
Environmental Signals Act as a Driving Force for Metabolic and Defense Responses in the Antarctic Plant Colobanthus quitensis
 ,
,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
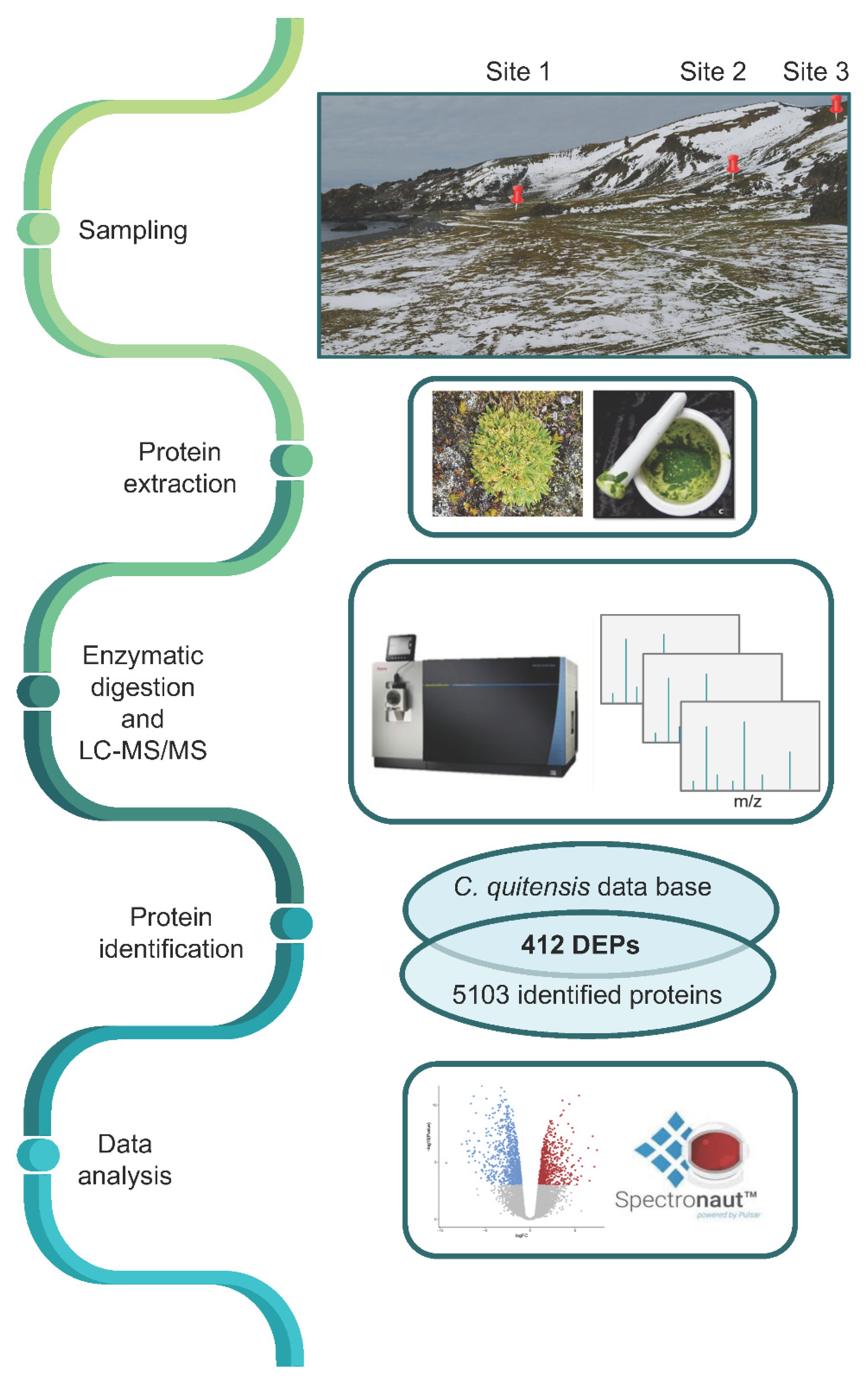
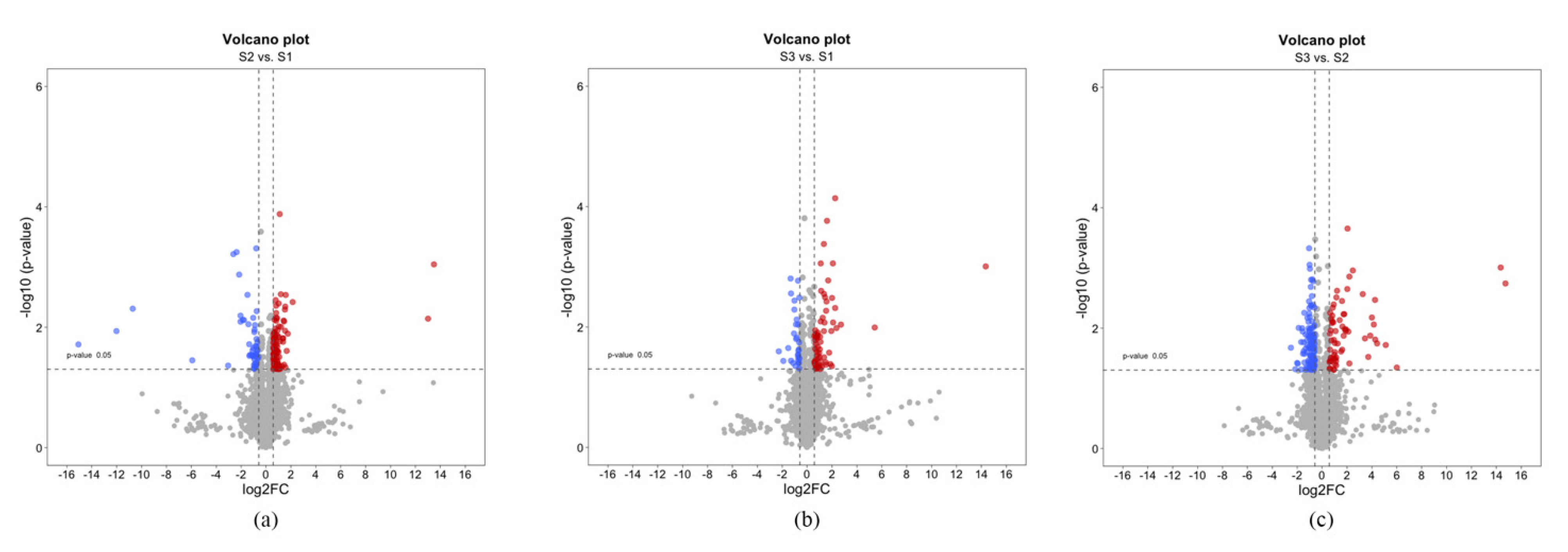
2.1. Determination of the Differentially Expressed Protein by a Proteomic Approach
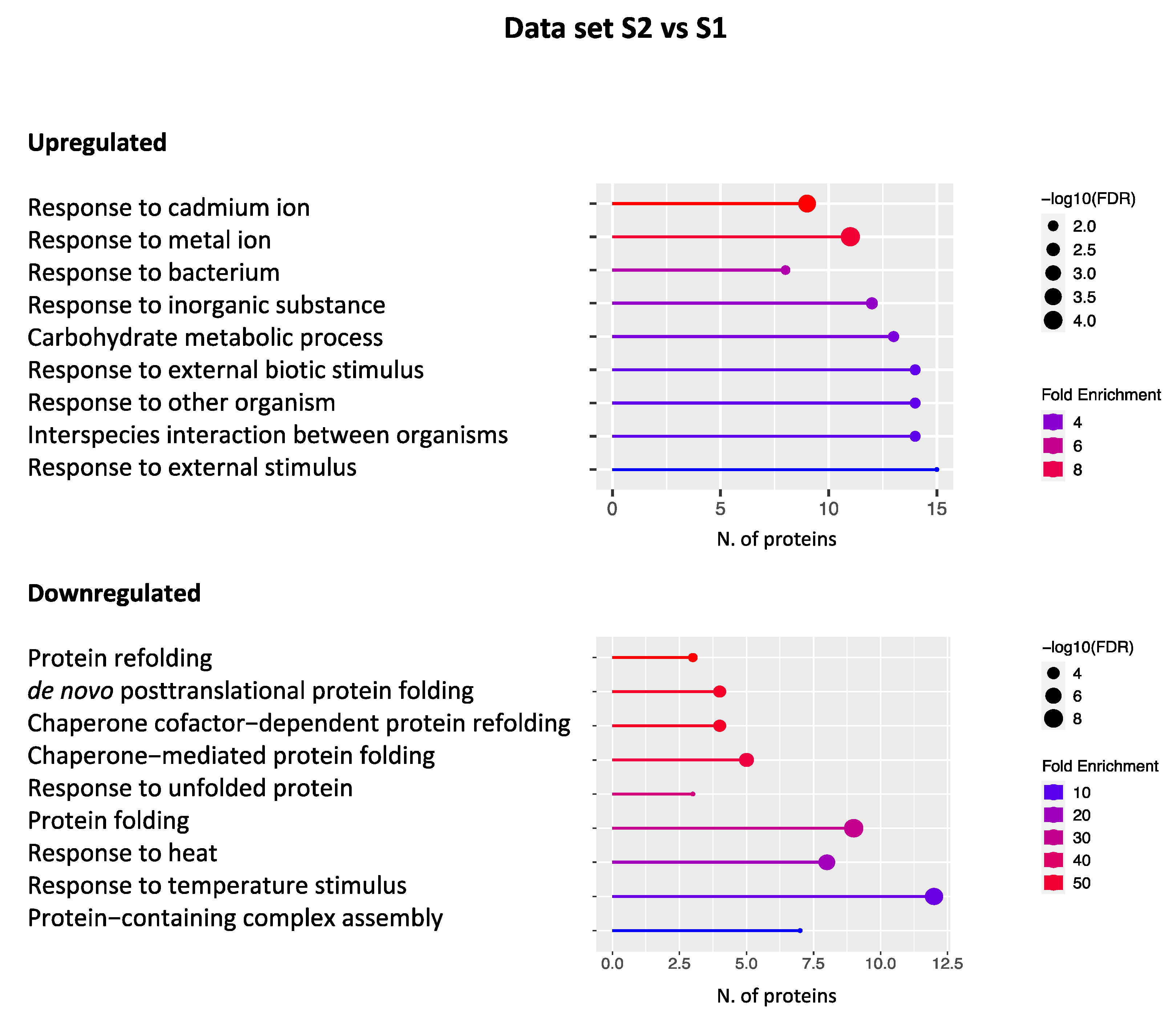
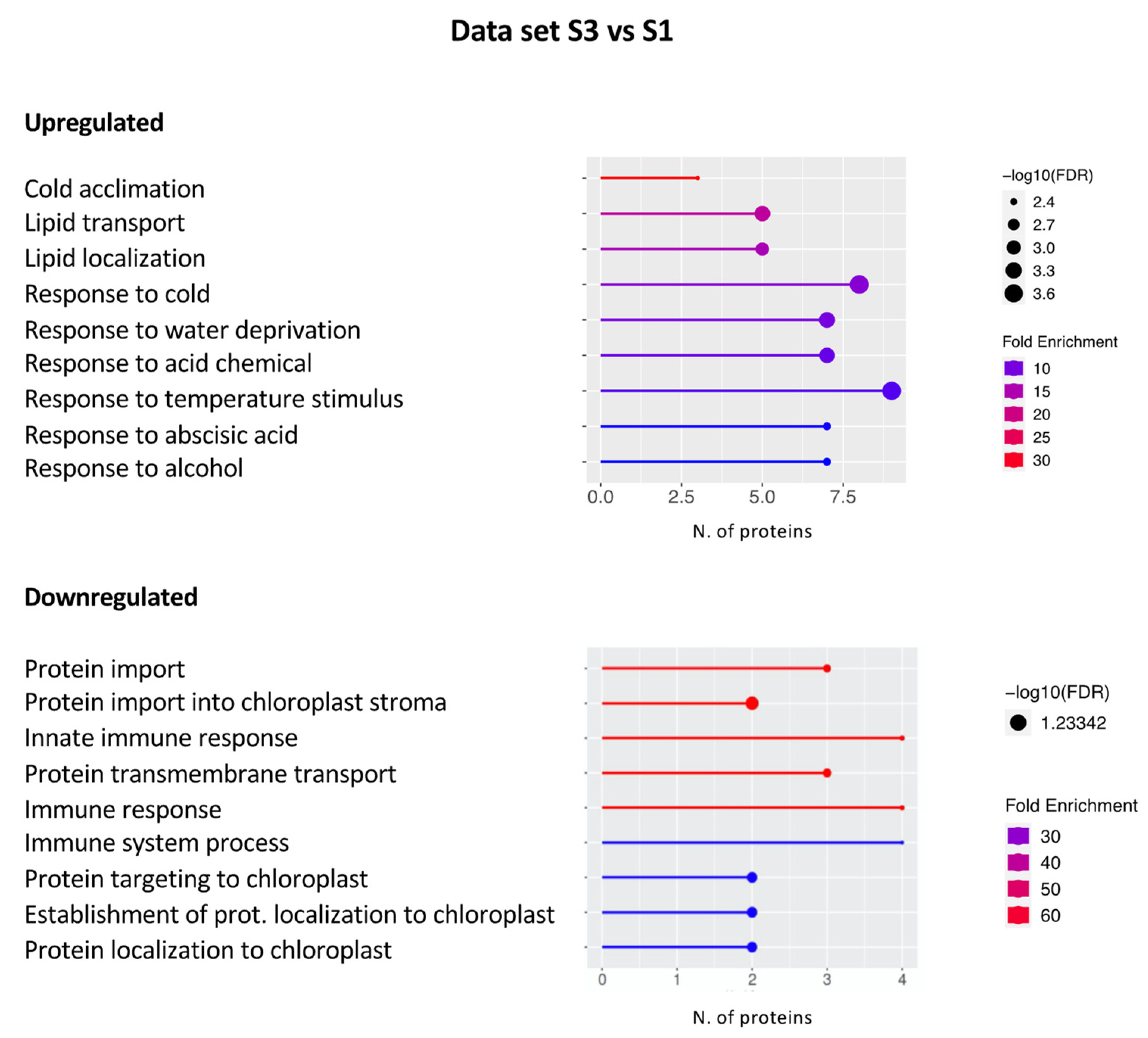
2.2. Gene Ontology Analysis
2.2.1. Data set S2 vs. S1
2.2.2. Data set S3 vs. S1
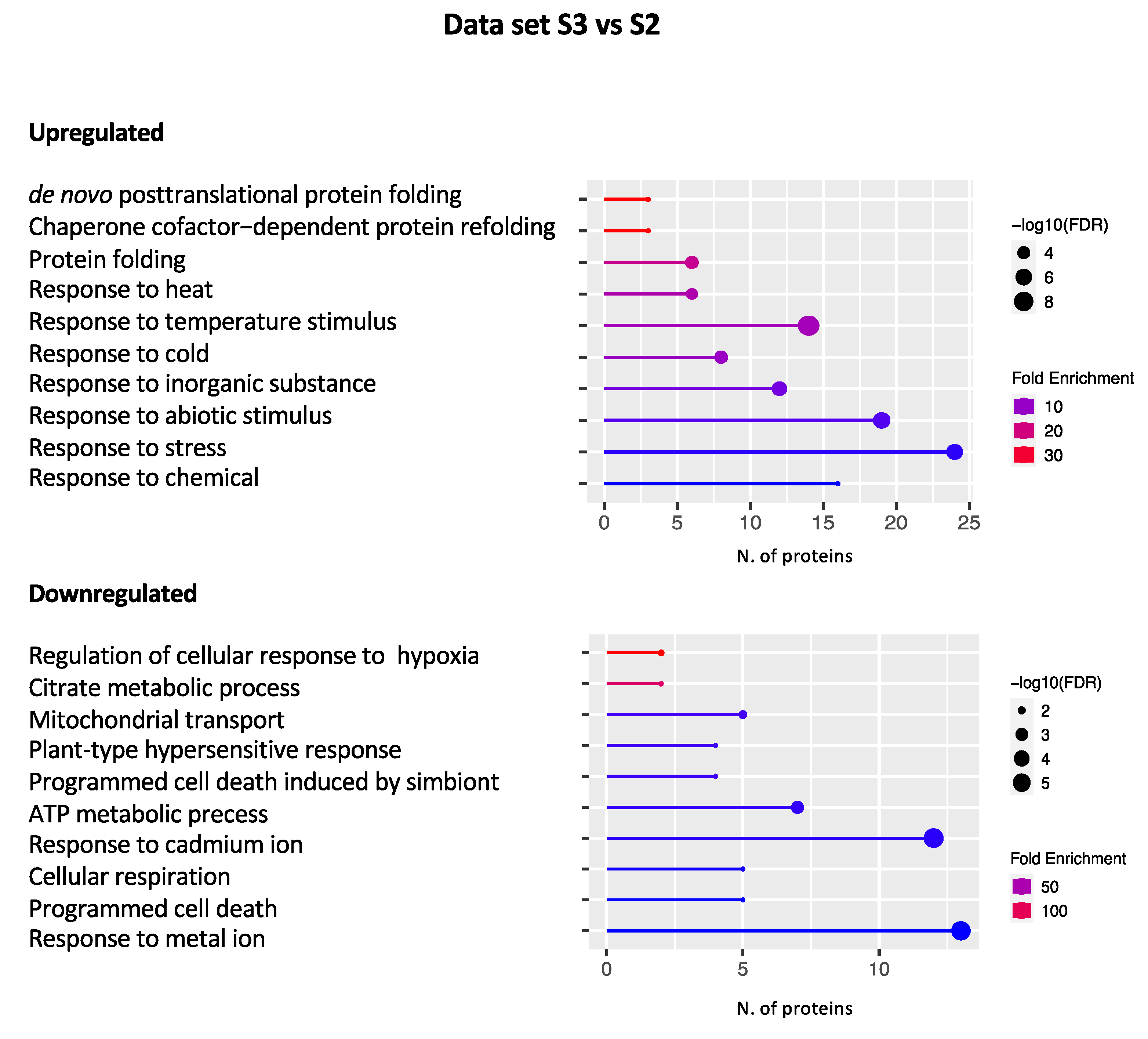
2.2.3. Data set S3 vs. S2
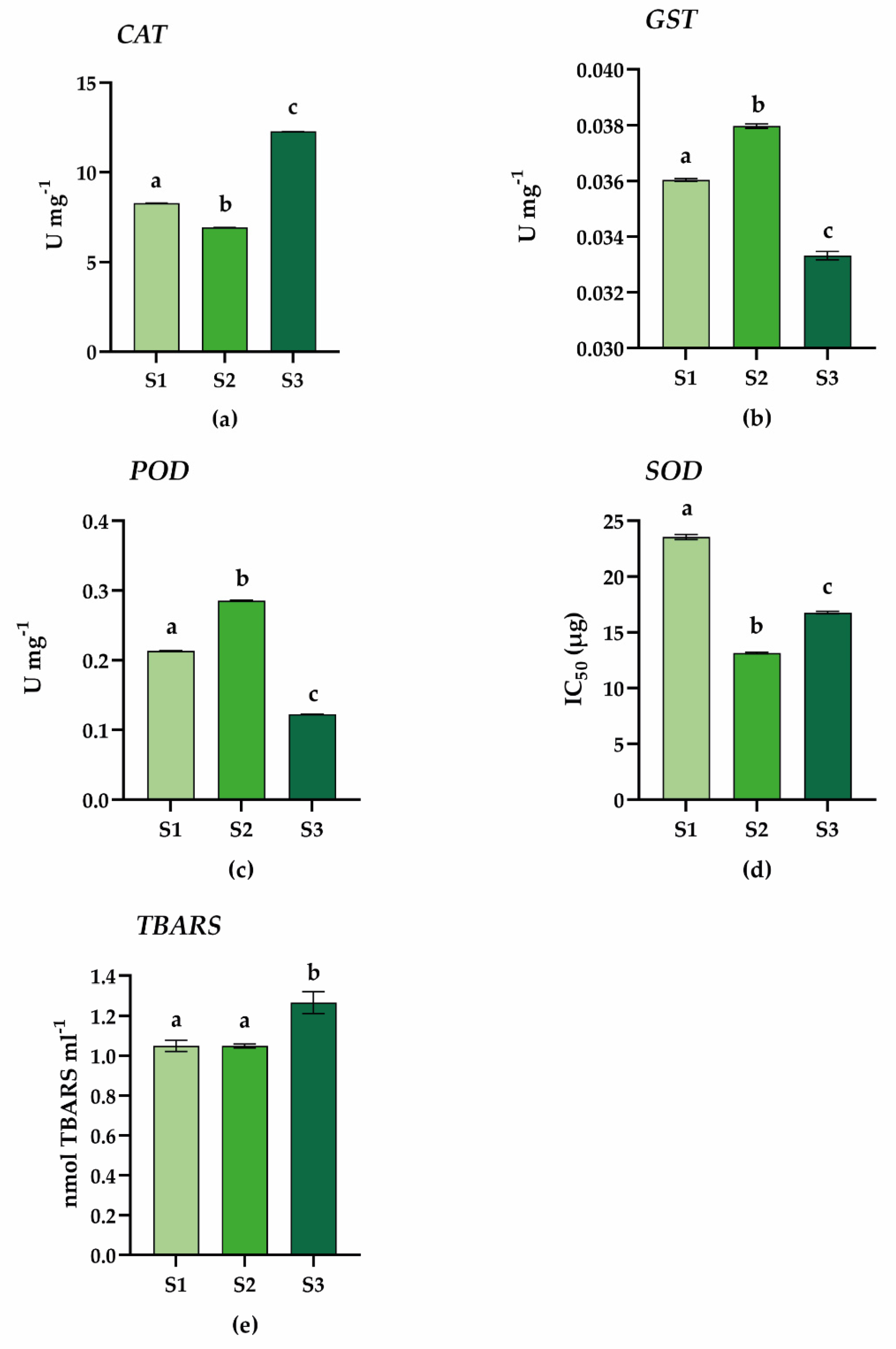
2.3. Antioxidant Enzyme Activity Assays
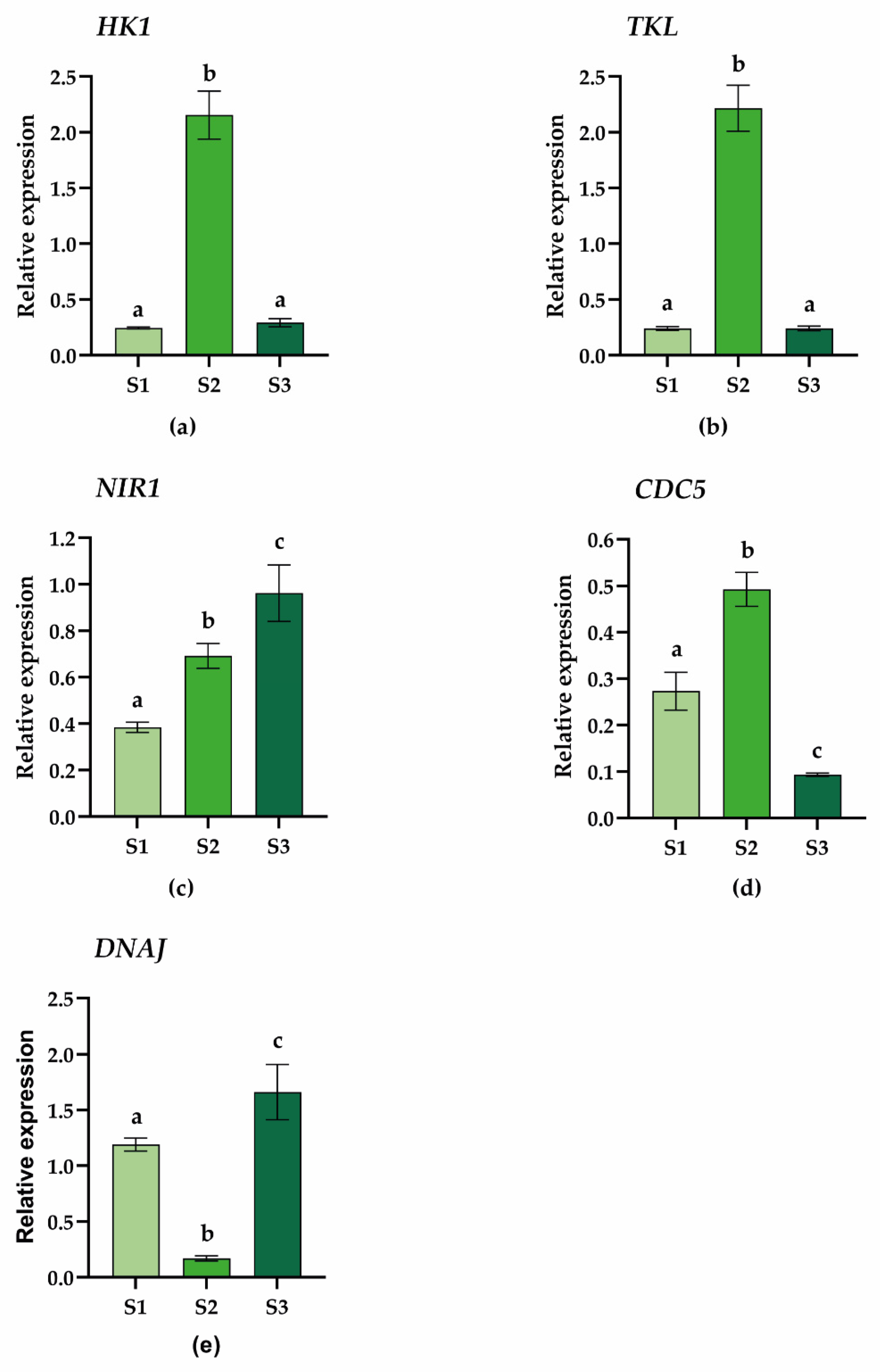
2.4. Gene Expression Analysis
3. Materials and Methods
3.1. Sample Collection
- Site 1 (62°9′43.33′′ S; 58°27′58.80′′ W), about 90 m from the coast. It is strongly influenced by fauna such as penguins, seals, and elephant seals which deposit guano and feces making the soil richer in nutrients. Furthermore, the proximity of the sea affects the soil salinity.
- Site 2 (62°9′49.15′′ S; 58°28′9.60′′ W), 300 m from the coast and 20 m above sea level; the soil of this site is well drained.
- Site 3 (62°9′52.90′′ S; 58°28′21.31′′ W), located about 550 m from the coast and 30 m above sea level. This site is very windy and scarcely covered by vegetation dominated by lichens and isolated individuals of C. quitensis and D. antarctica; the ground is stony and rocky.
3.2. Protein Sample Preparation
3.3. Mass Spectrometry
3.4. Bioinformatic Analysis
3.5. Enzymatic Activity Assays
3.6. Lipid Peroxidation Damage
3.7. RNA Extraction and Quantitative Reverse Transcriptase-PCR Analysis
3.8. Statistical Analysis
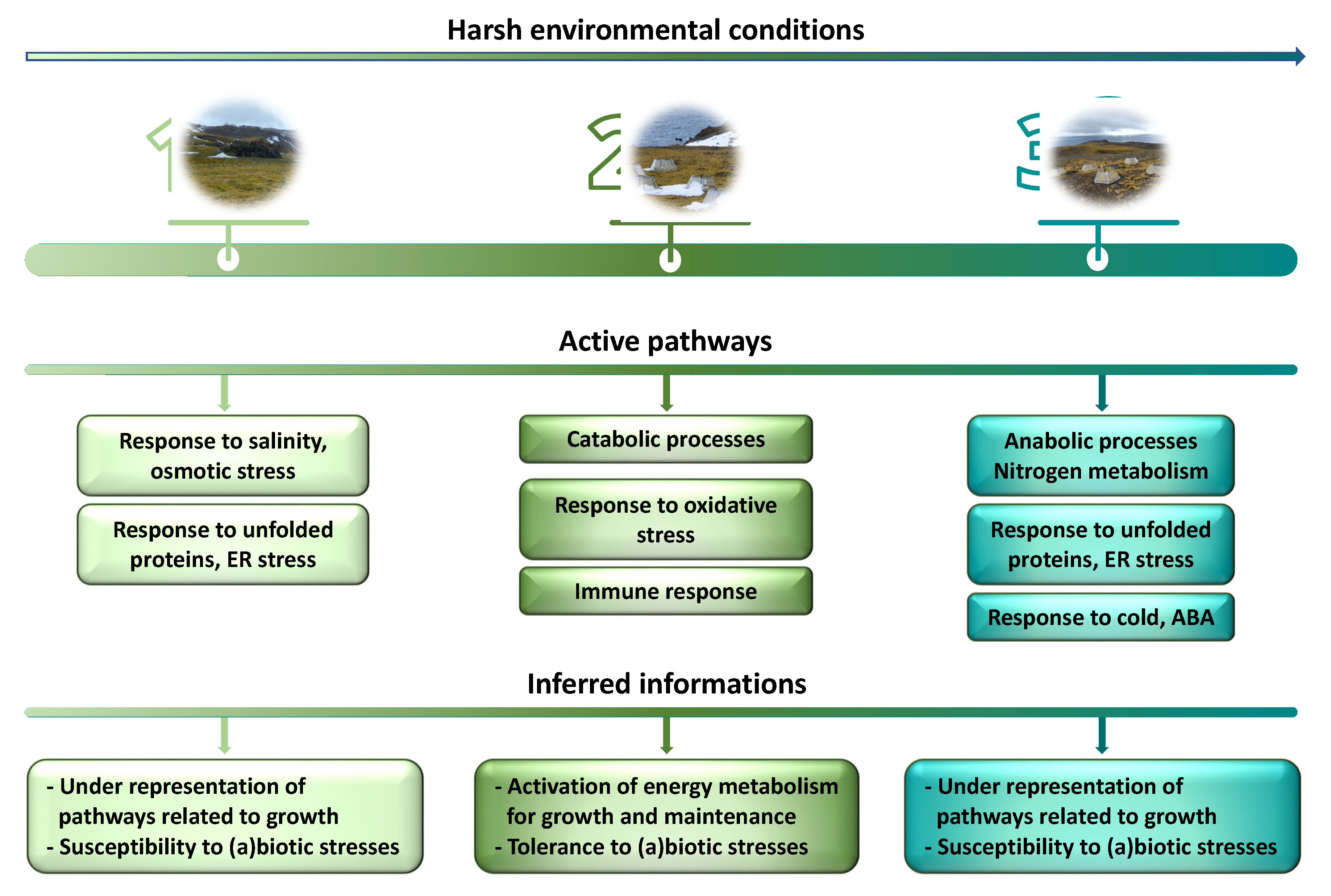
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Convey, P.; Coulson, S.J.; Worland, M.R.; Sjöblom, A. The importance of understanding annual and shorter-term temperature patterns and variation in the surface levels of polar soils for terrestrial biota. Polar. Biol. 2018, 41, 1587–1605. [Google Scholar] [CrossRef] [Green Version]
- Sierra-Almeida, A.; Cavieres, L.A.; Bravo, L.A. Warmer temperatures affect the in situ freezing resistance of the Antarctic vascular plants. Front. Plant. Sci. 2018, 9, 1456. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Moreno, M.J.; Omranian, N.; Sáez, P.; Figureueroa, C.M.; Del-Saez, N.; Elso, M.; Poblete, L.; Orf, I.; Cuadros-Inostroza, A.; Cavieres, L.; et al. Cytochrome respiration pathway and sulphur metabolism sustain stress tolerance to low temperature in the Antarctic species Colobanthus quitensis. New Phytol. 2020, 225, 754–768. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, M.; Bravo, L.A.; Gutiérrez, A.; Gidekel, M.; Corcuera, L.J. Ecophysiology of Antarctic vascular plants. Physiol. Plantarum. 2002, 115, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Wasley, J.; Robinson, S.A.; Lovelock, C.E.; Popp, M. Climate change manipulations show Antarctic flora is more strongly affected by elevated nutrients than water. Global. Change Biol. 2006, 12, 1800–1812. [Google Scholar] [CrossRef] [Green Version]
