
Construction of A GBS-Based High-Density Genetic Map and Flower Color-Related Loci Mapping in Grasspea (Lathyrus sativus L.)

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
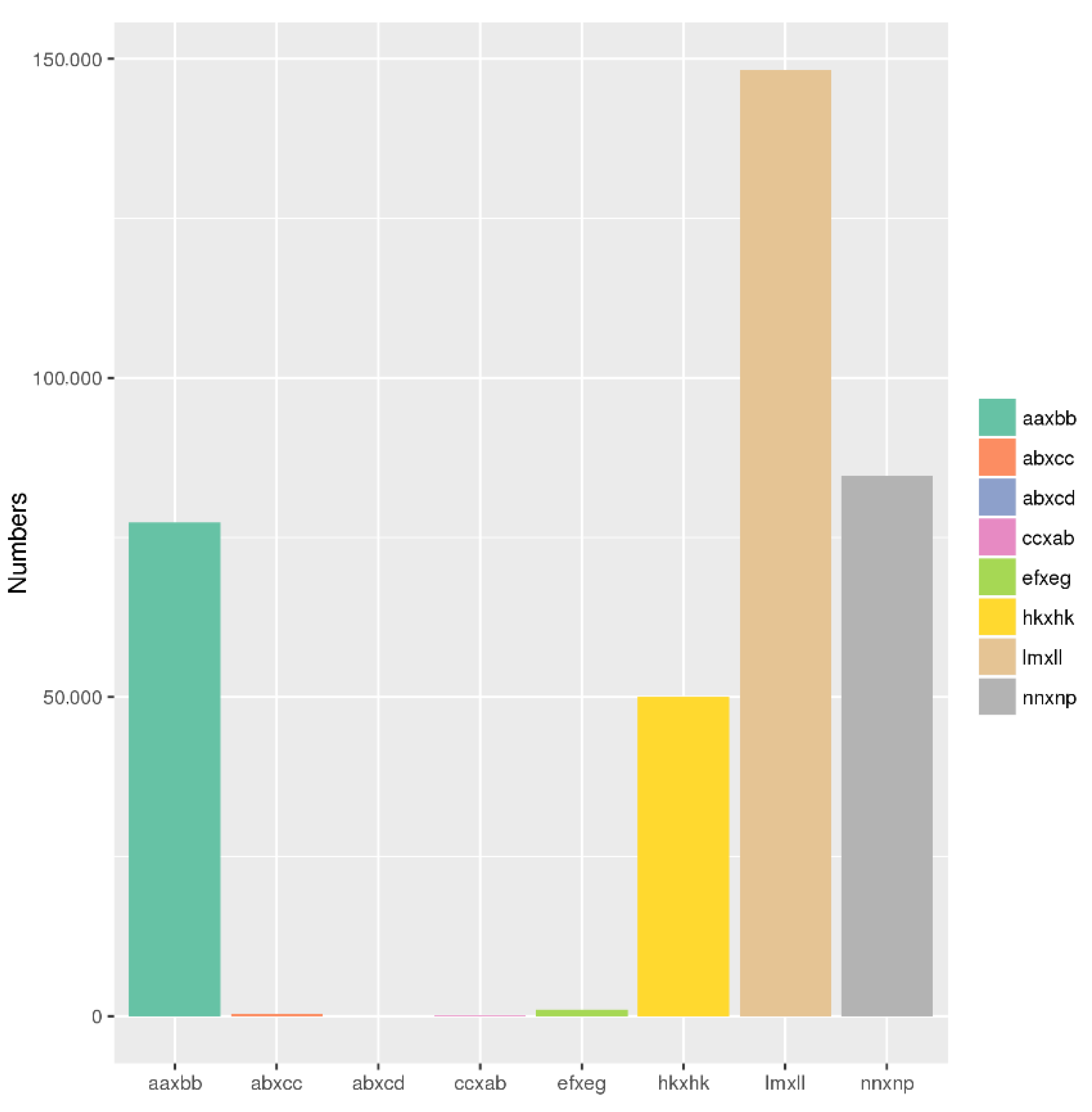
2.1. GBS Data Analysis and Assessment
2.2. SNP Calling and Genotyping
2.3. Genetic Linkage Map Constructing and Verification
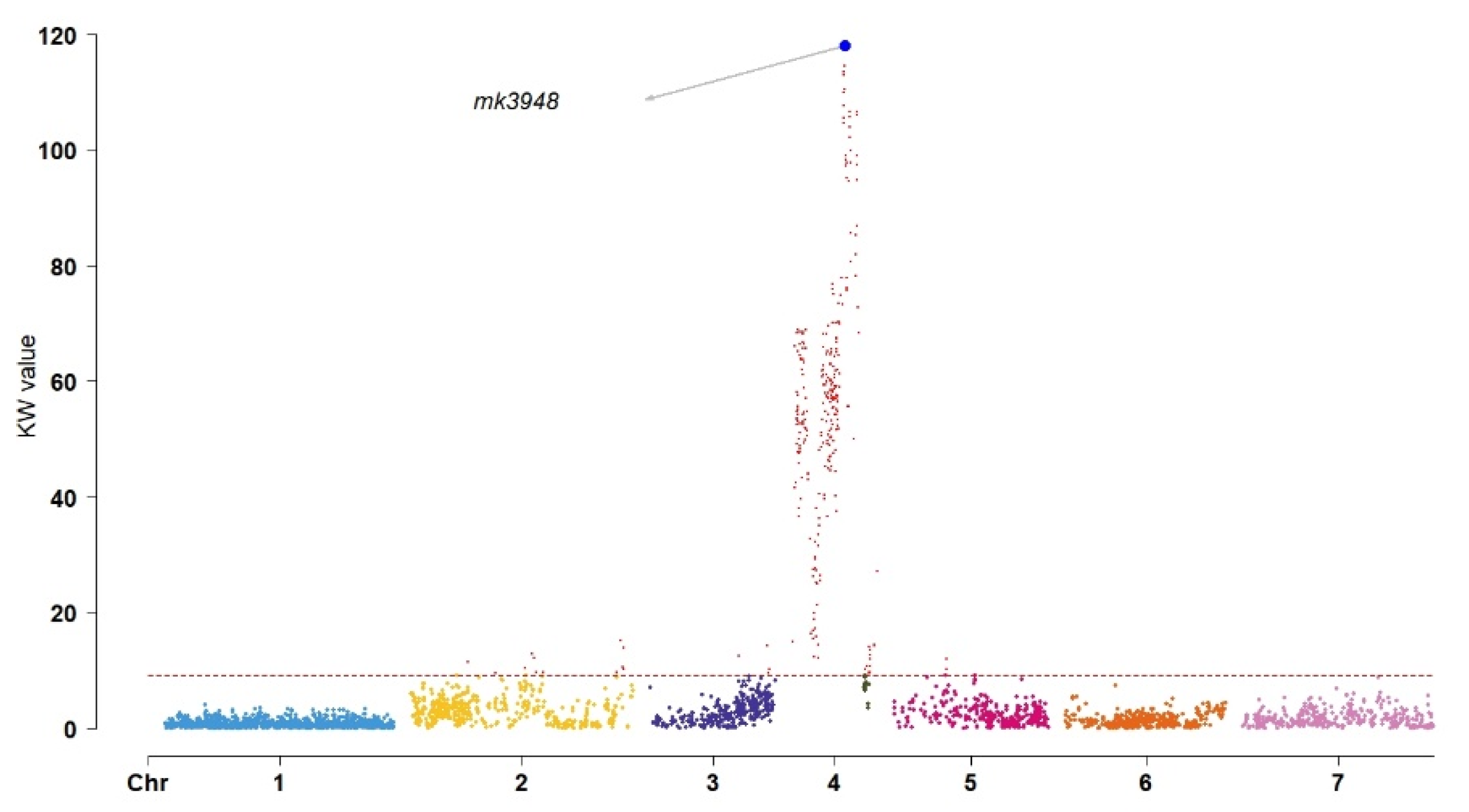
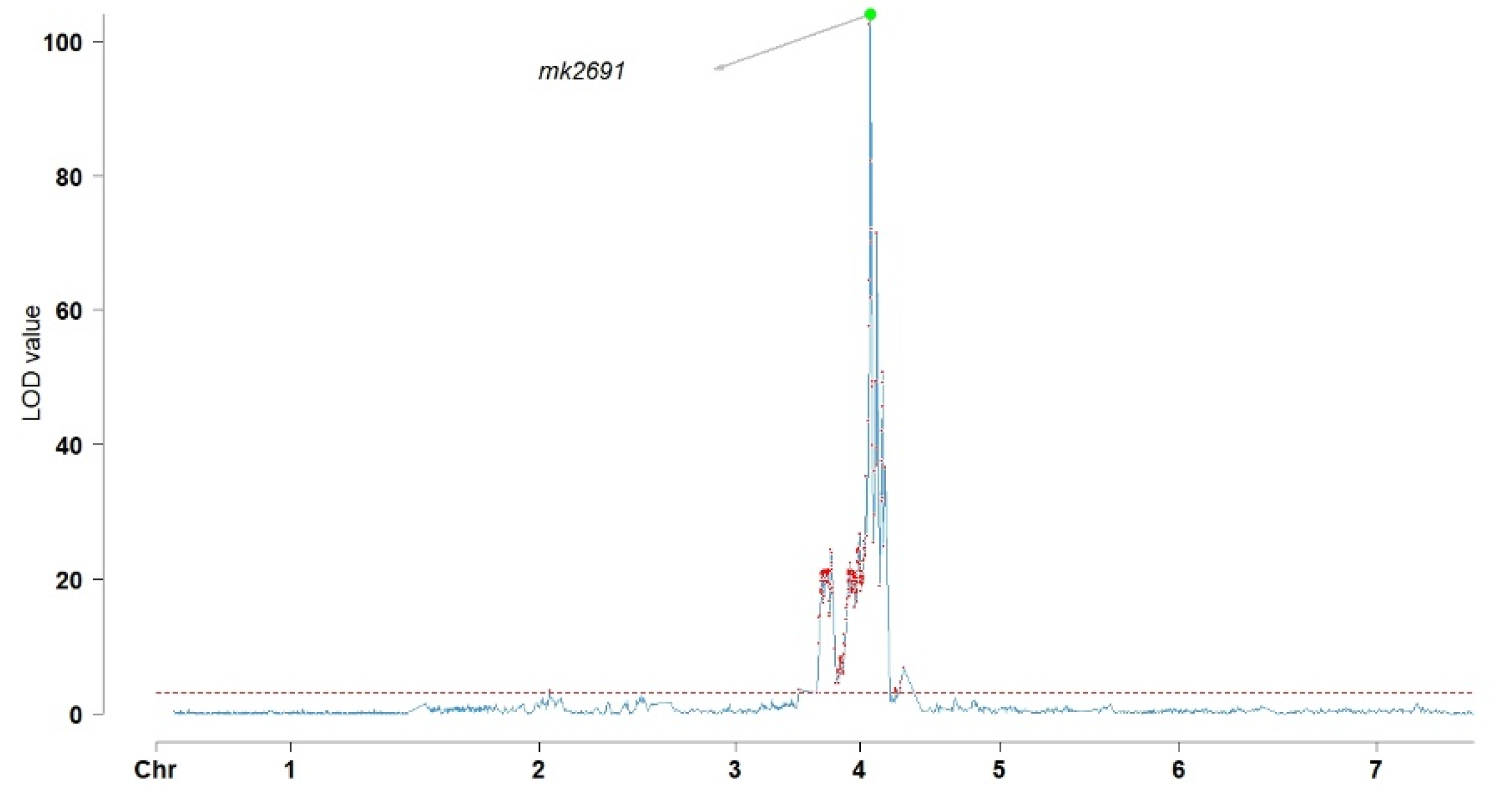
2.4. QTL Analysis of Flower Color-Related Traits
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Sample Preparation
4.3. Library Construction and GBS Sequencing
4.4. Sequencing Data Analysis and Quality Control
4.5. SNP Identification and Genotyping
4.6. Genetic Linkage Map Construction and Evaluation
4.7. Flower Color Data Collection and QTL Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Narayan, R.K.J. Discontinuous DNA variation in the evolution of plant species—The genus Lathyrus. Evol. 1982, 36, 877–891. [Google Scholar] [CrossRef] [PubMed]
- Roy, B. On the somatic chromosomes in Lathyrus. Cytologia 1936, 7, 424–430. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, D. Cytogenetic characterization of induced autotetraploids in grass pea (Lathyrus sativus L.). Caryologia 2010, 63, 62–72. [Google Scholar]
- Verma, S.C.; Ohri, D. Chromosome and nucclear phenotype in the legume Lathyrus sativus L. Cytologia 1979, 44, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Kislev, M.E. Origins of the cultivation of Lathyrus sativus and L. cicera (fabaceae). Econ. Bot. 1989, 43, 262–270. [Google Scholar] [CrossRef]
- Cullis, C.; Kunert, K.J. Unlocking the potential of orphan legumes. J. Exp. Bot. 2017, 68, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Emmrich, P.M.F.; Sarkar, A.; Njaci, I.; Kaithakottil, G.G.; Ellis, N.; Moore, C.; Edwards, A.; Heavens, D.; Waite, D.; Cheema, J.; et al. A draft genome of grass pea (Lathyrus sativus), a resilient diploid legume. bioRxiv 2020, 1–33. [Google Scholar] [CrossRef]
- Ramya, K.R.; Tripathi, K.; Pandey, A.; Barpete, S.; Gore, P.G.; Raina, A.P.; Khawar, K.M.; Swain, N.; Sarker, A. Rediscovering the potential of multifaceted orphan legume grasspea—A sustainable resource with high nutritional values. Front. Nutr. 2022, 8, 826208. [Google Scholar] [CrossRef]
