
Identification of Genes Associated with Nitrogen Stress Responses in Apple Leaves


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
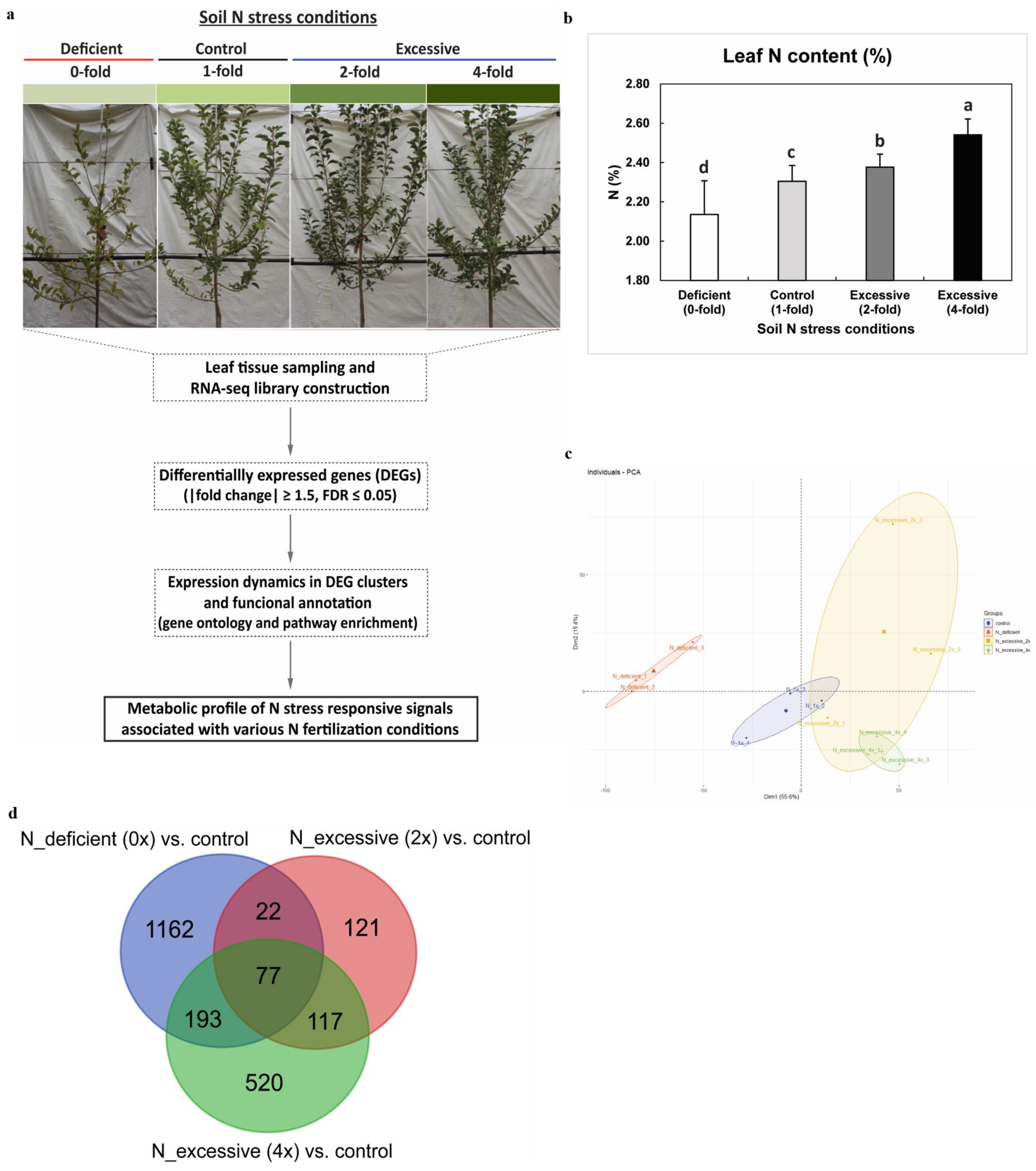
2.1. Transcriptome Profiling and the Leaf N Content
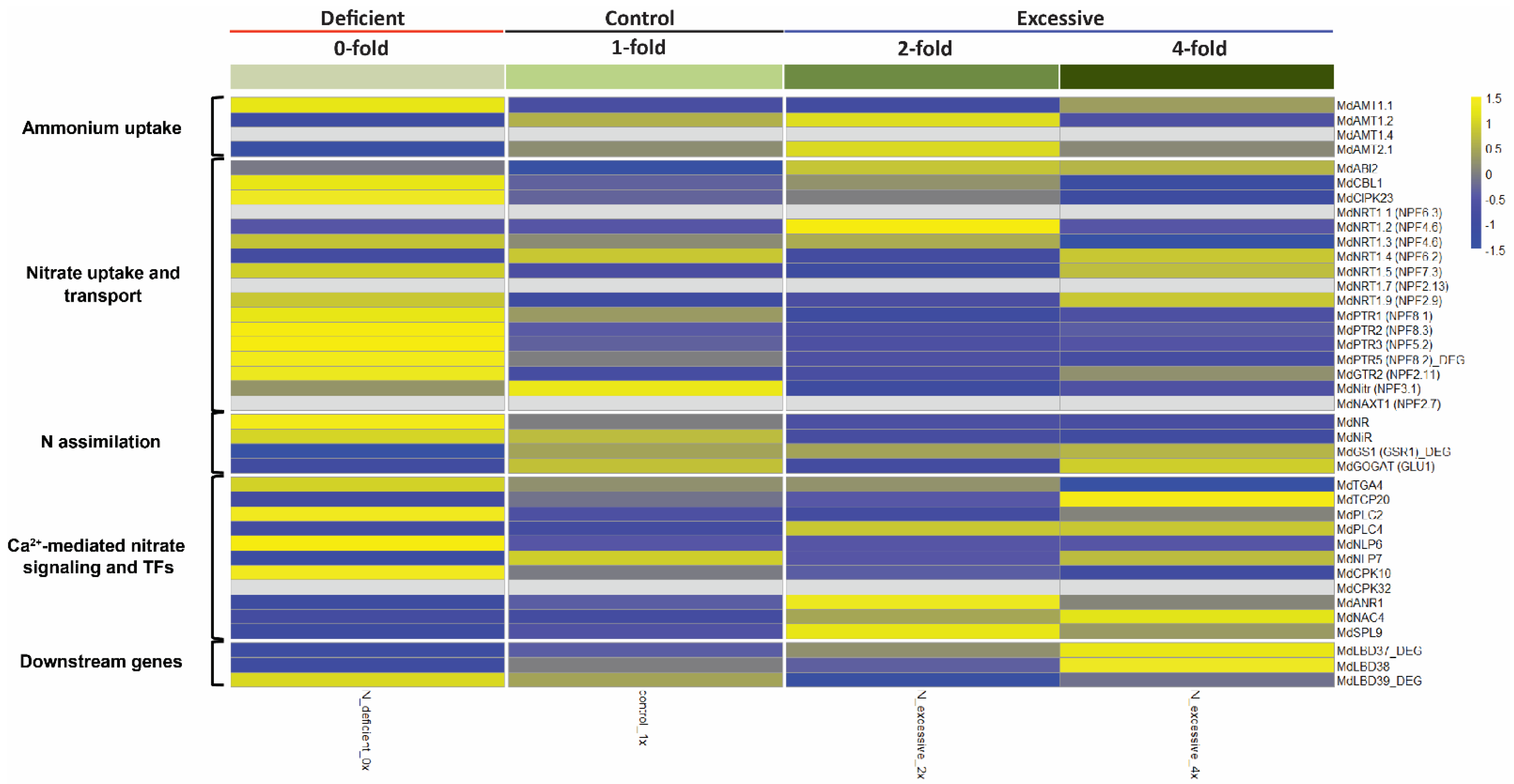
2.2. Analysis of Marker Genes Known to Be Associated with N Uptake, Assimilation, and Transport Pathways in Apple
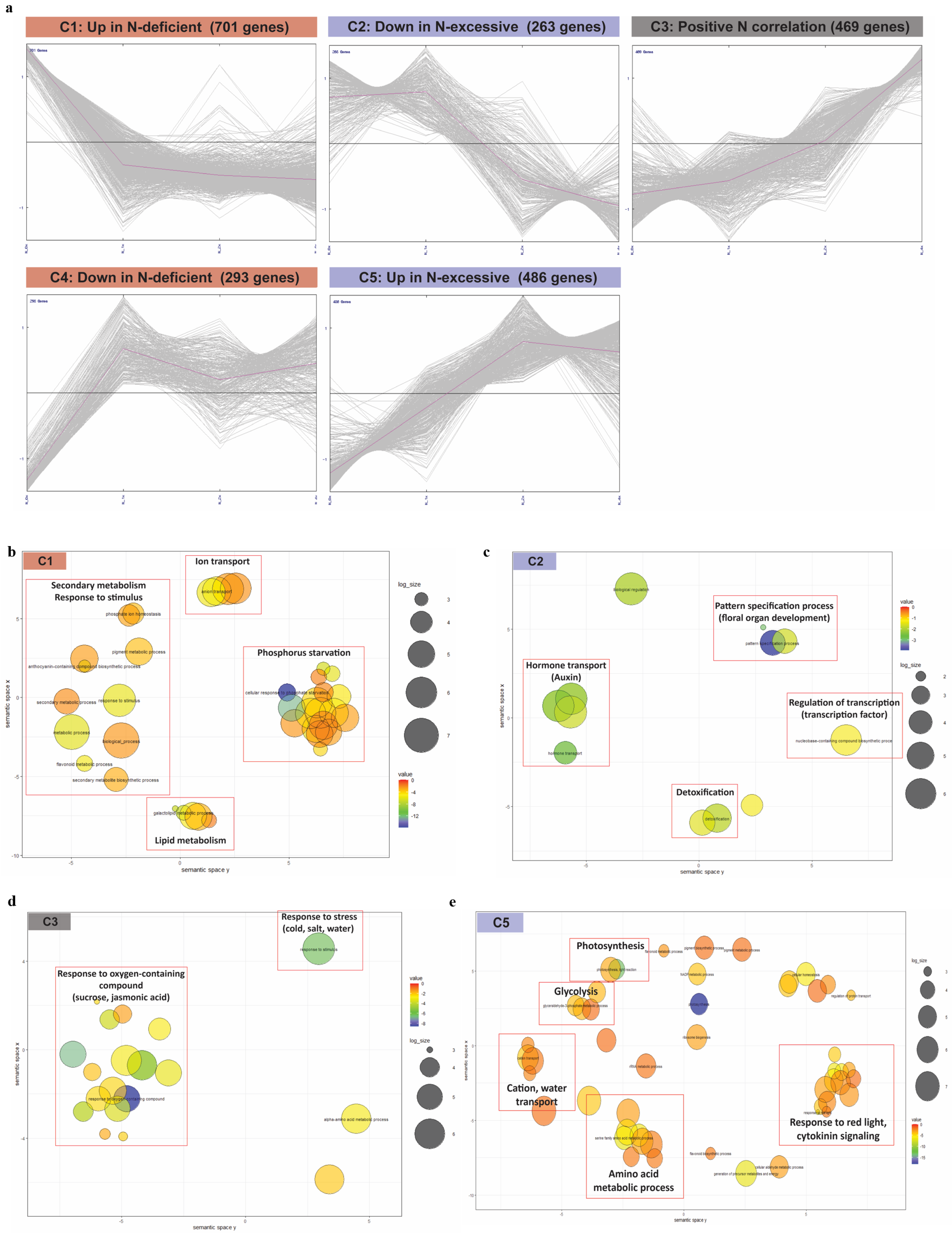
2.3. Gene Expression Dynamics in N Fertilization Groups Associated with Different N Stress Responses
2.4. Functional Annotation of DEGs in Five Clusters Associated with N Fertilization Stresses
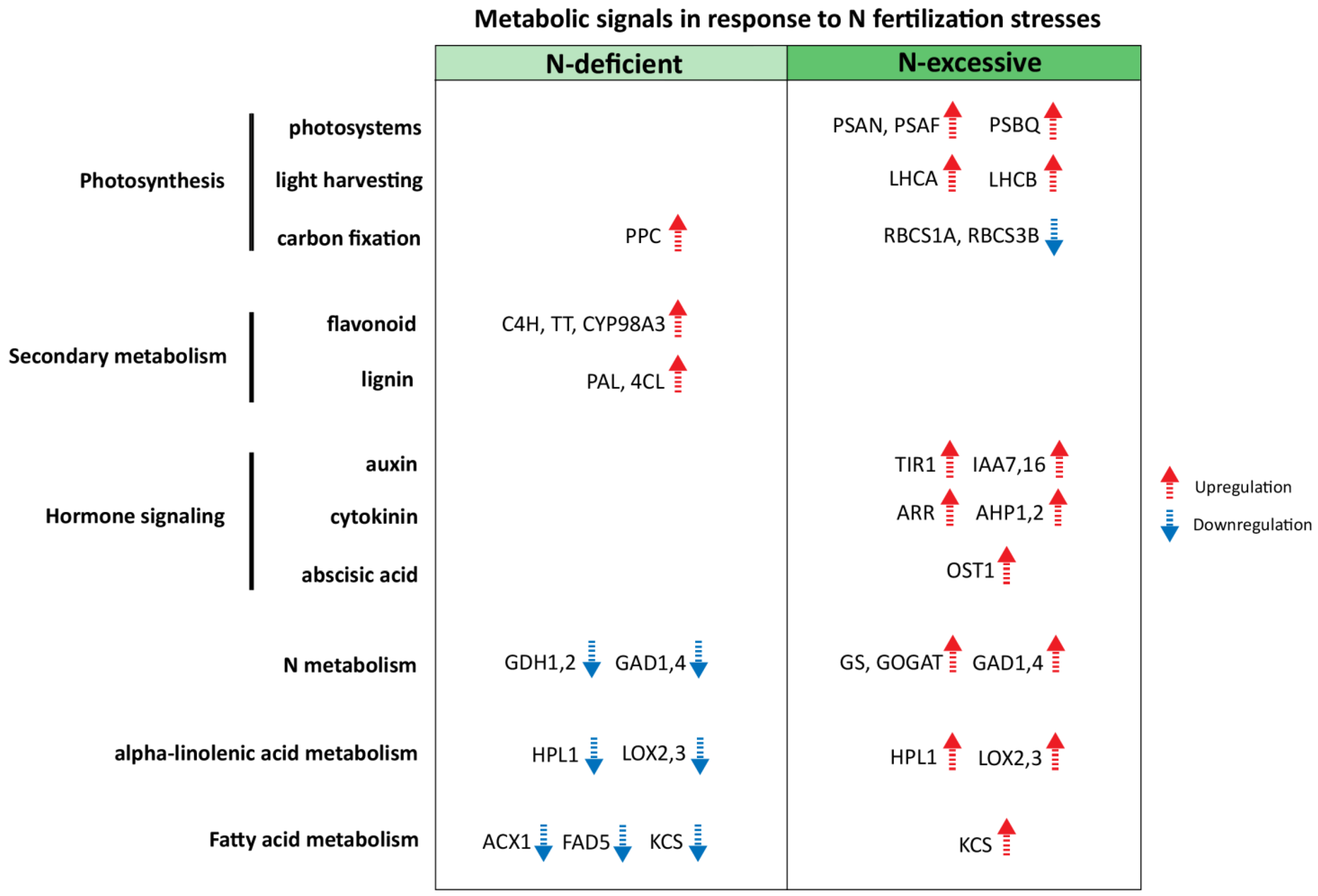
2.5. Identification of Candidate Metabolic Signals for N Stress Responses in Apple
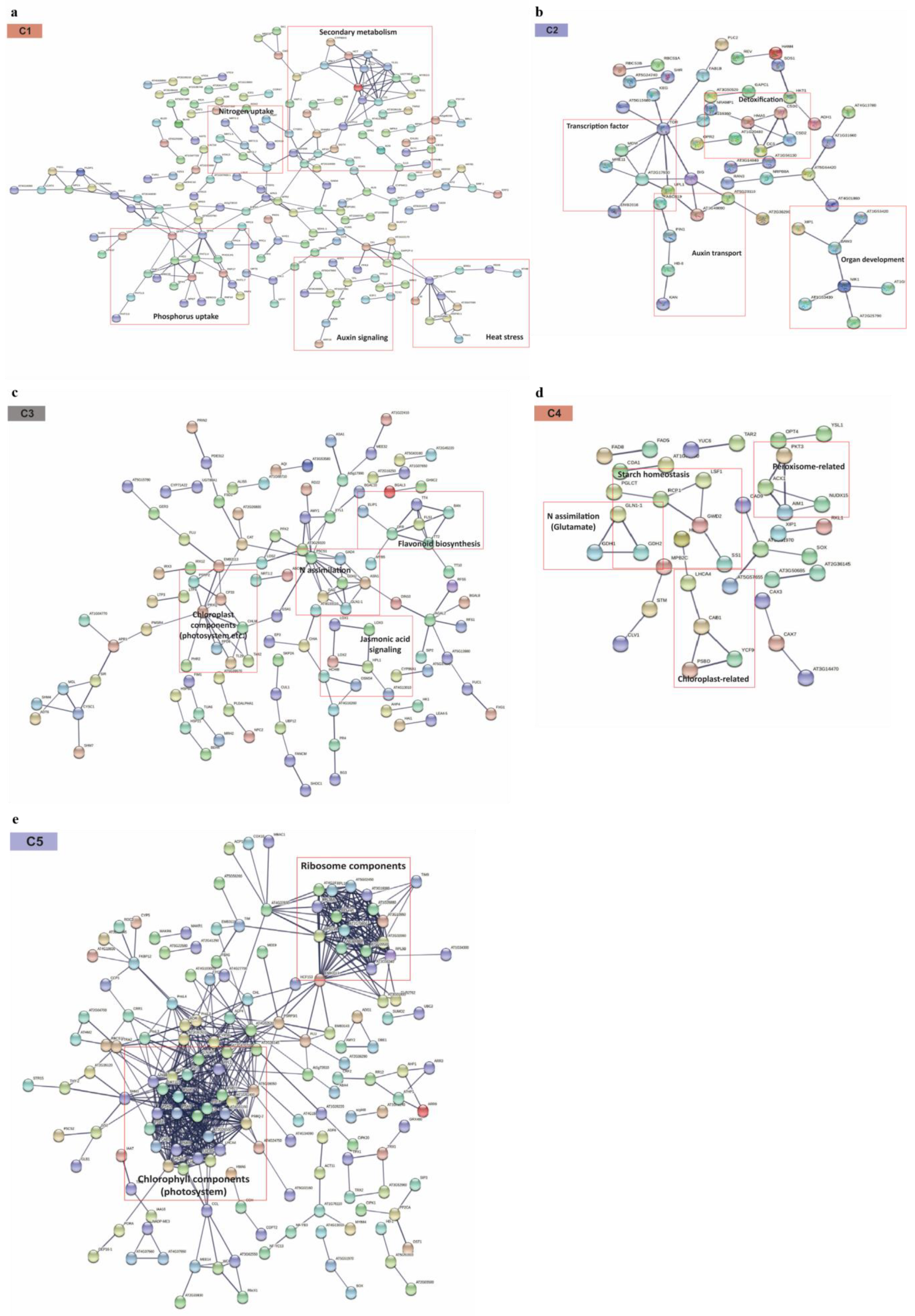
2.6. PPI Network Analysis Provides the Additional Clue for Functionally Important Metabolic Signals Responsive to N Stresses
3. Materials and Methods
3.1. Plant Materials and Sample Collection
3.2. Leaf N Content
3.3. RNA Extraction and Sequencing
3.4. Read Processing, Mapping, and Expression Data Analysis
3.5. Protein–Protein Interaction (PPI) Network Analysis
3.6. Quantitative Reverse Transcription PCR (qRT-PCR)
3.7. Quantification and Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lanquar, V.; Loqué, D.; Hörmann, F.; Yuan, L.; Bohner, A.; Engelsberger, W.R.; Lalonde, S.; Schulze, W.X.; von Wirén, N.; Frommer, W.B. Feedback inhibition of ammonium uptake by a phospho-Dependent allosteric mechanism in Arabidopsis. Plant Cell 2009, 21, 3610–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leran, S.; Varala, K.; Boyer, J.C.; Chiurazzi, M.; Crawford, N.; Daniel-Vedele, F.; David, L.; Dickstein, R.; Fernandez, E.; Forde, B.; et al. A unified nomenclature of NITRATE TRANSPORTER 1/PEPTIDE TRANSPORTER family members in plants. Trends Plant. Sci 2014, 19, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Kwang, W.J. International Review of Cell and Molecular Biology; Academic Press: Cambridge, MA, USA, 2014; Volume 310, pp. 1–37. [Google Scholar] [CrossRef]