- Convey, P. Antarctic terrestrial biodiversity in a changing world. Polar Biol. 2011, 34, 1629–1641. [Google Scholar] [CrossRef]
- Robinson, S.A.; King, D.H.; Bramley-Alves, J.; Waterman, M.J.; Ashcroft, M.B.; Wasley, J.; Turnbull, J.D.; Miller, R.E.; Ryan-Colton, E.; Benny, T.; et al. Rapid change in East Antarctic terrestrial vegetation in response to regional drying. Nat. Clim. Change 2018, 8, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Cavieres, L.A.; Sáez, P.; Sanhueza, C.; Sierra-Almeida, A.; Rabert, C.; Corcuera, L.J.; Alberdi, M.; Bravo, L.A. Ecophysiological traits of Antarctic vascular plants: Their importance in the responses to climate change. Plant Ecol. 2016, 217, 343–358. [Google Scholar] [CrossRef]
- Pertierra, L.R.; Lara, F.; Benayas, J.; Hughes, K.A. Poa pratensis L., current status of the longest-established non-native vascular plant in the Antarctic. Polar Biol. 2013, 36, 1473–1481. [Google Scholar] [CrossRef]
- Chwedorzewska, K.J.; Giełwanowska, I.; Olech, M.; Molina-Montenegro, M.A.; Wódkiewicz, M.; Galera, H. Poa annua L. in the maritime Antarctic: An overview. Polar Rec. 2015, 51, 637–643. [Google Scholar] [CrossRef]
- Cuba-Díaz, M.; Troncoso, J.; Cordero, C.; Finot, V.; Rondanelli-Reyes, M. Juncus bufonius, a new non-native vascular plant in King George Island, South Shetland Islands. Antarct. Sci. 2013, 25, 385–386. [Google Scholar] [CrossRef]
- Convey, P.; Hopkins, D.; Roberts, S.; Tyler, A. Global southern limit of flowering plants and moss peat accumulation. Polar Res. 2011, 30, 8929. [Google Scholar] [CrossRef]
- Smith, R.I.L. The enigma of Colobanthus quitensis and Deschampsia antarctica in Antarctica. In Antarctic Biology in a Global Context; Huiskes, A.H.L., Gieskes, W.W.C., Rozema, J., Schorno, R.M.L., van der Vies, S.M., Wolff, W.J., Eds.; Backhuys: Leiden, The Netherlands, 2003; pp. 234–239. [Google Scholar]
- Reyes-Bahamonde, C. Consecuencias del aumento de la temperatura y la sequía en la Resistencia al Congelamiento de Deschampsia antarctica Desv. (Poaceae) and Colobanthus quitensis (Kunth.) Bartl. (Caryophyllaceae). Undergraduate Thesis, Universidad de Concepción, Concepción, Chile, 2013. [Google Scholar]
- Giełwanowska, I.; Pastorczyk, M.; Kellmann-Sopyła, W.; Gorniak, D.; Gorecki, R.J. Morphological and ultrastructural changes of organelles in leaf mesophyll cells of the Arctic and Antarctic plants of Poaceae family under cold influence. Arct. Antarct. Alp. Res. 2015, 47, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Acuña-Rodríguez, I.S.; Torres-Díaz, C.; Hereme, R.; Molina-Montenegro, M.A. Asymmetric responses to simulated global warming by populations of Colobanthus quitensis along a latitudinal gradient. PeerJ 2017, 5, e3718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannone, N.; Guglielmin, M.; Convey, P.; Worland, M.R.; Favero Longo, S.E. Vascular plant changes in extreme environments: Effects of multiple drivers. Clim. Change 2016, 134, 651–665. [Google Scholar] [CrossRef] [Green Version]