- Sarkar, A.; Emmrich, P.M.F.; Sarker, A.; Zong, X.; Martin, C.; Wang, T.L. Grass pea: Remodeling an ancient insurance crop for climate resilience. In Genomic Designing of Climate-Smart Pulse Crops; Kole, C., Ramanna, R., Eds.; Springer Nature: Cham, Switzerland, 2019; pp. 426–434. [Google Scholar]
- Patto, M.C.V.; Fernández-Aparicio, M.; Moral, A.; Rubiales, D. Characterization of resistance to powdery mildew (Erysiphe pisi) in a germplasm collection of Lathyrus sativus. Plant Breed. 2006, 125, 308–310. [Google Scholar] [CrossRef]
- Patto, M.C.V.; Rubiales, D. Resistance to rust and powdery mildew in Lathyrus crops. Czech J. Genet. Plant 2014, 50, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, B.; Wójtowicz, T.; Makowski, W.; Jędrzejczyk, R.J.; Tokarz, K.M. What is the difference between the response of grass pea (Lathyrus sativus L.) to salinity and drought stress?—A physiological study. Agronomy 2020, 10, 833. [Google Scholar] [CrossRef]
- Wiraguna, E.; Malik, A.I.; Colmer, T.D.; Erskine, W. Tolerance of four grain legume species to waterlogging, hypoxia and anoxia at germination and recovery. AoB Plants 2021, 13, plab052. [Google Scholar] [CrossRef] [PubMed]
- Wiraguna, E.; Malik, A.I.C.; Timothy, D.; Erskine, W. Waterlogging tolerance of grass pea (Lathyrus sativus L.) at germination related to country of origin. Exp. Agr. 2020, 56, 837–850. [Google Scholar] [CrossRef]
- Zhou, L.; Cheng, W.; Hou, H.; Peng, R.; Hai, N.; Bian, Z.; Jiao, C.; Wang, C. Antioxidative responses and morpho-anatomical alterations for coping with flood-induced hypoxic stress in grass pea (Lathyrus sativus L.) in comparison with pea (Pisum sativum). J. Plant Growth Regul. 2016, 35, 690–700. [Google Scholar] [CrossRef]
- Lambein, F.; Travella, S.; Kuo, Y.-H.; Van Montagu, M.; Heijde, M. Grass pea (Lathyrus sativus L.): Orphan crop, nutraceutical or just plain food? Planta 2019, 250, 821–838. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Parihar, A.K.; Barpete, S.; Kumar, S.; Gupta, S. Current perspectives on reducing the β-ODAP content and improving potential agronomic traits in grass pea (Lathyrus sativus L.). Front. Plant Sci. 2021, 12, 703275. [Google Scholar] [CrossRef]
