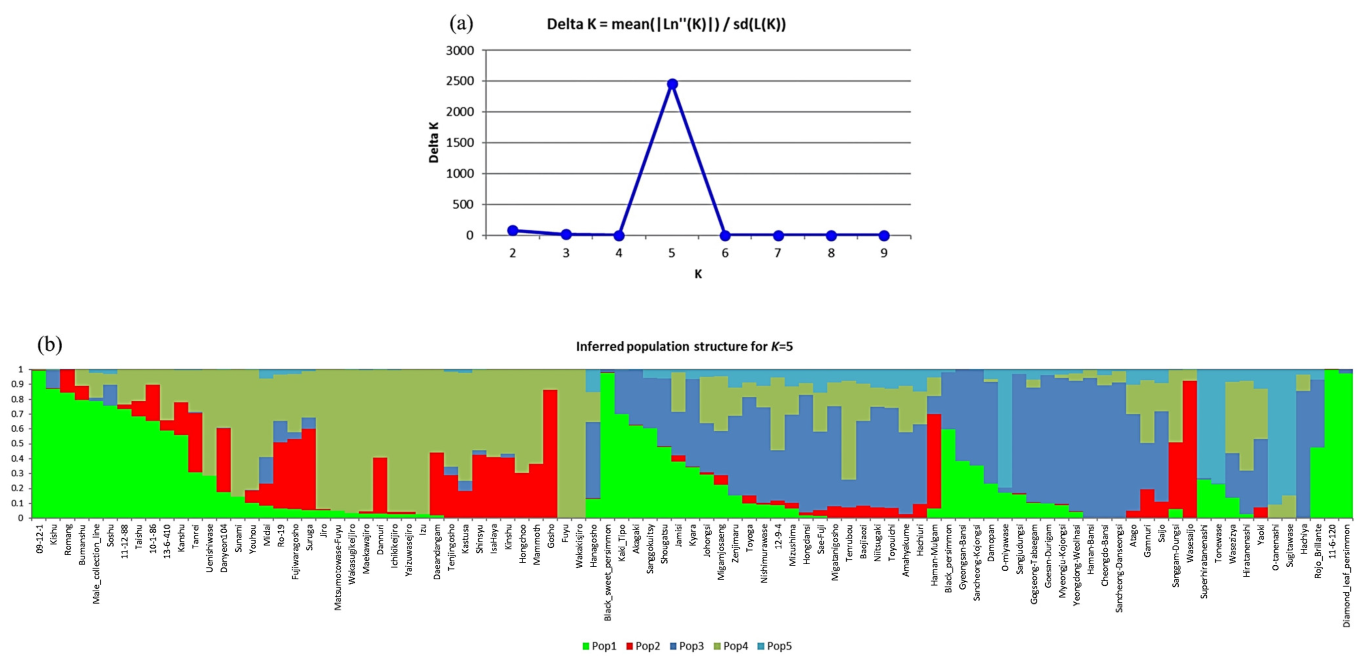
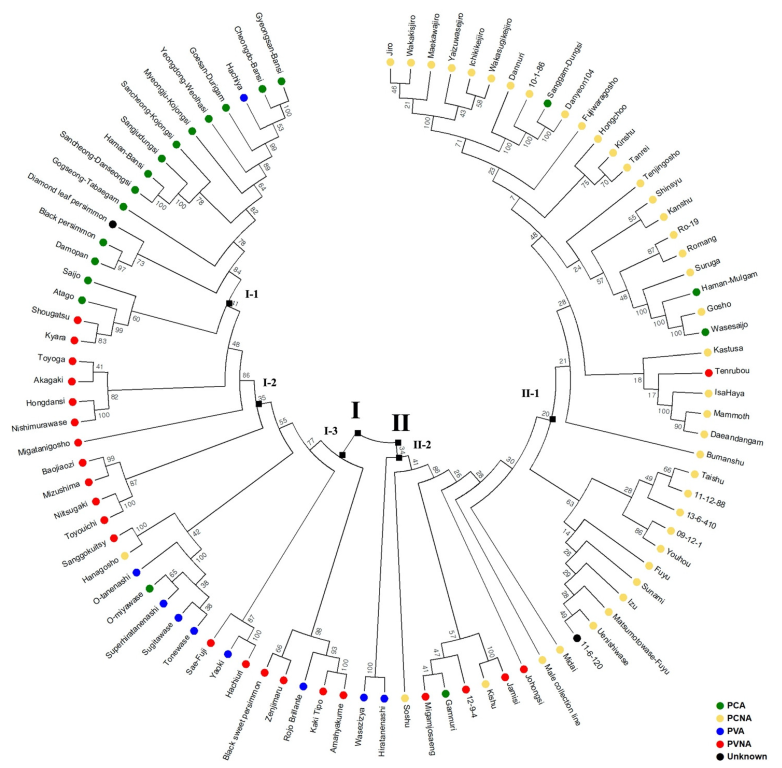
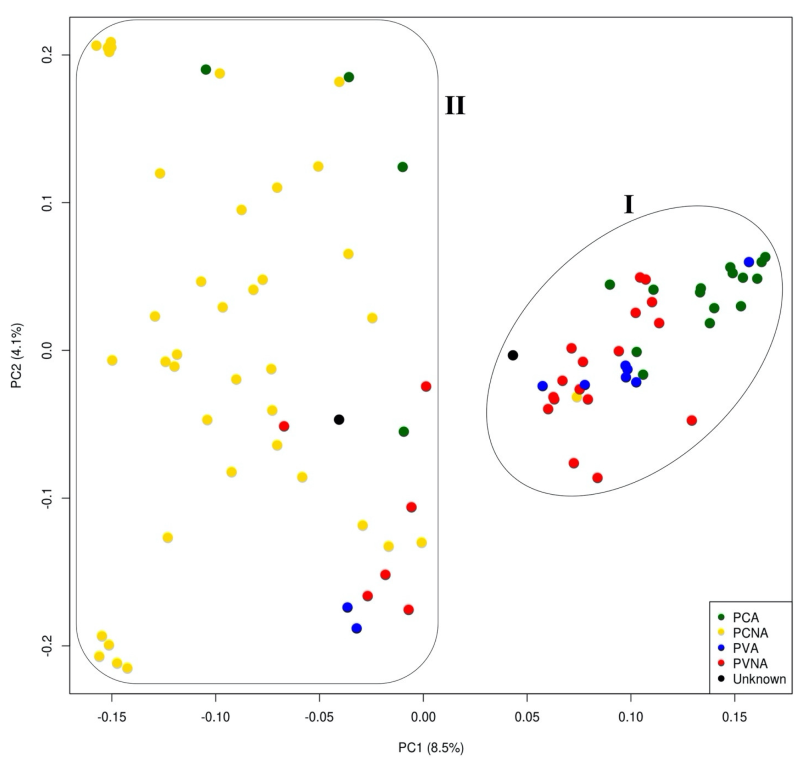
2.2. Population Structure and Phylogenetic Analysis
2.3. Population Genetic Diversity Based on SNPs
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Genomic DNA Extraction
4.2. GBS and SNP Detection
- -
- Homozygous: SNP read depth ≥ 90%
- -
- Heterozygous: 40% ≤ SNP read depth ≤ 60%
- -
- Others: 20% ≤ read rate < 40% or 60% < read rate < 90%.
4.3. Population Genetics Analysis
4.3.1. Population Genetic Diversity
4.3.2. Population Structure and Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ng, F.S.P. (Ed.) Ebenaceae. In Tree Flora of Malaya; Forest Res Inst.: Kepong, Malaysia, 1986; Volume 3, pp. 56–94. [Google Scholar]
- Yonemori, K.; Sugiura, A.; Yamada, M. Persimmon genetics and breeding. In Plant Breeding Reviews; John, J.J., Ed.; Wiley & Sons, Inc.: Oxford, UK, 2000; Volume 19. [Google Scholar]
- Seker, M.; Gur, E.; Ekinci, N.; Gundogdu, M.A. Comparison of genome sizes of persimmon (Diospyros kaki L.) and Caucasian persimmon (Diospyros lotus L.) seedling populations by using Flow Cytometry. J. Agri. Fac. Gaziosmanpasa Univ. 2018, 35, 286–289. [Google Scholar] [CrossRef]
- Stat. Food and Agriculture Organization. 2021. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 10 December 2022).