- Cannone, N.; Malfasi, F.; Favero-Longo, S.E.; Convey, P.; Guglielmin, M. Acceleration of climate warming and plant dynamics in Antarctica. Curr. Biol. 2022, 32, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Hereme, R.; Morales-Navarro, S.; Ballesteros, G.; Barrera, A.; Ramos, P.; Gundel, P.E.; Molina-Montenegro, M.A. Fungal endophytes exert positive effects on Colobanthus quitensis under water stress but neutral under a projected climate change scenario in Antarctica. Front Microbiol. 2020, 11, 264. [Google Scholar] [CrossRef] [PubMed]
- Torres-Díaz, C.; Gallardo-Cerda, J.; Lavin, P.; Oses, R.; Carrasco-Urra, F.; Atala, C.; Acuña-Rodríguez, I.S.; Convey, P.; Molina-Montenegro, M.A. Biological interactions and simulated climate change modulates the ecophysiological performance of Colobanthus quitensis in the Antarctic ecosystem. PLoS ONE 2016, 11, e0164844. [Google Scholar] [CrossRef] [Green Version]
- Bertini, L.; Perazzolli, M.; Proietti, S.; Capaldi, G.; Savatin, D.V.; Bigini, V.; Longa, C.M.O.; Basaglia, M.; Favaro, L.; Casella, S.; et al. Biodiversity and Bioprospecting of Fungal Endophytes from the Antarctic Plant Colobanthus quitensis. J. Fungi 2022, 8, 979. [Google Scholar] [CrossRef]
- Arthofer, W.; Bertini, L.; Caruso, C.; Cicconardi, F.; Delph, L.F.; Fields, P.D.; Ikeda, M.; Minegishi, Y.; Proietti, S.; Ritthammer, H.; et al. Transcriptome sequencing of the Antarctic Colobanthus quitensis (Kunth) Bartl (Caryophillaceae). Mol. Ecol. Resour. 2015, 15, 1014–1015. [Google Scholar] [CrossRef]
- Bertini, L.; Cozzolino, F.; Proietti, S.; Falconieri, G.S.; Iacobucci, I.; Salvia, R.; Falabella, P.; Monti, M.; Caruso, C. What Antarctic plants can tell us about climate changes: Temperature as a driver for metabolic reprogramming. Biomolecules 2021, 11, 1094. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.M.; Lee, H.; Jo, H.; Lee, H.; Kang, Y.; Park, H.; Lee, J. Comparative transcriptome analysis of field- and chamber-grown samples of Colobanthus quitensis (Kunth) Bartl, an Antarctic flowering plant. Sci. Rep. 2018, 8, 11049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertini, L.; Palazzi, L.; Proietti, S.; Pollastri, S.; Arrigoni, G.; Polverino de Laureto, P.; Caruso, C. Proteomic analysis of MeJa-induced defense responses in rice against wounding. Int. J. Mol. Sci. 2019, 20, 2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Samyshev, E.Z.; Minkina, N.I. Coastal Ecosystem Contamination by Heavy Metals as an Indicator of Climate Change in Antarctica. J. Comput. Theor. Nanosci. 2019, 16, 1. [Google Scholar] [CrossRef]
- Hughes, K.A. Threats to soil communities: Human impacts. In Antarctic Terrestrial Microbiology: Physical and Biological Properties of Antarctic Soils; Cowan, D.A., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 263–277. [Google Scholar] [CrossRef]
- Chmielowska-Bąk, J.; Gzyl, J.; Ruścińska-Sobkowiak, R.; Arasimowicz-Jelonek, M.; Deckert, J. The new insights into cadmium sensing. Front Plant Sci. 2014, 5, 245. [Google Scholar] [CrossRef]
- Dorta, D.J.; Leite, S.; Demarco, K.C.; Prado, I.M.; Rodrigues, T.; Mingatto, F.E.; Uyemura, S.A.; Santos, A.C.; Curti, C. A proposed sequence of events for cadmium-induced mitochondrial impairment. J. Inorg. Biochem. 2003, 97, 251–257. [Google Scholar] [CrossRef]