- Xu, G.; Fan, X.; Miller, A.J. Plant nitrogen assimilation and use efficiency. Annu. Rev. Plant Biol. 2012, 63, 153–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes-Nesi, A.; Fernie, A.R.; Stitt, M. Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 2010, 3, 973–996. [Google Scholar] [CrossRef]
- Huarancca Reyes, T.; Scartazza, A.; Pompeiano, A.; Ciurli, A.; Lu, Y.; Guglielminetti, L.; Yamaguchi, J. Nitrate reductase modulation in response to changes in C/N balance and nitrogen source in Arabidopsis. Plant. Cell Physiol. 2018, 59, 1248–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.; Cheung, C. A dynamic multi-Tissue flux balance model captures carbon and nitrogen metabolism and optimal resource partitioning during Arabidopsis growth. Front. Plant Sci. 2018, 9, 884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, Y.-M.; Wang, R.-L.; Zhu, T.; Rothstein, S.J. Global transcription profiling reveals differential responses to chronic nitrogen stress and putative nitrogen regulatory components in Arabidopsis. BMC Genom. 2007, 8, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krapp, A.; Berthomé, R.; Orsel, M.; Mercey-Boutet, S.; Yu, A.; Castaings, L.; Elftieh, S.; Major, H.; Renou, J.-P.; Daniel-Vedele, F. Arabidopsis roots and shoots show distinct temporal adaptation patterns toward nitrogen starvation. Plant Physiol. 2011, 157, 1255–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chardon, F.; Barthélémy, J.; Daniel-Vedele, F.; Masclaux-Daubresse, C. Natural variation of nitrate uptake and nitrogen use efficiency in Arabidopsis thaliana cultivated with limiting and ample nitrogen supply. J. Exp. Bot. 2010, 61, 2293–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menz, J.; Range, T.; Trini, J.; Ludewig, U.; Neuhäuser, B. Molecular basis of differential nitrogen use efficiencies and nitrogen source preferences in contrasting Arabidopsis accessions. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, T.B.; Soares, J.D.; Lima, J.E.; Silva, J.C.; Ivamoto, S.T.; Baba, V.Y.; Souza, S.G.; Lorenzetti, A.P.; Paschoal, A.R.; Meda, A.R. An integrated analysis of mRNA and sRNA transcriptional profiles in Coffea arabica L. roots: Insights on nitrogen starvation responses. Funct. Integr. Genom. 2019, 19, 151–169. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.; Arnink, K.J.; Hrazdina, G. Interaction of nitrogen availability during bloom and light intensity during veraison. I. Effects on grapevine growth, fruit development, and ripening. Am. J. Enol. Vitic. 1998, 49, 333–340. [Google Scholar]