- Sanford, J.C. Ploidy manupilations. In Methods in Fruit Breeding; Moore, J.N., Janick, J., Eds.; Purdue Univ. Press: West Lafayette, IN, USA, 1983; pp. 100–123. [Google Scholar]
- Zhuang, D.H.; Kitajima, A.; Ishida, M.; Sobajima, Y. Chromosome numbers of Diospyros kaki cultivars. J. Japan. Soc. Hortic. Sci. 1990, 59, 289–297. [Google Scholar] [CrossRef]
- Suo, Y.; Sun, P.; Cheng, H.; Han, W.; Diao, S.; Li, H.; Mai, Y.; Zhao, X.; Li, F.; Fu, J. A high-quality chromosomal genome assembly of Diospyros oleifera Cheng. Gigascience 2020, 1, 164. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.G.; Xu, Y.; Yang, Y.; Guan, C.F.; Zhang, Q.Y.; Huang, J.W.; Grierson, D.; Chen, K.S.; Gong, B.C.; Yin, X.R. The persimmon (Diospyros oleifera Cheng) genome provides new insights into the inheritance of astringency and ancestral evolution. Hortic. Res. 2019, 6, 138. [Google Scholar] [CrossRef]
- Akagi, T.; Shirasawa, K.; Nagasaki, H.; Hirakawa, H.; Tao, R.; Comai, L.; Henry, I.M. Correction: The persimmon genome reveals clues to the evolution of a lineage-specific sex determination system in plants. PLoS Genet. 2020, 16, e1008845. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Zhang, F.; Zhang, Q.L.; Guo, D.Y.; Luo, Z.R. Characterization and comparison of EST-SSR and TRAP markers for genetic analysis of the Japanese persimmon Diospyros kaki. Genet. Mol. Res. 2013, 12, 2841–2851. [Google Scholar] [CrossRef]
- Korea Seed and Variety Service. 2019. Available online: http://www.seed.go.kr/seed_eng/index (accessed on 12 December 2022).
- Je, H.; Park, Y.; Kim, S.; Hwang, J.; Lee, Y.; Son, B.; Park, Y. Evaluation of genetic relationships among persimmon cultivars introduced and indigenous in Korea using RAPD. Korean J. Hort. Sci. Technol. 2009, 27, 448–455. [Google Scholar]
- Yamagishi, M.; Matsumoto, S.; Nakatsuka, A.; Itamura, H. Identification of persimmon (Diospyros kaki) cultivars and phenetic relationships between Diospyros species by more effective RAPD analysis. Sci. Hortic. 2005, 105, 283–290. [Google Scholar] [CrossRef]
- Yonemori, K.; Honsho, C.; Kitajima, A.; Aradhya, M.; Giordani, E.; Bellini, E.; Parfitt, D.E. Relationship of European persimmon (Diospyros kaki Thunb.) cultivars to Asian cultivars, characterized using AFLPs. Genet. Resour. Crop Evol. 2008, 55, 81–89. [Google Scholar] [CrossRef]
- Parfitt, D.E.; Yonemori, K.; Honsho, C.; Nozaka, M.; Kanzaki, S.; Stao, A.; Yamada, M. Relationships among Asian persimmon cultivars, astringent and non-astringent types. Tree Genet. Genom. 2015, 11, 24. [Google Scholar] [CrossRef]
- Park, Y.; Park, D.; Son, J.; Choi, S.; Kim, S.; Hong, G.; Park, Y. Evaluation of genetic diversity among persimmon (Diospyros kaki Thunb.) collection lines and cultivars using simple sequence repeat markers. Korean J. Breed. Sci. 2012, 44, 127–135. [Google Scholar]
