
Single-Molecule Real-Time Sequencing of Full-Length Transcriptome and Identification of Genes Related to Male Development in Cannabis sativa
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatment Conditions
2.2. Library Preparation and SMRT Sequencing
2.3. Quality Filtering and Error Correction
2.4. Functional Annotation
2.5. Identification of Coding Sequences, Non-Coding RNAs, and Transcription Factors
2.6. Alternative Splicing Analysis and Validation
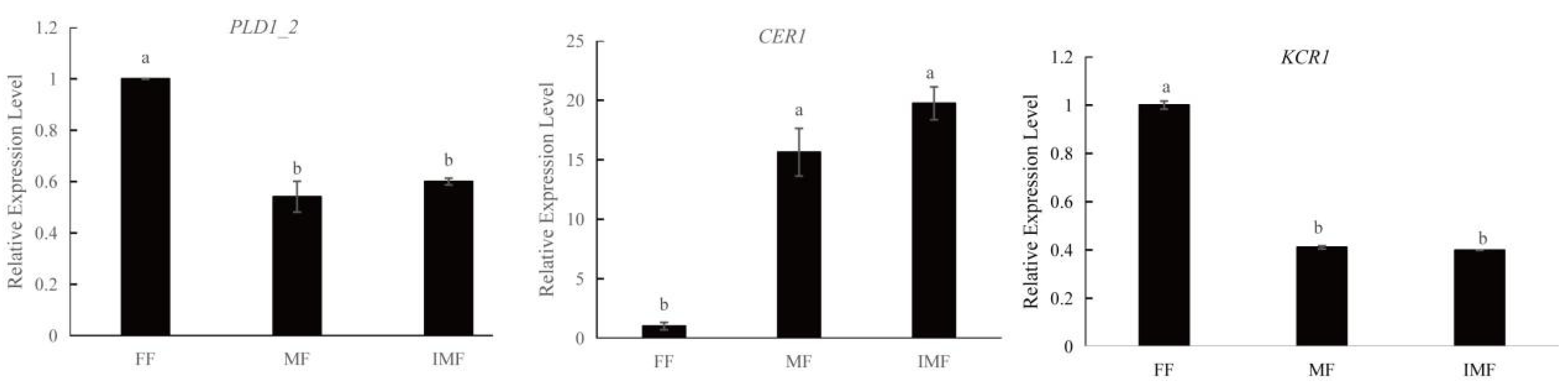
2.7. Real-Time Quantitative Reverse Transcription PCR Validation of Novel Genes
3. Results
3.1. Single-Molecule Real-Time Sequencing
3.2. Coding Sequence and lncRNA Identification
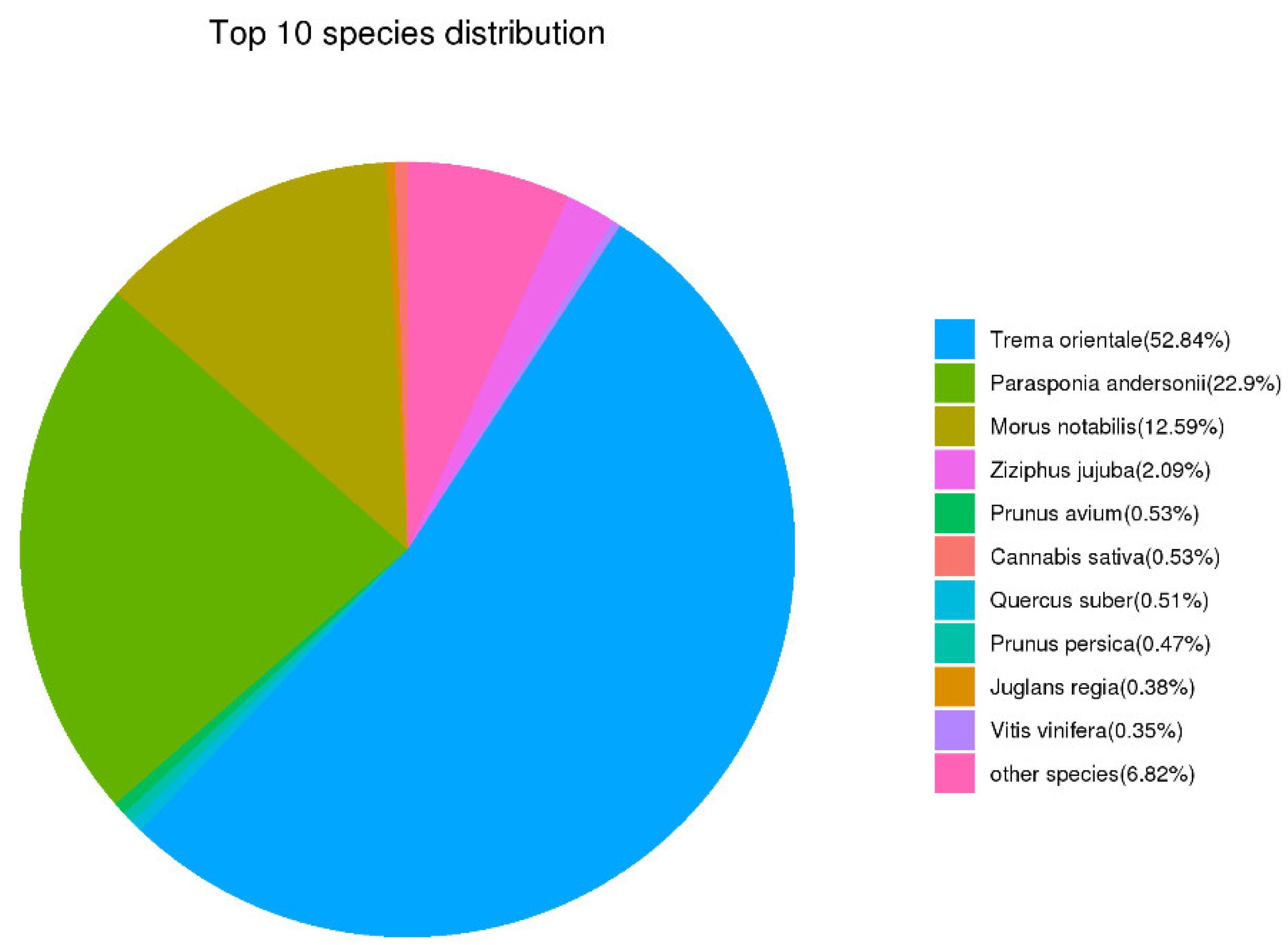
3.3. Functional Annotation of the Cannabis Full-Length Transcriptome
3.4. KEGG Enrichment Analysis
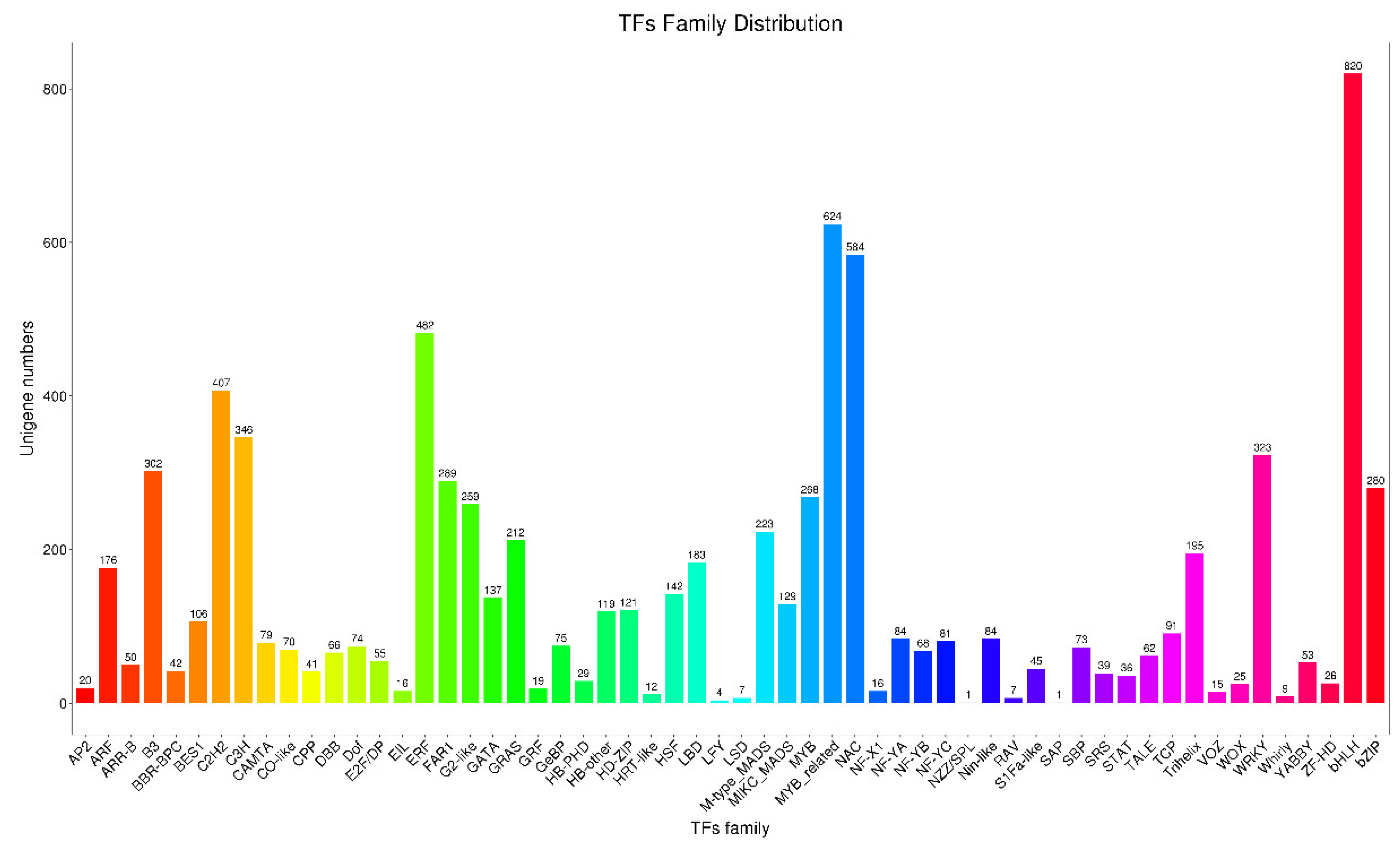
3.5. Identification and Analysis of TFs
3.6. Novel Gene Prediction and KEGG Enrichment Analysis
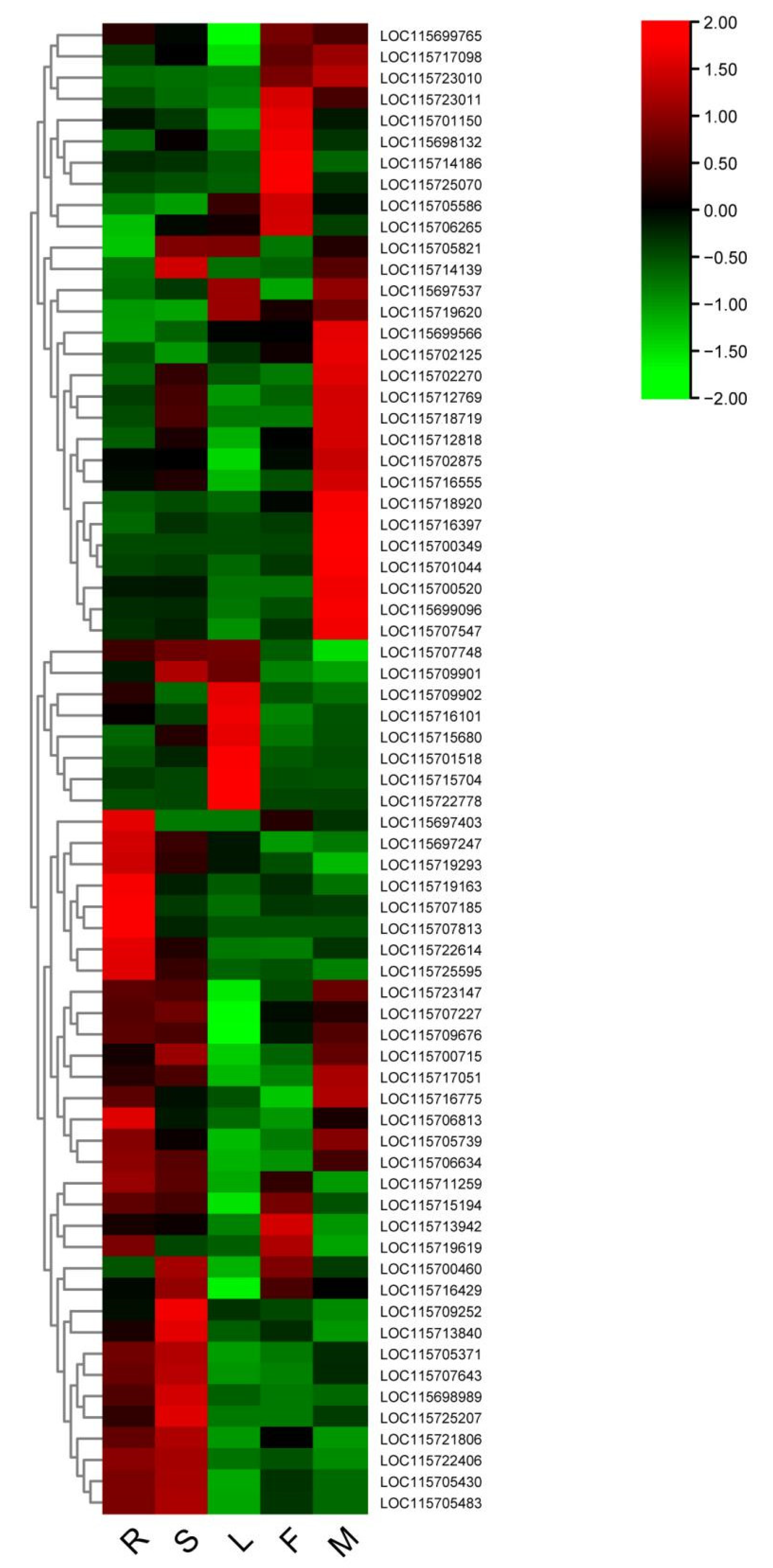
3.7. Identification and Validation of AS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rana, A.; Choudhary, N. Floral biology and pollination biology of Cannabis sativa L. Int J. Plant Reprod. Biol. 2010, 2, 191–195. [Google Scholar]
- Struik, P.C.; Amaducci, S.; Bullard, M.J.; Stutterheim, N.C.; Venturi, G.; Cromack, H.T.H. Agronomy of fibre hemp (Cannabis sativa L.) in Europe. Ind. Crop. Prod. 2000, 11, 107–118. [Google Scholar] [CrossRef]
- Faux, A.M.; Draye, X.; Flamand, M.C.; Occre, A.; Bertin, P. Identification of QTLs for sex expression in dioecious and monoecious hemp (Cannabis sativa L.). Euphytica 2016, 209, 357–376. [Google Scholar] [CrossRef]
- Salentijn, E.M.J.; Zhang, Q.; Amaducci, S.; Yang, M.; Trindade, L.M. New developments in fiber hemp (Cannabis sativa L.) breeding. Ind. Crop. Prod. 2015, 68, 32–41. [Google Scholar] [CrossRef]