- Weber, K.; Burow, M. Nitrogen–essential macronutrient and signal controlling flowering time. Physiol. Plant. 2018, 162, 251–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, A.; Spinardi, A.; Maggiore, T.; Testoni, A.; Gallina, P.M. Effect of nitrogen fertilisation levels on melon fruit quality at the harvest time and during storage. J. Sci. Food Agric. 2008, 88, 707–713. [Google Scholar] [CrossRef]
- Carranca, C.; Brunetto, G.; Tagliavini, M. Nitrogen nutrition of fruit trees to reconcile productivity and environmental concerns. Plants 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albornoz, F. Crop responses to nitrogen overfertilization: A review. Sci. Hortic. 2016, 205, 79–83. [Google Scholar] [CrossRef]
- Sete, P.B.; Comin, J.J.; Ciotta, M.N.; Salume, J.A.; Thewes, F.; Brackmann, A.; Toselli, M.; Nava, G.; Rozane, D.E.; Loss, A. Nitrogen fertilization affects yield and fruit quality in pear. Sci. Hortic. 2019, 258, 108782. [Google Scholar] [CrossRef]
- Neilsen, G.; Neilsen, D.; Peryea, F.; Fallahi, E.; Fallahi, B. Effects of mineral nutrition on fruit quality and nutritional disorders in apples. In Proceedings of the VI International Symposium on Mineral Nutrition of Fruit Crops 868, Faro, Portugal, 19–21 May 2008; pp. 49–60. [Google Scholar]
- Zhao, H.; Sun, S.; Zhang, L.; Yang, J.; Wang, Z.; Ma, F.; Li, M. Carbohydrate metabolism and transport in apple roots under nitrogen deficiency. Plant. Physiol. Biochem. 2020, 155, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Vimolmangkang, S.; Soria-Guerra, R.E.; Rosales-Mendoza, S.; Zheng, D.; Lygin, A.V.; Korban, S.S. Ectopic expression of apple F3′ H genes contributes to anthocyanin accumulation in the Arabidopsis tt7 mutant grown under nitrogen stress. Plant Physiol. 2010, 153, 806–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Zhang, J.; Zhang, Q.; Li, X.; Li, M.; Yang, Y.; Zhou, J.; Wei, Q.; Zhou, B. Integrative physiological, transcriptome, and metabolome analysis reveals the effects of nitrogen sufficiency and deficiency conditions in apple leaves and roots. Environ. Exp. Bot. 2021, 192, 104633. [Google Scholar] [CrossRef]