- Singh, S.S.; Rao, S.L.N. Lessons from neurolathyrism: A disease of the past & the future of Lathyrus sativus (Khesari dal). Indian J. Med. Res. 2013, 138, 32–37. [Google Scholar]
- Yan, Z.Y.; Spencer, P.S.; Li, Z.X.; Liang, Y.M.; Wang, Y.F.; Wang, C.Y.; Li, F.M. Lathyrus sativus (grass pea) and its neurotoxin ODAP. Phytochemistry 2006, 67, 107–121. [Google Scholar] [CrossRef]
- Sammour, R.H. Genetic diversity in Lathyrus sativus L. germplasm. Res. Rev. BioSciences 2014, 8, 325–336. [Google Scholar]
- Tadele, Z. African orphan crops under abiotic stresses: Challenges and opportunities. Hindawi Sci. 2018, 2018, 1451894. [Google Scholar] [CrossRef] [Green Version]
- Crow, J.F. A diamond anniversary: The first chromosomal map. Genetics 1988, 118, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.D.; Singh, A.K. Marker-Assisted Plant Breeding: Principles and Practices; Springer: New Delhi, India, 2015; pp. 19–25. [Google Scholar]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Qi, Z.M.; Huang, L.; Zhu, R.S.; Xin, D.W.; Liu, C.Y.; Han, X.; Jiang, H.W.; Hong, W.G.; Hu, G.H.; Zheng, H.K.; et al. A high-density genetic map for soybean based on specific length amplified fragment sequencing. PLoS ONE 2014, 9, e104871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddique, M.I.; Lee, J.H.; Ahn, J.H.; Kusumawardhani, M.K.; Safitri, R.; Harpenas, A.; Kwon, J.K.; Kang, B.C. Genotyping-by-sequencing-based QTL mapping reveals novel loci for Pepper yellow leaf curl virus (PepYLCV) resistance in Capsicum annuum. PLoS ONE 2022, 17, e0264026. [Google Scholar] [CrossRef]
- Wu, K.; Liu, H.; Yang, M.; Tao, Y.; Ma, H.; Wu, W.; Zuo, Y.; Zhao, Y. High-density genetic map construction and QTLs analysis of grain yield-related traits in sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Biol. 2014, 14, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yu, Z.; Qi, X.; Wang, Z.; Zheng, Y.; Ren, H.; Liang, S.; Zheng, X. Construction of a high-density genetic map and identification of leaf trait-related QTLs in chinese bayberry (Myrica rubra). Front. Plant Sci. 2021, 12, 675855. