
Absolute Quantification of Isoflavones in the Flowers of Pueraria lobata by qHNMR

 , and
, and
Abstract
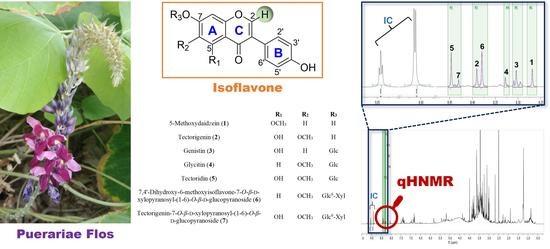
:
1. Introduction
2. Results and Discussion
2.1. 1H NMR Signal Assignment and Identification
2.2. Major Isoflavone Content Calculated by qHNMR
2.3. Validation Studies
2.4. Sensitivity
2.5. Accuracy
2.6. HPLC-UV Quantification Analysis
2.7. Comparison of Two Different Sample Preparation Methods, PLr and PLs
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Chemicals and Reagents
3.3. Plant Materials
3.4. Extraction and Isolation
3.4.1. MeOH Sonication of Puerariae Flos (PLs)
3.4.2. MeOH Reflux of Puerariae Flos (PLr)
3.4.3. Isolation of Compounds 1–7
3.5. Quantitative NMR Experiment
3.5.1. Sample Preparation
3.5.2. NMR Data Acquisition and Processing
3.5.3. Quantitative NMR Analysis
3.6. HPLC Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, S.; Ma, Y.; Zhang, Y.; Liu, L.; Liu, Q.; Peng, M.; Xiong, X. Systematic separation and purification of 18 antioxidants from Pueraria lobata flower using HSCCC target-guided by DPPH-HPLC experiment. Sep. Purif. Technol. 2012, 89, 225–233. [Google Scholar] [CrossRef]
- Li, J.; Mi, Q.-L.; Zhang, F.-M.; Yang, Y.-K.; Chen, J.-H.; Ye, L.; Zhang, C.-M.; Guangyu, Y.; Hu, Q.-F.; Liu, Z.-H.; et al. Two new isoflavones from Pueraria lobata and their bioactivities. Chem. Nat. Compd. 2018, 54, 851–855. [Google Scholar] [CrossRef]
- Wong, K.H.; Razmovski-Naumovski, V.; Li, K.M.; Li, G.Q.; Chan, K. Comparing morphological, chemical and anti-diabetic characteristics of Puerariae Lobatae Radix and Puerariae Thomsonii Radix. J. Ethnopharmacol. 2015, 164, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Rong, H.; Stevens, J.F.; Deinzer, M.L.; De Cooman, L.; De Keukeleire, D. Identification of isoflavones in the roots of Pueraria lobata. Planta Med. 1998, 64, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, M.; Sakano, K.; Nakamura, S.-I.; Kosaka, H. Antimutagenic activity of isoflavone from Pueraria lobata. J. Agric. Food Chem. 2001, 49, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Arao, T.; Udayama, M.; Kinjo, J.; Nohara, T.; Funakoshi, T.; Kojima, S. Preventive effects of saponins from puerariae radix (the root of Pueraria lobata Ohwi) on in vitro immunological injury of rat primary hepatocyte cultures. Biol. Pharm. Bull. 1997, 20, 988–991. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.C.; Guthrie, S.; Xie, C.Y.; Mai, K.; Lee, D.Y.; Lumeng, L.; Li, T.K. Isoflavonoid compounds extracted from Pueraria lobata suppress alcohol preference in a pharmacogenetic rat model of alcoholism. Alcohol. Clin. Exp. Res. 1996, 20, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Jun, M.; Fu, H.Y.; Hong, J.; Wan, X.; Yang, C.S.; Ho, C.T. Comparison of antioxidant activities of isoflavones from kudzu root (Pueraria lobata Ohwi). J. Food Sci. 2003, 68, 2117–2122. [Google Scholar] [CrossRef]
- Kinjo, J.; Takeshita, T.; Abe, Y.; Terada, N.; Yamashita, H.; Yamasaki, M.; Takeuchi, K.; Murakami, K.; Tomimatsu, T.; Nohara, T. Studies on the constituents of Pueraria lobata. IV. Chemical constituents in the flowers and the leaves. Chem. Pharm. Bull. 1988, 36, 1174–1179. [Google Scholar] [CrossRef] [Green Version]
- Bebrevska, L.; Bravo, L.; Vandervoort, J.; Pieters, L.; Vlietinck, A.; Apers, S. Development and validation of an HPLC method for quality control of Pueraria lobata flower. Planta Med. 2007, 73, 1606–1613. [Google Scholar] [CrossRef]