- Zhang, X.; Tahir, M.M.; Li, S.; Mao, J.; Nawaz, M.A.; Liu, Y.; Li, K.; Xing, L.; Niu, J.; Zhang, D. Transcriptome analysis reveals the inhibitory nature of high nitrate during adventitious roots formation in the apple rootstock. Physiol. Plant. 2021, 173, 867–882. [Google Scholar] [CrossRef]
- Lee, Y.; Kweon, H.J.; Park, M.-Y.; Lee, D. Field Assessment of Macronutrients and Nitrogen in Apple Leaves Using a Chlorophyll Meter. Hort. Technol. 2019, 29, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Daccord, N.; Celton, J.-M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; Van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R. High-Quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Armijo, G.; Gutierrez, R.A. Emerging Players in the Nitrate Signaling Pathway. Mol. Plant 2017, 10, 1019–1022. [Google Scholar] [CrossRef]
- Undurraga, S.F.; Ibarra-Henríquez, C.; Fredes, I.; Álvarez, J.M.; Gutiérrez, R.A. Nitrate signaling and early responses in Arabidopsis roots. J. Exp. Bot. 2017, 68, 2541–2551. [Google Scholar] [CrossRef]
- Wang, R.; Tischner, R.; Gutiérrez, R.A.; Hoffman, M.; Xing, X.; Chen, M.; Coruzzi, G.; Crawford, N.M. Genomic analysis of the nitrate response using a nitrate reductase-Null mutant of Arabidopsis. Plant Physiol. 2004, 136, 2512–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Sasaki, Y.; Ida, S.; Morikawa, H. Nitrite reductase gene enrichment improves assimilation of NO2 in Arabidopsis. Plant Physiol. 2001, 126, 731–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, N.; Ishiyama, K.; Matsuoka, K.; Maru, I.; Hayakawa, T.; Yamaya, T.; Kojima, S. NADH-dependent glutamate synthase plays a crucial role in assimilating ammonium in the Arabidopsis root. Physiol. Plant. 2014, 152, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Ullah, Z.; Xu, F.; An, L.; Aluko, O.O.; Wang, Q.; Liu, H. Nitrate Signaling, Functions, and Regulation of Root System Architecture: Insights from Arabidopsis Thaliana. Genes 2020, 11, 633. [Google Scholar] [CrossRef]