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Chowdry, M.A.; Slinkard, A.E. Natural outcrossing in grasspea. J. Hered. 1997, 88, 154–156. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Tian, L.; Zhang, J.; Huang, L.; Han, F.; Yan, S.; Wang, L.; Zheng, H.; Sun, J. Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content in Glycine max. BMC Genom. 2014, 15, 1086. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, M.A.; Slinkard, A.E. Linkage of random amplified polymorphic DNA, isozyme and morphological markers in grasspea (Lathyrus sativus). J. Agr. Sci. 1999, 133, 389–395. [Google Scholar] [CrossRef]
- Skiba, B.; Ford, R.; Pang, E.C.K. Construction of a linkage map based on a Lathyrus sativus backcross population and preliminary investigation of QTLs associated with resistance to ascochyta blight. Theor. Appl. Genet. 2004, 109, 1726–1735. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.; Polanco, C.; Rubiales, D.; Vaz Patto, M.C. The MLO1 powdery mildew susceptibility gene in Lathyrus species: The power of high-density linkage maps in comparative mapping and synteny analysis. Plant Genome 2021, 14, e20090. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.; Almeida, N.F.; Alves, M.L.; Horres, R.; Krezdorn, N.; Leitão, S.T.; Aznar-Fernández, T.; Rotter, B.; Winter, P.; Rubiales, D.; et al. First genetic linkage map of Lathyrus cicera based on RNA sequencing-derived markers: Key tool for genetic mapping of disease resistance. Hortic. Res. 2018, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, C.; Martins, D.; Rubiales, D.; Vaz Patto, M.C. Partial resistance against Erysiphe pisi and E. trifolii under different genetic control in Lathyrus cicera: Outcomes from a linkage mapping approach. Plant Dis. 2020, 104, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Ruggieri, V.; Pérez, S.; Alexiou, K.G.; Fernández, M.; Jahrmann, T.; Pujol, M.; Garcia-Mas, J. QTL mapping of melon fruit quality traits using a high-density GBS-based genetic map. BMC Plant Biol. 2018, 18, 324. [Google Scholar] [CrossRef] [PubMed]