- Chen, T.-R.; Shih, S.-C.; Ping, H.-P.; Wei, Q.-K. Antioxidant activity and isoflavonoid components in different sections of Pueraria lobata root. J. Food Drug Anal. 2012, 20, 681–685. [Google Scholar]
- Mun, S.-C.; Mun, G.-S. Dynamics of phytoestrogen, isoflavonoids, and its isolation from stems of Pueraria lobata (Willd.) Ohwi growing in Democratic People’s Republic of Korea. J. Food Drug Anal. 2015, 23, 538–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matkowski, A.; Wozniak, D.; Oszmianski, J.; Lamer-Zarawska, E. Flavonoids of Pueraria lobata: Chromatographic analysis of leaves and roots of cultivated plants. Pharmazie 2003, 58, 682–683. [Google Scholar] [PubMed]
- Caligiani, A.; Palla, G.; Maietti, A.; Cirlini, M.; Brandolini, V. 1H NMR fingerprinting of soybean extracts, with emphasis on identification and quantification of isoflavones. Nutrients 2010, 2, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foudah, A.I.; Abdel-Kader, M.S. Isoflavonoids. In Flavonoids-From Biosynthesis to Human Health; IntechOpen: London, UK, 2017. [Google Scholar]
- Napolitano, J.G.; Godecke, T.; Rodriguez-Brasco, M.F.; Jaki, B.U.; Chen, S.-N.; Lankin, D.C.; Pauli, G.F. The Tandem of Full Spin Analysis and qHNMR for the Quality Control of Botanicals Exemplified with Ginkgo biloba. J. Nat. Prod. 2012, 75, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, S.; Liu, C.; Liu, C.; Zhang, Y. Extraction and isolation of potential anti-stroke compounds from flowers of Pueraria lobata guided by in vitro PC12 cell model. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1048, 111–120. [Google Scholar] [CrossRef]
- Wu, Z.; Song, L.; Feng, S.; Liu, Y.; He, G.; Yioe, Y.; Liu, S.Q.; Huang, D. Germination dramatically increases isoflavonoid content and diversity in chickpea (Cicer arietinum L.) seeds. J. Agric. Food Chem. 2012, 60, 8606–8615. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.G.; Cheng, M.C.; Wang, Y.Q.; Xiao, H.B.; Liang, X.M. Simultaneous quantification of three isoflavonoid glycosides in rabbit plasma after oral administration of Astragalus mongholicus extract by high-performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry. Anal. Chim. Acta 2007, 602, 252–258. [Google Scholar] [CrossRef]
- Prokudina, E.; Havlíček, L.; Al-Maharik, N.; Lapčík, O.; Strnad, M.; Gruz, J. Rapid UPLC–ESI–MS/MS method for the analysis of isoflavonoids and other phenylpropanoids. J. Food Compos. Anal. 2012, 26, 36–42. [Google Scholar] [CrossRef]
- Phansalkar, R.S.; Simmler, C.; Bisson, J.; Chen, S.-N.; Lankin, D.C.; McAlpine, J.B.; Niemitz, M.; Pauli, G.F. Evolution of Quantitative Measures in NMR: Quantum Mechanical qHNMR Advances Chemical Standardization of a Red Clover (Trifolium pratense) Extract. J. Nat. Prod. 2017, 80, 634–647. [Google Scholar] [CrossRef]
- Wang, Z.-W.; Wang, J.-S.; Yang, M.-H.; Luo, J.-G.; Kong, L.-Y. Developmental changes in the composition of five anthraquinones from Rheum palmatum as quantified by 1H-NMR. Phytochem. Anal. 2013, 24, 329–335. [Google Scholar] [CrossRef]
- Hsieh, L.-Y.; Chan, H.-H.; Kuo, P.-C.; Hung, H.-Y.; Li, Y.-C.; Kuo, C.-L.; Peng, Y.; Zhao, Z.-Z.; Kuo, D.-H.; Sun, I.W.; et al. A feasible and practical 1H NMR analytical method for the quality control and quantification of bioactive principles in Lycii Fructus. J. Food Drug Anal. 2018, 26, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Pauli, G.F.; Chen, S.-N.; Simmler, C.; Lankin, D.C.; Godecke, T.; Jaki, B.U.; Friesen, J.B.; McAlpine, J.B.; Napolitano, J.G. Importance of Purity Evaluation and the Potential of Quantitative 1H NMR as a Purity Assay. J. Med. Chem. 2014, 57, 9220–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulesh, N.I.; Vasilevskaya, N.A.; Veselova, M.V.; Denisenko, V.A.; Fedoreev, S.A. Minor polyphenols from Maackia amurensis wood. Chem. Nat. Compd. 2008, 44, 712–714. [Google Scholar] [CrossRef]