- Pastuszak, J.; Kopeć, P.; Płażek, A.; Gondek, K.; Szczerba, A.; Hornyák, M.; Dubert, F. Antioxidant activity as a response to cadmium pollution; in three durum wheat genotypes differing in salt-tolerance. Open Chem. 2020, 18, 1230–1241. [Google Scholar] [CrossRef]
- Ohi, R.; Feoktistova, A.; McCann, S.; Virginia Valentine, V.; Look, A.T.; Lipsick, J.S.; Gould, L.K. Myb-related Schizosaccharomyces pombe cdc5p is structurally and functionally conserved in eukaryotes. Mol. Cell. Biol. 1998, 18, 4097–4108. [Google Scholar] [CrossRef]
- Hirayama, T.; Shinozaki, K. A cdc5+ homolog of a higher plant, Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 1996, 93, 13371–13376. [Google Scholar] [CrossRef] [Green Version]
- Burns, C.G.; Ohi, R.; Krainer, A.R.; Gould, K.L. Evidence that Myb-related CDC5 proteins are required for pre-mRNA splicing. Proc. Natl. Acad. Sci. USA 1999, 96, 13789–13794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, W.H.; Ohi, R.; Smelkova, N.; Frendewey, D.; Gould, K.L. Myb-related fission yeast CDC5p is a component of a 40S snRNP-containing complex and is essential for pre-mRNA splicing. Mol. Cell. Biol. 1999, 19, 5352–5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, K.; Zhao, Q.; Cheng, Y.T.; Bi, D.; Monaghan, J.; Cheng, W.; Zhang, Y.; Li, X. Regulation of plant innate immunity by three proteins in a complex conserved across the plant and animal kingdoms. Genes Dev. 2007, 21, 1484–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Yin, Z.; Zhu, D.; Chen, Z.; Gu, H.; Qu, L.J. AtCDC5 regulates the G2 to M transition of the cell cycle and is critical for the function of Arabidopsis shoot apical meristem. Cell Res. 2007, 17, 815–828. [Google Scholar] [CrossRef] [Green Version]
- Ikram, Z.; Mouna, G.; Faiçal, B. Pathogenesis Related Proteins (PRs): From Cellular Mechanisms to Plant Defense. Curr. Protein Pept. Sci. 2021, 22, 396–412. [Google Scholar] [CrossRef]
- Bertini, L.; Leonardi, L.; Caporale, C.; Tucci, M.; Cascone, A.; Di Berardino, I.; Buonocore, V.; Caruso, C. Pathogen-responsive wheat PR4 genes are induced by activators of systemic acquired resistance and wounding. Plant Sci. 2003, 164, 1067–1078. [Google Scholar] [CrossRef]
- De Lorenzo, G.; D’Ovidio, R.; Felice Cervone, F. The role of polygalacturonase-inhibiting proteins (PGIPs) in defense against pathogenic fungi. Annu. Rev. Phytopathol. 2001, 39, 313–335. [Google Scholar] [CrossRef]
- Cecchini, N.; Steffes, K.; Schläppi, M.R.; Gifford, A.N.; Greenberg, J.T. Arabidopsis AZI1 family proteins mediate signal mobilization for systemic defence priming. Nat. Commun. 2015, 6, 7658. [Google Scholar] [CrossRef] [Green Version]
- Carrera, D.A.; George, G.M.; Fischer-Stettler, M.; Galbier, F.; Eicke, S.; Truernit, E.; Streb, S.; Zeeman, S.C. Distinct plastid fructose bisphosphate aldolases function in photosynthetic and non-photosynthetic metabolism in Arabidopsis. J. Exp. Bot. 2021, 72, 3739–3755. [Google Scholar] [CrossRef]