- Andre, C.M.; Hausman, J.; Guerriero, G. Cannabis sativa: The Plant of the Thousand and One Molecules. Front. Plant Sci. 2016, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Van Bakel, H.; Stout, J.M.; Cote, A.G.; Tallon, C.M.; Sharpe, A.G.; Hughes, T.R.; Page, J.E. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011, 12, R102. [Google Scholar] [CrossRef] [Green Version]
- Livingston, S.J.; Quilichini, T.D.; Booth, J.K.; Wong, D.C.J.; Rensing, K.H.; Laflamme Yonkman, J.; Castellarin, S.D.; Bohlmann, J.; Page, J.E.; Samuels, A.L. Cannabis glandular trichomes alter morphology and metabolite content during flower maturation. Plant J. 2020, 101, 37–56. [Google Scholar] [CrossRef]
- Amaducci, S.; Scordia, D.; Liu, F.H.; Zhang, Q.; Guo, H.; Testa, G.; Cosentino, S.L. Key cultivation techniques for hemp in Europe and China. Ind. Crop. Prod. 2015, 68, 2–16. [Google Scholar] [CrossRef]
- Thomson, B.; Wellmer, F. Molecular regulation of flower development. Curr. Top. Dev. Biol. 2019, 131, 185–210. [Google Scholar]
- Stewart, D.; Graciet, E.; Wellmer, F. Molecular and regulatory mechanisms controlling floral organ development. FEBS J. 2016, 283, 1823–1830. [Google Scholar] [CrossRef]
- Adal, A.M.; Doshi, K.; Holbrook, L.; Mahmoud, S.S. Comparative RNA-Seq analysis reveals genes associated with masculinization in female Cannabis sativa. Planta 2021, 253, 17. [Google Scholar] [CrossRef]
- Yanofsky, M.F.; Pelaz, S.; Ditta, G.S.; Baumann, E.; Wisman, E. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 2000, 405, 200–203. [Google Scholar]
- Ruelens, P.; Zhang, Z.; Mourik, V.H.; Maere, S.; Kaufmann, K.; Geuten, K. The Origin of Floral Organ Identity Quartets. Plant Cell 2017, 29, 229–242. [Google Scholar] [CrossRef]