- Guan, C.; Chachar, S.; Zhang, P.; Hu, C.; Wang, R.; Yang, Y. Inter- and intra-specific genetic diversity in Diospyros using SCoT and IRAP markers. Hortic. Plant J. 2020, 6, 71–80. [Google Scholar] [CrossRef]
- Van Dijk, E.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Chung, Y.S.; Choi, S.C.; Jun, T.H.; Kim, C. Genotyping-by-sequencing: A promising tool for plant genetics research and breeding. Hortic. Environ. Biotechnol. 2017, 58, 425–431. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef]
- Hyma, K.E.; Barba, P.; Wang, M.; Londo, J.P.; Acharya, C.B.; Mitchell, S.E.; Sun, Q.; Reisch, B.; Cadle-Davidson, L. Heterzygous mapping strategy (HetMapps) for high resolution genotyping-by-sequencing markers: A case study in grapevine. PLoS ONE 2010, 10, e0134880. [Google Scholar] [CrossRef]
- LaFlamme, B. Persimmon sex determination. Nat. Genet. 2014, 46, 1257. [Google Scholar] [CrossRef]
- Ma, K.; Yang, S.; Jo, Y.; Kang, S.; Nam, M. Development of kompetitive allele specific PCR markers for identification of persimmon varieties using genotyping-by-sequencing. Electron. J. Biotechnol. 2021, 49, 72–81. [Google Scholar] [CrossRef]
- Badenes, M.; Garcés, A.; Romero, C.; Romero, M.; Clave, J.; Rovira, M.; Llacer, G. Genetic diversity of introduced and local Spanish persimmon cultivars revealed by RAPD markers. Genet. Resour. Crop Evol. 2003, 50, 579–585. [Google Scholar] [CrossRef]
- Guo, D.L.; Luo, Z.R. Microsatellite isolation and characterization in Japanese persimmon (Diospyros kaki). Biochem. Genet. 2008, 46, 323. [Google Scholar] [CrossRef] [PubMed]
- Naval, M.d.; Zuriaga, E.; Pecchioli, S.; Llacer, G.; Giordani, E.; Badnes, M. Analysis of genetic diversity among persimmon cultivars using microsatellite markers. Tree Genet. Genomes 2010, 6, 677–687. [Google Scholar] [CrossRef]
- Jing, Z.; Ruan, X.; Wang, R.; Yang, Y. Genetic diversity and relationships between and within persimmon (Diospyros L.) wild species and cultivated varieties by SRAP markers. Plant Syst. Evol. 2013, 299, 1485–1492. [Google Scholar] [CrossRef]
- Guan, C.; Zhang, P.; Hu, C.; Chachar, S.; Riaz, A.; Wang, R.; Yang, Y. Genetic diversity, germplasm identification and population structure of Diospyros kaki Thunb. from different geographic regions in China using SSR markers. Sci. Hort. 2019, 251, 233–240. [Google Scholar] [CrossRef]
- Thomson, M.J. High-Throughput SNP genotyping to accelerate crop improvement plant breeding and biotechnology. Plant Breed. Biotech. 2014, 2, 195–212. [Google Scholar] [CrossRef]
- Mason, A.S. Challenges of genotyping polyploid Species. Methods Mol. Biol. 2015, 1245, 161–168. [Google Scholar] [CrossRef]
- Kim, C.; Guo, H.; Kong, W.; Chandnani, R.; Shuang, L.S.; Paterson, A.H. Application of genotyping by sequencing technology to a variety of crop breeding programs. Plant Sci. 2016, 242, 14–22. [Google Scholar] [CrossRef]