- Shi, J.; Yi, K.; Liu, Y.; Xie, L.; Zhou, Z.; Chen, Y.; Hu, Z.; Zheng, T.; Liu, R.; Chen, Y.; et al. Phosphoenolpyruvate Carboxylase in Arabidopsis Leaves Plays a Crucial Role in Carbon and Nitrogen Metabolism. Plant Physiol. 2015, 167, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.; Samraj, S.; Juan Jiménez, J.; Gómez, C.; Liu, T.; Begcy, K. Genome-wide identification of heat shock factors and heat shock proteins in response to UV and high intensity light stress in lettuce. BMC Plant Biol. 2021, 21, 185. [Google Scholar] [CrossRef] [PubMed]
- Volkov, R.A.; Panchuk, I.; Mullineaux, P.M.; Schöffl, F. Heat stress-induced H2O2 is required for effective expression of heat shock genes in Arabidopsis. Plant Mol. Biol. 2006, 61, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Driedonks, N.; Xu, J.; Peters, J.L.; Park, S.; Ivo Rieu, I. Multi-Level Interactions between Heat Shock Factors, Heat Shock Proteins, and the Redox System Regulate Acclimation to Heat. Front. Plant Sci. 2015, 6, 999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, S.H. Endoplasmic Reticulum Stress Responses in Plants. Annual. Rev. Plant Bio. 2013, 64, 477–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Hennessy, F.; Nicoll, W.S.; Zimmermann, R.; Cheetham, M.E.; Blatch, G.L. Not all J domains are created equal: Implications for the specificity of Hsp40-Hsp70 interactions. Protein Sci. 2005, 14, 1697–1709. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [Green Version]
- Anken, E.; Braakman, I.; Craig, E. Versatility of the endoplasmic reticulum protein folding factory. Crit. Rev. Biochem. Mol. 2005, 40, 191–228. [Google Scholar] [CrossRef]
- Lu, D.P.; Christopher, D.A. Endoplasmic reticulum stress activates the expression of a sub-group of protein disulfide isomerase genes and AtbZIP60 modulates the response in Arabidopsis thaliana. Mol. Genet. Genom. 2008, 280, 199–210. [Google Scholar] [CrossRef]
- Irfan, M.; Kumar, P.; Ahmad, I.; Datta, A. Unraveling the role of tomato Bcl-2-associated athanogene (BAG) proteins during abiotic stress response and fruit ripening. Sci. Rep. 2021, 11, 21734. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Naik, D.; Reddy, A.R. Plant aldo-keto reductases (AKRs) as multi-tasking soldiers involved in diverse plant metabolic processes and stress defense: A structure-function update. J. Plant Physiol. 2015, 179, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Chakrabarty, D. Dehydrin in the past four decades: From chaperones to transcription co-regulators in regulating abiotic stress response. Curr. Res. Biotechnol. 2021, 3, 249–259. [Google Scholar] [CrossRef]
- Puhakainen, T.; Hess, M.W.; Mäkelä, P.; Svensson, J.; Heino, P.; Tapio, P.E. Overexpression of multiple dehydrin genes enhances tolerance to freezing stress in Arabidopsis. Plant Mol. Biol. 2004, 54, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, S.J.; Jang, B.; Jung, C.H.; Ahn, S.J.; Goh, C.H.; Cho, K.; Han, O.; Kang, H. Functional characterization of a glycine-rich RNA-binding protein 2 in Arabidopsis thaliana under abiotic stress conditions. Plant J. 2007, 50, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Jung, H.J.; Kim, B.M.; Xu, T.; Lee, K.; Kim, Y.-O.; Kang, H. A chloroplast-localized S1 domain-containing protein SRRP1 plays a role in Arabidopsis seedling growth in the presence of ABA. J. Plant Physiol. 2015, 189, 34–41. [Google Scholar] [CrossRef]