- Liu, K.-H.; Niu, Y.; Konishi, M.; Wu, Y.; Du, H.; Chung, H.S.; Li, L.; Boudsocq, M.; McCormack, M.; Maekawa, S. Discovery of nitrate–CPK–NLP signalling in central nutrient–growth networks. Nature 2017, 545, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Forde, B.G. An Arabidopsis MADS box gene that controls nutrient-Induced changes in root architecture. Science 1998, 279, 407–409. [Google Scholar] [CrossRef]
- Krouk, G.; Mirowski, P.; LeCun, Y.; Shasha, D.E.; Coruzzi, G.M. Predictive network modeling of the high-Resolution dynamic plant transcriptome in response to nitrate. Genome Biol. 2010, 11, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, E.A.; Moyano, T.C.; Riveras, E.; Contreras-López, O.; Gutiérrez, R.A. Systems approaches map regulatory networks downstream of the auxin receptor AFB3 in the nitrate response of Arabidopsis thaliana roots. Proc. Natl. Acad. Sci. USA 2013, 110, 12840–12845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, G.; Tohge, T.; Matsuda, F.; Saito, K.; Scheible, W.-R.D. Members of the LBD family of transcription factors repress anthocyanin synthesis and affect additional nitrogen responses in Arabidopsis. Plant Cell 2009, 21, 3567–3584. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Konishi, M.; Kiba, T.; Sakuraba, Y.; Sawaki, N.; Kurai, T.; Ueda, Y.; Sakakibara, H.; Yanagisawa, S. A NIGT1-Centred transcriptional cascade regulates nitrate signalling and incorporates phosphorus starvation signals in Arabidopsis. Nat. Commun. 2018, 9, 1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Jiang, Z.; Wang, W.; Qiu, Y.; Zhang, Z.; Liu, Y.; Li, A.; Gao, X.; Liu, L.; Qian, Y. Nitrate–NRT1. 1B–SPX4 cascade integrates nitrogen and phosphorus signalling networks in plants. Nat. Plants 2019, 5, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Chu, C. Nitrogen–Phosphorus interplay: Old story with molecular tale. New Phytol. 2020, 225, 1455–1460. [Google Scholar] [CrossRef] [Green Version]
- Krouk, G.; Lacombe, B.; Bielach, A.; Perrine-Walker, F.; Malinska, K.; Mounier, E.; Hoyerova, K.; Tillard, P.; Leon, S.; Ljung, K. Nitrate-Regulated auxin transport by NRT1. 1 defines a mechanism for nutrient sensing in plants. Dev. Cell 2010, 18, 927–937. [Google Scholar] [CrossRef]