- Kaur, S.; Francki, M.G.; Forster, J.W. Identification, characterization and interpretation of single-nucleotide sequence variation in allopolyploid crop species. Plant Biotechnol. J. 2012, 10, 125–138. [Google Scholar] [CrossRef]
- Griffin, P.C.; Robin, C.; Hoffmann, A.A. A next-generation sequencing method for overcoming the multiple gene copy problem in polyploid phylogenetics, applied to Poa grasses. BMC Biol. 2011, 9, 19. [Google Scholar] [CrossRef]
- Campa, A.; Ferreira, J.J. Genetic diversity assessed by genotyping by sequencing (GBS) and for phenological traits in blueberry cultivars. PLoS ONE 2018, 13, e0206361. [Google Scholar] [CrossRef]
- Yang, X.; Tan, B.; Liu, H.; Zhu, W.; Xu, L.; Wang, Y.; Fan, X.; Sha, L.; Zhang, H.; Zeng, J.; et al. Genetic diversity and population structure of Asian and European common wheat accessions based on genotyping-by-sequencing. Front. Genet. 2010, 11, 580782. [Google Scholar] [CrossRef]
- Mourad, A.M.I.; Belamkar, V.; Baenziger, P.S. Molecular genetic analysis of spring wheat core collection using genetic diversity, population structure, and linkage disequilibrium. BMC Genom. 2020, 21, 434. [Google Scholar] [CrossRef]
- Alipour, H.; Bihamta, M.R.; Mohammadi, V.; Peyghambari, S.A.; Bai, G.; Zhang, G. Genotyping-by-sequencing (GBS) revealed molecular genetic diversity of Iranian wheat landraces and cultivars. Front. Plant Sci. 2017, 8, 1293. [Google Scholar] [CrossRef]
- Malmberg, M.M.; Shi, F.; Spangenberg, G.C.; Daetwyler, H.D.; Cogan, N.O.I. Diversity and genome analysis of Australian and global oilseed Brassica napus L. germplasm using transcriptomics and whole genome re-sequencing. Front. Plant Sci. 2018, 9, 508. [Google Scholar] [CrossRef]
- Hamon, P.; Grover, C.E.; Davis, A.P.; Rakotomalala, J.J.; Raharimalala, N.E.; Albert, V.A.; Sreenath, H.L.; Stoffelen, P.; Mitchell, S.E.; Couturon, E.; et al. Genotyping-by-sequencing provides the first well-resolved phylogeny for coffee (Coffea) and insights into the evolution of caffeine content in its species: GBS coffee phylogeny and the evolution of caffeine content. Mol. Phylogenet. Evol. 2017, 109, 351–361. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef]
- Heng, L.I.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Kim, J.E.; Oh, S.K.; Lee, J.H.; Jo, S.H. Genome-wide SNP calling using next generation sequencing data in tomato. Mol. Cells 2014, 37, 36–42. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 14–31. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Cultivar | Type 1 | Origin |
|---|---|---|---|
| 1 | Akagaki | PVNA | Japan |
| 2 | Amahyakume | PVNA | Japan |
| 3 | Atago | PCA | Japan |
| 4 | Baojiaozi | PVNA | China |
| 5 | Black persimmon | PCA | China |
| 6 | Black sweet persimmon | PVNA | China |
| 7 | Bumanshu | PCNA | Korea |
| 8 | Cheongdo-Bansi | PCA | Korea |
| 9 | Daeandangam | PCNA | Korea |