- Stanton, M.L. Reproductive biology of petal color variants in wild populations of raphanus sativus: I. Pollinator response to color morphs. Am. J. Bot. 1987, 74, 178–187. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, Z.; Jiang, Y.; Wang, L.; Wu, Y.; Liao, J.; Yang, R.; Zhang, L. Inheritance and QTL mapping for flower color in salvia miltiorrhiza bunge. J. Hered. 2022, 113, esac012. [Google Scholar] [CrossRef]
- Rahman, M.M.; Kumar, J.; Rahman, M.A.; Afzal, M.A. Natural outcrossing in Lathyrus satlvus L. Indian J. Genet. Plant Breed. 1995, 55, 204–207. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Torkamaneh, D.; Laroche, J.; Belzile, F. Genome-Wide SNP Calling from Genotyping by Sequencing (GBS) Data: A Comparison of Seven Pipelines and Two Sequencing Technologies. PLoS ONE 2016, 11, e0161333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Pathak, M.; Singla, D.; Sharma, A.; Chhuneja, P.; Sarao, N.K. High-density gbs-based genetic linkage map construction and QTL identification associated with yellow mosaic disease resistance in bitter gourd (Momordica charantia L.). Front. Plant Sci. 2021, 12, 671620. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Kim, C.W.; Han, J.W.; Choi, H.J.; Han, K.; Lee, E.S.; Kim, D.S.; Lee, J.; Siddique, M.I.; Lee, H.E. Genotyping-by-sequencing derived genetic linkage map and quantitative trait loci for sugar content in onion (Allium cepa L.). Plants 2021, 10, 2267. [Google Scholar] [CrossRef] [PubMed]
- Brhane, H.; Haileselassie, T.; Tesfaye, K.; Ortiz, R.; Hammenhag, C.; Abreha, K.B.; Geleta, M. Novel gbs-based snp markers for finger millet and their use in genetic diversity analyses. Front. Genet. 2022, 13, 848627. [Google Scholar] [CrossRef]
- Li, L.; Yang, X.; Wang, Z.; Ren, M.; An, C.; Zhu, S.; Xu, R. Genetic mapping of powdery mildew resistance genes in wheat landrace Guizi 1 via genotyping by sequencing. Mol. Biol. Rep. 2022, 49, 4461–4468. [Google Scholar] [CrossRef]
- Nyirahabimana, F.; Shimira, F.; Zahid, G.; Solmaz, I. Recent status of genotyping by sequencing (GBS) technology in cucumber (Cucumis sativus L.): A review. Mol. Biol. Rep. 2022, 49, 5547–5554. [Google Scholar] [CrossRef]