- Teng, Y.; Liang, Y.; Wang, M.; Mai, H.; Ke, L. Nitrate Transporter 1.1 is involved in regulating flowering time via transcriptional regulation of FLOWERING LOCUS C in Arabidopsis thaliana. Plant Sci. 2019, 284, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tillard, P.; Gojon, A.; Maurel, C. Dual regulation of root hydraulic conductivity and plasma membrane aquaporins by plant nitrate accumulation and high-Affinity nitrate transporter NRT2. 1. Plant Cell Physiol. 2016, 57, 733–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araus, V.; Swift, J.; Alvarez, J.M.; Henry, A.; Coruzzi, G.M. A balancing act: How plants integrate nitrogen and water signals. J. Exp. Bot. 2020, 71, 4442–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verly, C.; Djoman, A.C.R.; Rigault, M.; Giraud, F.; Rajjou, L.; Saint-Macary, M.-E.; Dellagi, A. Plant Defense Stimulator Mediated Defense Activation Is Affected by Nitrate Fertilization and Developmental Stage in Arabidopsis thaliana. Front. Plant Sci. 2020, 11, 583. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-l.; Acedo, G.N.; Cristinsin, M.; Conkling, M.A. Sucrose mimics the light induction of Arabidopsis nitrate reductase gene transcription. Proc. Natl. Acad. Sci. USA 1992, 89, 1861–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, C.; Palacios-Rojas, N.; Feil, R.; Stitt, M. Regulation of secondary metabolism by the carbon–Nitrogen status in tobacco: Nitrate inhibits large sectors of phenylpropanoid metabolism. Plant J. 2006, 46, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Izumi, M.; Tsunoda, H.; Suzuki, Y.; Makino, A.; Ishida, H. RBCS1A and RBCS3B, two major members within the Arabidopsis RBCS multigene family, function to yield sufficient Rubisco content for leaf photosynthetic capacity. J. Exp. Bot. 2012, 63, 2159–2170. [Google Scholar] [CrossRef]
- Warren, C.R.; Dreyer, E.; Adams, M.A. Photosynthesis-Rubisco relationships in foliage of Pinus sylvestris in response to nitrogen supply and the proposed role of Rubisco and amino acids as nitrogen stores. Trees 2003, 17, 359–366. [Google Scholar] [CrossRef]