- Finkina, E.I.; Melnikova, D.N.; Bogdanov, I.V.; Ovchinnikova, T.V. Lipid Transfer Proteins as Components of the Plant Innate Immune System: Structure, Functions, and Applications. Acta Nat. 2016, 8, 47–61. [Google Scholar] [CrossRef]
- Jülke, S.; Müller-Ludwig, J. Response of Arabidopsis thaliana Roots with Altered Lipid Transfer Protein (LTP) Gene Expression to the Clubroot Disease and Salt Stress. Plants 2016, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Miho Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucl. Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Solomonson, I.P.; Barber, M.J. Assimilatory nitrate reductase: Functional properties and regulation. Ann. Rev. Plant Physiol. Plant Mol. Biol. 1990, 41, 225–253. [Google Scholar] [CrossRef]
- Knaff, D.B.; Hirasawa, M. Ferredoxin-dependent chloroplast enzymes. Biochim. Biophys. Acta 1991, 1056, 93–125. [Google Scholar] [CrossRef]
- Gupta, K.J.; Igamberdiev, A.U. The anoxic plant mitochondrion as a nitrite: NO reductase. Mitochondrion 2011, 11, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Bender, D.; Schwarz, G. Nitrite-dependent nitric oxide synthesis by molybdenum enzymes. FEBS Lett. 2018, 592, 2126–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolbert, Z.; Barroso, J.B.; Brouquisse, R.; Corpas, F.J.; Gupta, K.J.; Lindermayr, C.; Loake, G.J.; Palma, J.M.; Petrivalský, M.; Wendehenne, D.; et al. A forty-year journey: The generation and roles of NO in plants. Nitric Oxide 2019, 93, 53–70. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Chung, E.H.; Eitas, T.K.; Dangl, J.L. Plant intracellular innate immune receptor Resistance to Pseudomonas syringae pv. maculicola 1 (RPM1) is activated at, and functions on, the plasma membrane. Proc. Natl. Acad. Sci. USA 2011, 108, 7619–7624. [Google Scholar] [CrossRef] [Green Version]
- Paila, Y.D.; Richardson, L.G.L.; Schnell, D.J. New insights into the mechanism of chloroplast protein import and its integration with protein quality control, organelle biogenesis and development. J. Mol. Biol. 2015, 427, 1038–1060. [Google Scholar] [CrossRef] [Green Version]
- Bédard, J.; Trösch, R.; Wu, F.; Ling, Q.; Flores-Pérez, Ú.; Töpel, M.; Nawaz, F.; Paul Jarvis, P. Suppressors of the Chloroplast Protein Import Mutant tic40 Reveal a Genetic Link between Protein Import and Thylakoid Biogenesis. Plant Cell 2017, 29, 1726–1747. [Google Scholar] [CrossRef] [Green Version]
- Samol, I.; Rossig, C.; Buhr, F.; Springer, A.; Pollmann, S.; Lahroussi, A.; von Wettstein, D.; Reinbothe, C.; Reinbothe, S. The outer chloroplast envelope protein OEP16-1 for plastid import of NADPH:protochlorophyllide oxidoreductase A in Arabidopsis thaliana. Plant Cell Physiol. 2011, 52, 96–111. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Carrie, C.; Duncan, O.; Ho, L.H.; Howell, K.A.; Murcha, M.W.; Whelan, J. Functional definition of outer membrane proteins involved in preprotein import into mitochondria. Plant Cell 2007, 19, 3739–3759. [Google Scholar] [CrossRef]
- Das, A.; Pramanik, K.; Sharma, R.; Gantait, S.; Banerjee, J. In-silico study of biotic and abiotic stress-related transcription factor binding sites in the promoter regions of rice germin-like protein genes. PLoS ONE 2019, 14, e0211887. [Google Scholar] [CrossRef]