- Ali, Z.; Raza, Q.; Atif, R.M.; Aslam, U.; Ajmal, M.; Chung, G. Genetic and Molecular Control of Floral Organ Identity in Cereals. Int. J. Mol. Sci. 2019, 20, 12743. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Wang, L.; Jia, R.; Liang, W.; Zhang, X.; Xu, J.; Chen, X.; Lu, D.; Chen, M.; Luo, Z.; et al. Rice transcription factor MADS32 regulates floral patterning through interactions with multiple floral homeotic genes. J. Exp. Bot. 2021, 72, 2434–2449. [Google Scholar] [CrossRef]
- Chang, T.; An, B.; Liang, M.; Duan, X.; Du, L.; Cai, W.; Zhu, B.; Gao, X.; Chen, Y.; Xu, L.; et al. PacBio Single-Molecule Long-Read Sequencing Provides New Light on the Complexity of Full-Length Transcripts in Cattle. Front. Genet. 2021, 12, 664974. [Google Scholar] [CrossRef]
- Hurgobin, B.; Tamiru Oli, M.; Welling, M.T.; Doblin, M.S.; Bacic, A.; Whelan, J.; Lewsey, M.G. Recent advances in Cannabis sativa genomics research. New Phytol. 2021, 230, 73–89. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Sharma, T.R.; Devanna, B.N.; Kiran, K.; Singh, P.K.; Arora, K.; Jain, P.; Tiwari, I.M.; Dubey, H.; Saklani, B.; Kumari, M.; et al. Status and Prospects of Next Generation Sequencing Technologies in Crop Plants. Curr. Issues Mol. Biol. 2018, 27, 1–36. [Google Scholar] [CrossRef]
- Comin, M.; Schimd, M. Assembly-free genome comparison based on next-generation sequencing reads and variable length patterns. BMC Bioinform. 2014, 15 (Suppl. S9), S1. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Flajšman, M.; Slapnik, M.; Murovec, J. Production of Feminized Seeds of High CBD Cannabis sativa L. by Manipulation of Sex Expression and Its Application to Breeding. Front. Plant Sci. 2021, 12, 718092. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F. Third-generation sequencing techniques and applications to drug discovery. Expert Opin. Drug Discov. 2012, 7, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. Lond B Biol. Sci. 2019, 374, 0097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Barnabás, B.; Jäger, K.; Fehér, A. The effect of drought and heat stress on reproductive processes in cereals. Plant Cell Environ. 2008, 31, 11–38. [Google Scholar] [CrossRef]