- Shi, J.; Yi, K.; Liu, Y.; Xie, L.; Zhou, Z.; Chen, Y.; Hu, Z.; Zheng, T.; Liu, R.; Chen, Y. Phospho enol pyruvate carboxylase in Arabidopsis leaves plays a crucial role in carbon and nitrogen metabolism. Plant Physiol. 2015, 167, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Kiba, T.; Kudo, T.; Kojima, M.; Sakakibara, H. Hormonal control of nitrogen acquisition: Roles of auxin, abscisic acid, and cytokinin. J. Exp. Bot. 2011, 62, 1399–1409. [Google Scholar] [CrossRef]
- Vega, A.; O’Brien, J.A.; Gutiérrez, R.A. Nitrate and hormonal signaling crosstalk for plant growth and development. Curr. Opin. Plant Biol. 2019, 52, 155–163. [Google Scholar] [CrossRef]
- Gray, W.M.; Kepinski, S.; Rouse, D.; Leyser, O.; Estelle, M. Auxin regulates SCF TIR1-Dependent degradation of AUX/IAA proteins. Nature 2001, 414, 271–276. [Google Scholar] [CrossRef]
- Poitout, A.; Crabos, A.; Petřík, I.; Novák, O.; Krouk, G.; Lacombe, B.; Ruffel, S. Responses to systemic nitrogen signaling in Arabidopsis roots involve trans-Zeatin in shoots. Plant Cell 2018, 30, 1243–1257. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Wang, T.; Ju, C.; Deng, J.; Zhang, T.; Li, M.; Tian, H.; Wang, C. Abscisic acid signaling negatively regulates nitrate uptake via phosphorylation of NRT1. 1 by SnRK2s in Arabidopsis. J. Integr. Plant Biol. 2021, 63, 597–610. [Google Scholar] [CrossRef]
- Fait, A.; Nesi, A.N.; Angelovici, R.; Lehmann, M.; Pham, P.A.; Song, L.; Haslam, R.P.; Napier, J.A.; Galili, G.; Fernie, A.R. Targeted enhancement of glutamate-To-γ-Aminobutyrate conversion in Arabidopsis seeds affects carbon-Nitrogen balance and storage reserves in a development-Dependent manner. Plant Physiol. 2011, 157, 1026–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouche, N.; Fromm, H. GABA in plants: Just a metabolite? Trends Plant Sci. 2004, 9, 110–115. [Google Scholar] [CrossRef]
- Barbosa, J.M.; Singh, N.K.; Cherry, J.H.; Locy, R.D. Nitrate uptake and utilization is modulated by exogenous γ-Aminobutyric acid in Arabidopsis thaliana seedlings. Plant Physiol. Biochem. 2010, 48, 443–450. [Google Scholar] [CrossRef]