- Ma, L.; Cheng, K.; Li, J.; Deng, Z.; Zhang, C.; Zhu, H. Roles of Plant Glycine-Rich RNA-Binding Proteins in Development and Stress Responses. Int. J. Mol. Sci. 2021, 22, 5849. [Google Scholar] [CrossRef] [PubMed]
- Rapala-Kozik, M.; Wolak, N.; Kujda, M.; Banas, A.K. The upregulation of thiamine (vitamin B1) biosynthesis in Arabidopsis thaliana seedlings under salt and osmotic stress conditions is mediated by abscisic acid at the early stages of this stress response. BMC Plant Biol. 2012, 12, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Tian, Y.-S.; Xing, X.-J.; Peng, R.-H.; Zhu, B.; Gao, J.-J.; Yao, Q.-H. Over-expression of AtGSTU19 provides tolerance to salt, drought and methyl viologen stresses in Arabidopsis. Physiol. Plantarum. 2016, 156, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Kriegshauser, L.; Knosp, S.; Grienenberger, E.; Tatsumi, K.; Gütle, D.D.; Sørensen, I.; Herrgott, L.; Zumsteg, J.; Rose, J.K.C.; Reski, R.; et al. Function of the HYDROXYCINNAMOYL-CoA:SHIKIMATE HYDROXYCINNAMOYL TRANSFERASE is evolutionarily conserved in embryophytes. Plant Cell 2021, 33, 1472–1491. [Google Scholar] [CrossRef]
- Ye, N.; Zhu, G.; Liu, Y.; Yingxuan, L.; Zhang, J. ABA Controls H2O2 Accumulation Through the Induction of OsCATB in Rice Leaves Under Water Stress. Plant Cell Physiol. 2011, 52, 689–698. [Google Scholar] [CrossRef] [Green Version]
- de Dios, A. A concise appraisal of lipid oxidation and lipoxidation in higher plants. Redox Biol. 2019, 23, 101136. [Google Scholar] [CrossRef]
- Biswas, M.S.; Mano, J. Lipid Peroxide-Derived Short-Chain Carbonyls Mediate Hydrogen Peroxide-Induced and Salt-Induced Programmed Cell Death in Plants. Plant Physiol. 2015, 168, 885–898. [Google Scholar] [CrossRef] [Green Version]
- Costa-Broseta, Á.; Castillo, M.; León, J. Nitrite Reductase 1 Is a Target of Nitric Oxide-Mediated Post-Translational Modifications and Controls Nitrogen Flux and Growth in Arabidopsis. Int. J. Mol. Sci. 2020, 21, 7270. [Google Scholar] [CrossRef]
- Christen, P.; Han, W. Cis-Effect of DnaJ on DnaK in ternary complexes with chimeric DnaK/DnaJ-binding peptides. FEBS Lett. 2004, 563, 146–150. [Google Scholar] [CrossRef]
- Cavieres, L.A.; Vivas, M.; Mihoc, M.A.K.; Osses, D.A.; Ortiz-Gutiérrez, J.M.; Sáez, P.; Bravo, L.A. The importance of facilitative interactions on the performance of Colobanthus quitensis in an Antarctic tundra. J. Veg. Sci. 2018, 29, 236–244. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Hewapathirana, S.; García-Seisdedos, D.; Kamatchinathan, S.; Kundu, D.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Bertini, L.; Focaracci, F.; Proietti, S.; Papetti, P.; Caruso, C. Physiological response of Posidonia oceanica to heavy metal pollution along the Tyrrhenian coast. Funct. Plant Biol. 2019, 46, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid sensitive method for the quantification of microgram quantities of protein utilizing the principle of 778 protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Claiborne, A. Catalase activity. In Handbook of Methods for Oxygen Radical Research; Greenwald, R.A., Ed.; CRC Press: Boca Raton, FL, USA, 1984; pp. 283–284. [Google Scholar]