- Al-Maharik, N.; Botting, N.P. An efficient method for the glycosylation of isoflavones. Eur. J. Org. Chem. 2008, 33, 5622–5629. [Google Scholar] [CrossRef]
- Park, H.-J.; Park, J.-H.; Moon, J.-O.; Lee, K.-T.; Jung, W.-T.; Oh, S.-R.; Lee, H.-K. Isoflavone glycosides from the flowers of Pueraria thunbergiana. Phytochemistry 1999, 51, 147–151. [Google Scholar] [CrossRef]
- Pauli, G.F.; Jaki, B.U.; Lankin, D.C. Quantitative 1H NMR: Development and potential of a method for natural products analysis. J. Nat. Prod. 2005, 68, 133–149. [Google Scholar] [CrossRef]
- Schoenberger, T.; Menges, S.; Bernstein, M.A.; Perez, M.; Seoane, F.; Sykora, S.; Cobas, C. Improving the performance of high-precision qNMR measurements by a double integration procedure in practical cases. Anal. Chem. 2016, 88, 3836–3843. [Google Scholar] [CrossRef]
- Guideline, I.H.T. Validation of analytical procedures: Text and methodology. Q2 (R1) 2005, 1, 5. [Google Scholar]
- Borman, P.; Elder, D. Q2(R1) Validation of analytical procedures. ICH Qual. Guidel. 2018, 5, 127–166. [Google Scholar]
- Cui, H.-Y.; Murthy, H.N.; Moh, S.H.; Cui, Y.Y.; Lee, E.-J.; Paek, K.-Y. Comparison of conventional and ultrasound-assisted methods for extraction of nutraceutical compounds from Dendrobium candidum. CyTA—J. Food 2014, 12, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Woo, J.-H.; Kim, H.M.; Oh, M.S.; Jang, D.S.; Choi, J.-H. Anti-endometriotic effects of Pueraria flower extract in human endometriotic cells and mice. Nutrients 2017, 9, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | % w/w (Mean ± Std Dev) of PLs | % w/w (Mean ± Std Dev) of PLr | ||
|---|---|---|---|---|
| qHNMR | HPLC-UV | qHNMR | HPLC-UV | |
| 5-Methoxydaidzein (1) | 0.95 ± 0.03 | - | 0.92 ± 0.01 | - |
| Tectorigenin (2) | 0.67 ± 0.01 | 0.65 ± 0.00 | 0.59 ± 0.02 | 0.49 ± 0.01 |
| Genistin (3) | 0.43 ± 0.02 | - | 0.53 ± 0.02 | - |
| Glycitin (4) | 1.96 ± 0.06 | - | 3.02 ± 0.05 | - |
| Tectoridin (5) | 1.86 ± 0.09 | 1.60 ± 0.01 | 2.42 ± 0.04 | 1.92 ± 0.02 |
| 7,4′-Dihydroxy-6-methoxyisoflavone 7-O-β-d-xylopyranosyl-(1-6)-O-β-d-glucopyranoside (6) | 0.57 ± 0.03 | - | 0.98 ± 0.03 | - |
| Tectorigenin-7-O-β-d-xylopyranosyl-(1-6)-O-β-d-glucopyranoside (7) | 1.55 ± 0.07 | 1.32 ± 0.01 | 2.10 ± 0.05 | 1.75 ± 0.02 |
| Total | 7.99 ± 0.11 | 10.57 ± 0.10 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thapa, P.; Kim, H.M.; Hong, J.-P.; Kim, R.; Paudel, S.B.; Choi, H.; Jang, D.S.; Nam, J.-W. Absolute Quantification of Isoflavones in the Flowers of Pueraria lobata by qHNMR. Plants 2022, 11, 548. https://doi.org/10.3390/plants11040548
Thapa P, Kim HM, Hong J-P, Kim R, Paudel SB, Choi H, Jang DS, Nam J-W. Absolute Quantification of Isoflavones in the Flowers of Pueraria lobata by qHNMR. Plants. 2022; 11(4):548. https://doi.org/10.3390/plants11040548
Chicago/Turabian StyleThapa, Punam, Hye Mi Kim, Joon-Pyo Hong, Ranhee Kim, Sunil Babu Paudel, Hyukjae Choi, Dae Sik Jang, and Joo-Won Nam. 2022. "Absolute Quantification of Isoflavones in the Flowers of Pueraria lobata by qHNMR" Plants 11, no. 4: 548. https://doi.org/10.3390/plants11040548