- Morikawa, H.; Takahashi, M.; Sakamoto, A.; Matsubara, T.; Arimura, G.-I.; Kawamura, Y.; Fukunaga, K.; Fujita, K.; Sakurai, N.; Hirata, T. Formation of unidentified nitrogen in plants: An implication for a novel nitrogen metabolism. Planta 2004, 219, 14–22. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Report. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Liu, C.-H.; Di, Y.P. Analysis of RNA sequencing data using CLC Genomics Workbench. In Molecular Toxicology Protocols; Springer: Berlin/Heidelberg, Germany, 2020; pp. 61–113. [Google Scholar]
- Howe, E.A.; Sinha, R.; Schlauch, D.; Quackenbush, J. RNA-Seq analysis in MeV. Bioinformatics 2011, 27, 3209–3210. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Lee, T.; Cheng, C.-H.; Buble, K.; Zheng, P.; Yu, J.; Humann, J.; Ficklin, S.P.; Gasic, K.; Scott, K. 15 years of GDR: New data and functionality in the Genome Database for Rosaceae. Nucleic Acids Res. 2019, 47, D1137–D1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P. The STRING database in 2021: Customizable protein–Protein networks, and functional characterization of user-Uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.; Ireland, H.S.; Crowhurst, R.; Luo, Z.; Watson, A.E.; Foster, T.; Gapper, N.; Giovanonni, J.J.; Mattheis, J.P.; Watkins, C. Selection of low-Variance expressed Malus x domestica (apple) genes for use as quantitative PCR reference genes (housekeepers). Tree Genet. Genomes 2014, 10, 751–759. [Google Scholar] [CrossRef]
- Tellmann, G. The E-Method: A highly accurate technique for gene-Expression analysis. Nat. Methods 2006, 3, i–ii. [Google Scholar] [CrossRef]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A Fast and Simple Web Server for Genome Scale Functional Annotation of Plant Sequence Data. Plant Cell Environ. 2014, 5, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.; Do, V.G.; Kim, S.; Kweon, H. Identification of Genes Associated with Nitrogen Stress Responses in Apple Leaves. Plants 2021, 10, 2649. https://doi.org/10.3390/plants10122649
Lee Y, Do VG, Kim S, Kweon H. Identification of Genes Associated with Nitrogen Stress Responses in Apple Leaves. Plants. 2021; 10(12):2649. https://doi.org/10.3390/plants10122649
Chicago/Turabian StyleLee, Youngsuk, Van Giap Do, Seonae Kim, and Hunjoong Kweon. 2021. "Identification of Genes Associated with Nitrogen Stress Responses in Apple Leaves" Plants 10, no. 12: 2649. https://doi.org/10.3390/plants10122649