- Clément, C.; Chavant, L.; Burrus, M.; Audran, J. Anther starch variations in Lilium during pollen development. Sex. Plant Reprod. 1994, 7, 347–356. [Google Scholar] [CrossRef]
- Fu, G.; Jian, S.; Xiong, J.; Li, Y.; Chen, H.; Le, M.; Tao, L. Changes of oxidative stress and soluble sugar in anthers involve in rice pollen abortion under drought stress. Agric. Sci. China. 2011, 10, 1016–1025. [Google Scholar] [CrossRef]
- Hu, W.; Coomer, T.D.; Loka, D.A.; Oosterhuis, D.M.; Zhou, Z. Potassium deficiency affects the carbon-nitrogen balance in cotton leaves. Plant Physiol. Biochem. 2017, 115, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.L.; Liu, L.; Niu, Q.K.; Xia, C.; Yang, K.Z.; Li, R.; Chen, L.Q.; Zhang, X.Q.; Zhou, Y.; Ye, D. Male gametophyte defective 4 encodes a rhamnogalacturonan II xylosyltransferase and is important for growth of pollen tubes and roots in Arabidopsis. Plant J. Cell Mol. Biol. 2011, 65, 647–660. [Google Scholar] [CrossRef]
- Caffall, K.H.; Mohnen, D. The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydr. Res. 2009, 344, 1879–1900. [Google Scholar] [CrossRef]
- Wang, L.; Wang, W.; Wang, Y.Q.; Liu, Y.Y.; Wang, J.X.; Zhang, X.Q.; Ye, D.; Chen, L.Q. Arabidopsis galacturonosyltransferase (GAUT) 13 and GAUT14 have redundant functions in pollen tube growth. Mol. Plant 2013, 6, 1131–1148. [Google Scholar] [CrossRef] [Green Version]
- Hord, C.L.; Chen, C.; Deyoung, B.J.; Clark, S.E.; Ma, H. The BAM1/BAM2 receptor-like kinases are important regulators of Arabidopsis early anther development. Plant Cell 2006, 18, 1667–1680. [Google Scholar] [CrossRef]