- Proietti, S.; Bertini, L.; Falconieri, G.S.; Baccelli, I.; Timperio, A.M.; Caruso, C. A Metabolic Profiling Analysis Revealed a Primary Metabolism Reprogramming in Arabidopsis glyI4 Loss-of-Function Mutant. Plants 2021, 10, 2464. [Google Scholar] [CrossRef] [PubMed]
- Bertini, L.; Proietti, S.; Focaracci, F.; Canini, F.; Bravo, L.A.; Rabert, C.; Caruso, C. Identification and validation of new reference genes for accurate qRT-PCR normalization in the Antarctic plant Colobanthus quitensis under abiotic stress conditions. Polar Biol. 2021, 44, 389–405. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Site 1 | Site 2 | Site 3 |
|---|---|---|---|
| Mean Air Temperature (°C) | 2.7 ± 0.1 | 2.1 ± 0.1 | 2.1 ± 0.1 |
| Mean Max Air T (°C) | 5.3 ± 0.4 | 4.9 ± 0.4 | 5.1 ± 0.4 |
| Mean Min Air T (°C) | 0.6 ± 0.3 | 0 ± 0.3 | 0 ± 0.3 |
| Ground Temperature | 2–4 °C higher than air T | ||
| Wind (m/s) | 5 | 7 | 10 |
| Distance from the Coastline (m) | 90 | 300 | 550 |
| Altitude (masl) | 0 | 20 | 30 |
| Soil composition % | |||
| Sand | 73 | 83 | 57 |
| Silt | 19 | 11 | 27 |
| Clay | 9 | 6 | 16 |
| Textural Class | Sandy loam | Loamy sand | Sandy loam |
| pH | 4.77 | 5.37 | 6.11 |
| Classification | Very strongly acidic | Strongly acidic | Slightly acidic |
| Gene Target | Acronym | Primers | Annealing Temperature (°C) | Amplicon Size (bp) |
|---|---|---|---|---|
| Heat shock protein 40 | DNAJ | For: GCCTCAACAAGTGATGCTTTC | 50 | 103 |
| Rev: CTCCAGCCGACTTAGTCTTTATT | ||||
| Cell division cycle 5 | CDC5 | For: ACTTGAGAGACCATAGGCATTAC | 50 | 102 |
| Rev: GGACATGAAGAGGACTCACTTG | ||||
| Nitrite reductase 1 | NIR1 | For: CCGTCACAAACTGCGAAATAAG | 54 | 118 |
| Rev: CTTCAGAGTGGCATGGACAA | ||||
| Hexokinase 1 | HK1 | For: GCTTCTGAAGGCGGTTCTAA | 50 | 100 |
| Rev: CACCAAGGTCCAAAGCATAGA | ||||
| Transketolase | TKL | For: GACCCAGCTTCGATGCTAAC | 50 | 151 |
| Rev: CCCAAGCAGGTGATGAACTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertini, L.; Proietti, S.; Fongaro, B.; Holfeld, A.; Picotti, P.; Falconieri, G.S.; Bizzarri, E.; Capaldi, G.; Polverino de Laureto, P.; Caruso, C. Environmental Signals Act as a Driving Force for Metabolic and Defense Responses in the Antarctic Plant Colobanthus quitensis. Plants 2022, 11, 3176. https://doi.org/10.3390/plants11223176
Bertini L, Proietti S, Fongaro B, Holfeld A, Picotti P, Falconieri GS, Bizzarri E, Capaldi G, Polverino de Laureto P, Caruso C. Environmental Signals Act as a Driving Force for Metabolic and Defense Responses in the Antarctic Plant Colobanthus quitensis. Plants. 2022; 11(22):3176. https://doi.org/10.3390/plants11223176
Chicago/Turabian StyleBertini, Laura, Silvia Proietti, Benedetta Fongaro, Aleš Holfeld, Paola Picotti, Gaia Salvatore Falconieri, Elisabetta Bizzarri, Gloria Capaldi, Patrizia Polverino de Laureto, and Carla Caruso. 2022. "Environmental Signals Act as a Driving Force for Metabolic and Defense Responses in the Antarctic Plant Colobanthus quitensis" Plants 11, no. 22: 3176. https://doi.org/10.3390/plants11223176