| 10 | Damopan | PCA | Japan |
| 11 | Dannuri | PCNA | Korea |
| 12 | Danyeon104 | PCNA | Korea |
| 13 | Diamond leaf persimmon | Unknown | China |
| 14 | Fujiwaragosho | PCNA | Japan |
| 15 | Fuyu | PCNA | Japan |
| 16 | Gamnuri | PCA | Korea |
| 17 | Goesan-Durigam | PCA | Korea |
| 18 | Gogseong-Tabaegam | PCA | Korea |
| 19 | Gosho | PCNA | Japan |
| 20 | Gyeongsan-Bansi | PCA | Korea |
| 21 | Hachiuri | PVNA | Japan |
| 22 | Hachiya | PVA | Japan |
| 23 | Haman-Bansi | PCA | Korea |
| 24 | Haman-Mulgam | PCA | Korea |
| 25 | Hanagosho | PCNA | Japan |
| 26 | Hiratanenashi | PVA | Japan |
| 27 | Hongchoo | PCNA | Korea |
| 28 | Hongdansi | PVNA | Korea |
| 29 | Ichikikeijiro | PCNA | Japan |
| 30 | IsaHaya | PCNA | Japan |
| 31 | Izu | PCNA | Japan |
| 32 | Jamisi | PVNA | Korea |
| 33 | Jiro | PCNA | Japan |
| 34 | Johongsi | PVNA | Japan |
| 35 | Kaki Tipo | PVNA | Italy |
| 36 | Kanshu | PCNA | Japan |
| 37 | Kastusa | PCNA | Japan |
| 38 | Kinshu | PCNA | Japan |
| 39 | Kinshu-2 | PCNA | Japan |
| 40 | Kyara | PVNA | Japan |
| 41 | Maekawajiro | PCNA | Japan |
| 42 | Male collection line | PCNA | Korea |
| 43 | Mammoth | PCNA | Japan |
| 44 | Matsumotowase-Fuyu | PCNA | Japan |
| 45 | Midai | PCNA | Japan |
| 46 | Migamjosang | PVNA | Korea |
| 47 | Migatanigosho | PVNA | Japan |
| 48 | Mizushima | PVNA | Japan |
| 49 | Myeongju-Kojongsi | PCA | Korea |
| 50 | Niitsugaki | PVNA | Japan |
| 51 | Nishimurawase | PVNA | Japan |
| 52 | O-miyawase | PCA | Korea |
| 53 | O-tanenashi | PVA | Japan |
| 54 | Ro-19 | PCNA | Japan |
| 55 | Rojo Brillante | PVA | Spain |
| 56 | Romang | PCNA | Korea |
| 57 | Sae-Fuji | PVNA | Japan |
| 58 | Saijo | PCA | Korea |
| 59 | Sancheong-Danseongsi | PCA | Korea |
| 60 | Sancheong-Kojongsi | PCA | Korea |
| 61 | Sanggam-Dungsi | PCA | Korea |
| 62 | Sanggokuitsy | PVNA | Japan |
| 63 | Sangjudungsi | PCA | Korea |
| 64 | Shinsyu | PCNA | Japan |
| 65 | Shougatsu | PVNA | Japan |
| 66 | Soshu | PCNA | Japan |
| 67 | Sugitawase | PVA | Japan |
| 68 | Sunami | PCNA | Japan |
| 69 | Superhiratanenashi | PVA | Japan |
| 70 | Suruga | PCNA | Japan |
| 71 | Taishu | PCNA | Japan |
| 72 | Tanrei | PCNA | Japan |
| 73 | Tenjingosho | PCNA | Japan |
| 74 | Tenrubou | PVNA | Japan |
| 75 | Tonewase | PVA | Japan |
| 76 | Toyoga | PVNA | Japan |
| 77 | Toyouichi | PVNA | Japan |
| 78 | Uenishiwase | PCNA | Japan |
| 79 | Wakakisjiro | PCNA | Japan |
| 80 | Wakasugikeijiro | PCNA | Japan |
| 81 | Wasesaijo | PCA | Japan |
| 82 | Wasezizya | PVA | Japan |
| 83 | Yaizuwasejiro | PCNA | Japan |
| 84 | Yaoki | PVA | Japan |
| 85 | Yeongdong-Weolhasi | PCA | Korea |
| 86 | Youhou | PCNA | Japan |
| 87 | Zenjimaru | PVNA | Japan |
| 88 | 09-12-1 | PCNA | Korea |
| 89 | 10-1-86 | PCNA | Korea |
| 90 | 11-12-88 | PCNA | Korea |
| 91 | 11-6-120 | Unknown | Korea |