- DeYoung, B.J.; Bickle, K.L.; Schrage, K.J.; Muskett, P.; Patel, K.; Clark, S.E. The CLAVATA1-related BAM1, BAM2 and BAM3 receptor kinase-like proteins are required for meristem function in Arabidopsis. Plant J. 2006, 45, 1–16. [Google Scholar] [CrossRef]
- De Lorenzo, L.; Merchan, F.; Laporte, P.; Thompson, R.; Clarke, J.; Sousa, C.; Crespi, M. A novel plant leucine-rich repeat receptor kinase regulates the response of Medicago truncatula roots to salt stress. Plant Cell 2009, 21, 668–680. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Salman, A.; Guo, C.; Yu, J.; Cao, S.; Gao, X.; Li, W.; Li, H.; Guo, Y. Identification and Characterization of LRR-RLK Family Genes in Potato Reveal Their Involvement in Peptide Signaling of Cell Fate Decisions and Biotic/Abiotic Stress Responses. Cells 2018, 7, 120. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.Y.; Osborne, E.; Poindexter, P.D.; Somerville, C.R. Microspore separation in the quartet 3 mutants of Arabidopsis is impaired by a defect in a developmentally regulated polygalacturonase required for pollen mother cell wall degradation. Plant Physiol. 2003, 133, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- Aarts, M.G.; Keijzer, C.J.; Stiekema, W.J.; Pereira, A. Molecular characterization of the CER1 gene of arabidopsis involved in epicuticular wax biosynthesis and pollen fertility. Plant Cell 1995, 7, 2115–2127. [Google Scholar]
- Wang, W.; Liu, X.; Gai, X.; Ren, J.; Liu, X.; Cai, Y.; Wang, Q.; Ren, H. Cucumis sativus L. WAX2 Plays a Pivotal Role in Wax Biosynthesis, Influencing Pollen Fertility and Plant Biotic and Abiotic Stress Responses. Plant Cell Physiol. 2015, 56, 1339–1354. [Google Scholar] [CrossRef] [Green Version]