- Jia, G.; Wang, H.; Tang, S.; Zhi, H.; Liu, S.; Wen, Q.; Qiao, Z.; Diao, X. Detection of genomic loci associated with chromosomal recombination using high-density linkage mapping in Setaria. Sci. Rep. 2017, 7, 15180. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Wang, W.; Gong, S.; Zuo, J.; Li, S.; Xu, S. High density linkage map construction and mapping of yield trait QTLs in maize (Zea mays) using the genotyping-by-sequencing (GBS) technology. Front. Plant Sci. 2017, 8, 706. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wei, T.; Zhong, Y.; Li, X.; Huang, J. Construction of a high-density genetic map of Ziziphus jujuba Mill. using genotyping by sequencing technology. Tree Genet. Genomes 2016, 12, 76. [Google Scholar] [CrossRef]
- Lavania, U.C. Chromosomal instability in Lathyrus sativus L. Theor. Appl. Genet. 1982, 62, 135–138. [Google Scholar] [CrossRef]
- Mollel, M.; Haileselassie, T. Flower colour inheritance in Nicotiana alata (Solanaceae) and its use as a genetic marker for gene flow studies. Huria. J. Open Univ. Tanzan. 2012, 12, 94–101. [Google Scholar]
- Nieuwhof, M.; Eijk, J.P.; Garretsen, G.; Eikelboom, W. Inheritance of flower colour in relation to inheritance of flower pigments in tulip (Tulipa L.). J. Genet. Breed. 1990, 44, 277–280. [Google Scholar]
- Lo, S.; Muñoz-Amatriaín, M.; Boukar, O.; Herniter, I.; Cisse, N.; Guo, Y.; Roberts, P.A.; Xu, S.; Fatokun, C.; Close, T.J. Identifcation of QTL controlling domestication-related traits in cowpea (Vigna unguiculata L. Walp). Sci. Rep. 2018, 8, 6216. [Google Scholar] [CrossRef] [Green Version]
- Nesi, N.; Debeaujon, I.; Jond, C.; Pelletier, G.; Caboche, M.; Lepiniec, L. The TT8 gene encodes a basic helix-loop-helix domain protein required for expression of DFR and BAN genes in Arabidopsis siliques. Plant Cell 2000, 12, 1863–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, A.A.; Clayton, M.K.; Brunet, J. Floral traits infl uencing plant attractiveness to three bee species: Consequences for plant reproductive success. Am. J. Bot. 2017, 104, 772–781. [Google Scholar] [CrossRef] [Green Version]
- Campbell, D.R.; Bischoff, M.; Lord, J.M.; Robertson, A.W. Flower color influences insect visitation in alpine New Zealand. Ecology 2010, 91, 2638–2649. [Google Scholar] [CrossRef]