| 92 | 12-9-4 | PVNA | Korea |
| 93 | 13-6-410 | PCNA | Korea |
| Category | Total | Average per Cultivar |
|---|---|---|
| No. of total raw reads | 751,086,680 | 7,823,820 |
| No. of trimmed reads | 712,437,918 | 7,421,228 |
| Total length of raw reads (bp) | 113,414,088,680 | 1,181,396,757 |
| Total length of trimmed reads (bp) | 92,565,980,293 | 964,228,961 |
| No. of mapped reads | 301,735,646 | 3,143,080 |
| Total length of mapped regions (bp) | 379,067,691 | 3,948,622 |
| Population | Category | N | Na | Ne | I | Ho | He | uHe | F |
|---|---|---|---|---|---|---|---|---|---|
| Total | Max | 93 | 2.000 | 2.000 | 0.693 | 0.765 | 0.500 | 0.504 | 1.00 |
| Min | 66 | 2.000 | 1.119 | 0.217 | 0.000 | 0.106 | 0.107 | −0.536 | |
| Mean | 78.904 | 2.000 | 1.711 | 0.588 | 0.350 | 0.403 | 0.405 | 0.169 | |
| SE | 0.083 | 0.000 | 0.003 | 0.001 | 0.002 | 0.001 | 0.001 | 0.003 |
| Population | N | Na | Ne | I | Ho | He | uHe | F | |
|---|---|---|---|---|---|---|---|---|---|
| PCA | Mean | 16.617 | 1.989 | 1.666 | 0.559 | 0.319 | 0.380 | 0.392 | 0.176 |
| SE | 0.023 | 0.001 | 0.003 | 0.002 | 0.002 | 0.001 | 0.001 | 0.004 | |
| PCNA | Mean | 34.393 | 1.976 | 1.661 | 0.546 | 0.368 | 0.373 | 0.378 | 0.062 |
| SE | 0.042 | 0.002 | 0.003 | 0.002 | 0.002 | 0.002 | 0.002 | 0.004 | |
| PVA | Mean | 8.039 | 1.941 | 1.634 | 0.531 | 0.377 | 0.361 | 0.386 | −0.024 |
| SE | 0.013 | 0.003 | 0.003 | 0.002 | 0.003 | 0.002 | 0.002 | 0.005 | |
| PVNA | Mean | 19.347 | 1.990 | 1.680 | 0.566 | 0.339 | 0.386 | 0.397 | 0.145 |
| SE | 0.026 | 0.001 | 0.003 | 0.002 | 0.002 | 0.001 | 0.001 | 0.004 |
| Source | df | Sum of Squares | Estimated Variation | Percent of Variation | Fixation Index (Fst) |
|---|---|---|---|---|---|
| Among groups | 3 | 16221.286 | 57.668 | 3% | 0.026 |
| Among individuals | 87 | 260542.626 | 873.294 | 40% | |
| Within individuals | 91 | 113582.000 | 1248.154 | 57% | |
| Total | 181 | 390345.912 | 2179.117 | 100% |
| PCNA | PVNA | PCA | PVA | |
|---|---|---|---|---|
| PCNA | 0.0000 | 0.0000 | 0.0000 | |
| PVNA | 0.07184 | 0.0000 | 0.01074 | |
| PCA | 0.09416 | 0.02762 | 0.00098 | |
| PVA | 0.08467 | 0.01566 | 0.03575 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Park, Y.-O.; Park, Y. Population Genetic Analysis in Persimmons (Diospyros kaki Thunb.) Based on Genome-Wide Single-Nucleotide Polymorphisms. Plants 2023, 12, 2097. https://doi.org/10.3390/plants12112097
Park S, Park Y-O, Park Y. Population Genetic Analysis in Persimmons (Diospyros kaki Thunb.) Based on Genome-Wide Single-Nucleotide Polymorphisms. Plants. 2023; 12(11):2097. https://doi.org/10.3390/plants12112097
Chicago/Turabian StylePark, Seoyeon, Ye-Ok Park, and Younghoon Park. 2023. "Population Genetic Analysis in Persimmons (Diospyros kaki Thunb.) Based on Genome-Wide Single-Nucleotide Polymorphisms" Plants 12, no. 11: 2097. https://doi.org/10.3390/plants12112097