- Ni, E.; Zhou, L.; Li, J.; Jiang, D.; Wang, Z.; Zheng, S.; Qi, H.; Zhou, Y.; Wang, C.; Xiao, S.; et al. OsCER1 Plays a Pivotal Role in Very-Long-Chain Alkane Biosynthesis and Affects Plastid Development and Programmed Cell Death of Tapetum in Rice (Oryza sativa L.). Front. Plant Sci. 2018, 9, 1217. [Google Scholar] [CrossRef] [Green Version]
- Ariel, F.; Jegu, T.; Latrasse, D.; Romero-Barrios, N.; Christ, A.; Benhamed, M.; Crespi, M. Noncoding transcription by alternative RNA polymerases dynamically regulates an auxin-driven chromatin loop. Mol. Cell 2014, 55, 383–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariel, F.; Romero-Barrios, N.; Jégu, T.; Benhamed, M.; Crespi, M. Battles and hijacks: Noncoding transcription in plants. Trends Plant Sci. 2015, 20, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Bohra, A. Non-coding RNAs and plant male sterility: Current knowledge and future prospects. Plant Cell Rep. 2018, 37, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, M.; Li, N.; Wang, H.; Qiu, P.; Pei, L.; Xu, Z.; Wang, T.; Gao, E.; Liu, J.; et al. Long noncoding RNAs involve in resistance to Verticillium dahliae, a fungal disease in cotton. Plant Biotechnol. J. 2018, 16, 1172–1185. [Google Scholar] [CrossRef]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CCS Reads | Full Length | FLNC Number | Percentage (%) |
|---|---|---|---|---|
| GYDM | 347,387 | 301,937 | 290,886 | 83.74 |
| Annotated Database | Annotated Number | 300 ≤ Length < 1000 | Length ≥ 1000 |
|---|---|---|---|
| NR | 13,538 (99.13%) | 1246 (9.12%) | 12,288 (89.98%) |
| Swissprot | 11,587 (84.84%) | 965 (7.07%) | 10,620 (77.76%) |
| KEGG | 6025 (44.12%) | 575 (4.21%) | 5449 (39.90%) |
| KOG | 8552 (62.62%) | 664 (4.86%) | 7887 (57.75%) |
| eggNOG | 13,379 (97.96%) | 1190 (8.71%) | 12,185 (89.22%) |
| GO | 10,896 (79.78%) | 942 (6.90%) | 9952 (72.87%) |
| Pfam | 12,844 (94.05%) | 1038 (7.60%) | 11,806 (86.45%) |
| Classification | Pathway Definition | Pathway | Gene Number |
|---|---|---|---|