- Ooijen, J. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 2011, 93, 343–349. [Google Scholar] [CrossRef]
- Narasimha, R. Kosambi and proper orthogonal decomposition. Resonance 2011, 16, 574–581. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| LG a | Number of SNPs | Genetic Distance (cM)/LG | Avg. Genetic Distance (cM)/LG | Max. Gap (cM)/LG |
|---|---|---|---|---|
| LG1 | 1240 | 1367.47 | 1.1 | 10.41 |
| LG2 | 476 | 1328.02 | 2.79 | 29.15 |
| LG3 | 354 | 751.70 | 2.12 | 18.29 |
| LG4 | 290 | 504.11 | 1.74 | 31.47 |
| LG5 | 367 | 924.76 | 2.52 | 29.16 |
| LG6 | 427 | 959.59 | 2.25 | 18.42 |
| LG7 | 382 | 1140.03 | 2.98 | 25.22 |
| Total | 3536 | 6975.68 | 2.21 | 31.47 |
| LG a | Number of SNPs in <5 cM | Number of SNPs in 5–10 cM | Number of SNPs in 10–20 cM | Number of SNPs in >20 cM | Number of SNPs in <5 cM Ratio (%) |
|---|---|---|---|---|---|
| LG1 | 1228 | 11 | 1 | 0 | 99.03 |
| LG2 | 399 | 51 | 22 | 4 | 83.82 |
| LG3 | 316 | 25 | 13 | 0 | 89.27 |
| LG4 | 272 | 11 | 5 | 2 | 93.79 |
| LG5 | 306 | 39 | 20 | 2 | 83.38 |
| LG6 | 380 | 35 | 12 | 0 | 88.99 |
| LG7 | 304 | 60 | 17 | 1 | 79.58 |
| Total | 3205 | 232 | 90 | 9 | 90.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, X.; Yang, T.; Wang, Y.; Liu, R.; Dong, X.; Zhao, J.; Han, J.; Zong, X.; Chang, J.; Liu, H. Construction of A GBS-Based High-Density Genetic Map and Flower Color-Related Loci Mapping in Grasspea (Lathyrus sativus L.). Plants 2022, 11, 2172. https://doi.org/10.3390/plants11162172
Hao X, Yang T, Wang Y, Liu R, Dong X, Zhao J, Han J, Zong X, Chang J, Liu H. Construction of A GBS-Based High-Density Genetic Map and Flower Color-Related Loci Mapping in Grasspea (Lathyrus sativus L.). Plants. 2022; 11(16):2172. https://doi.org/10.3390/plants11162172
Chicago/Turabian StyleHao, Xiaopeng, Tao Yang, Yan Wang, Rong Liu, Xue Dong, Jiandong Zhao, Jucai Han, Xuxiao Zong, Jianwu Chang, and Huiping Liu. 2022. "Construction of A GBS-Based High-Density Genetic Map and Flower Color-Related Loci Mapping in Grasspea (Lathyrus sativus L.)" Plants 11, no. 16: 2172. https://doi.org/10.3390/plants11162172