| Metabolism–Carbohydrate metabolism | Glycolysis/Gluconeogenesis | ko00010 | 138 |
| Starch and sucrose metabolism | ko00500 | 173 | |
| Amino sugar and nucleotide sugar metabolism | ko00520 | 141 | |
| Metabolism–Global and overview maps | Carbon metabolism | ko01200 | 321 |
| Biosynthesis of amino acids | ko01230 | 253 | |
| Genetic Information Processing–Translation | Ribosome | ko03010 | 253 |
| RNA transport | ko03013 | 148 | |
| mRNA surveillance pathway | ko03015 | 125 | |
| Genetic Information Processing–Transcription | Spliceosome | ko03040 | 209 |
| Environmental Information Processing–Signal transduction | Plant hormone signal transduction | ko04075 | 174 |
| Genetic Information Processing–Folding, sorting and degradation | Protein processing in endoplasmic reticulum | ko04141 | 180 |
| Sample | Total Reads | Total Base (bp) | Min Length | Max Length | Mean Length |
|---|---|---|---|---|---|
| Novel genes | 232 | 417,131 | 346 | 4706 | 1797.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, H.; Li, Y.; Luan, M.; Huang, S.; Zhao, L.; Yang, G.; Pan, G. Single-Molecule Real-Time Sequencing of Full-Length Transcriptome and Identification of Genes Related to Male Development in Cannabis sativa. Plants 2022, 11, 3559. https://doi.org/10.3390/plants11243559
Jiang H, Li Y, Luan M, Huang S, Zhao L, Yang G, Pan G. Single-Molecule Real-Time Sequencing of Full-Length Transcriptome and Identification of Genes Related to Male Development in Cannabis sativa. Plants. 2022; 11(24):3559. https://doi.org/10.3390/plants11243559
Chicago/Turabian StyleJiang, Hui, Ying Li, Mingbao Luan, Siqi Huang, Lining Zhao, Guang Yang, and Gen Pan. 2022. "Single-Molecule Real-Time Sequencing of Full-Length Transcriptome and Identification of Genes Related to Male Development in Cannabis sativa" Plants 11, no. 24: 3559. https://doi.org/10.3390/plants11243559