
Attenuation of Dopaminergic Neurodegeneration in a C. elegans Parkinson’s Model through Regulation of Xanthine Dehydrogenase (XDH-1) Expression by the RNA Editase, ADR-2
 , and
, and 
Abstract
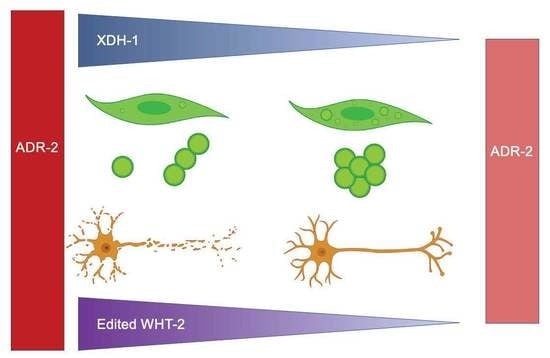
:
1. Introduction
2. Materials and Methods
2.1. C. elegans Strains
2.2. RNAi and Analysis of Protein Misfolding
2.3. GO Term Analysis of Candidate Genes
2.4. Analysis of Neurodegeneration
2.5. Body Wall Muscle and Dopaminergic Neuron Image Acquisition
2.6. RT-qPCR of α-syn Expression
- α-syn Forward: 5′-ATGTAGGCTCCAAAACCAAGG-3′
- α-syn Reverse: 5′-ACTGCTCCTCCAACATTTGTC-3′
- snb-1 Forward: 5′-CCGGATAAGACCATCTTGACG-3′
- snb-1 Reverse: 5′-GACGACTTCATCAACCTGAGC-3′
- tba-1 Forward: 5′-ATCTCTGCTGACAAGGCTTAC-3′
- tba-1 Reverse: 5′-GTACAAGAGGCAAACAGCCAT-3′
- ama-1 Forward: 5′-TCCTACGATGTATCGAGGCAA-3′
- ama-1 Reverse: 5′-CTCCCTCCGGTGTAATAATGA-3′
2.7. Quantification of ROS
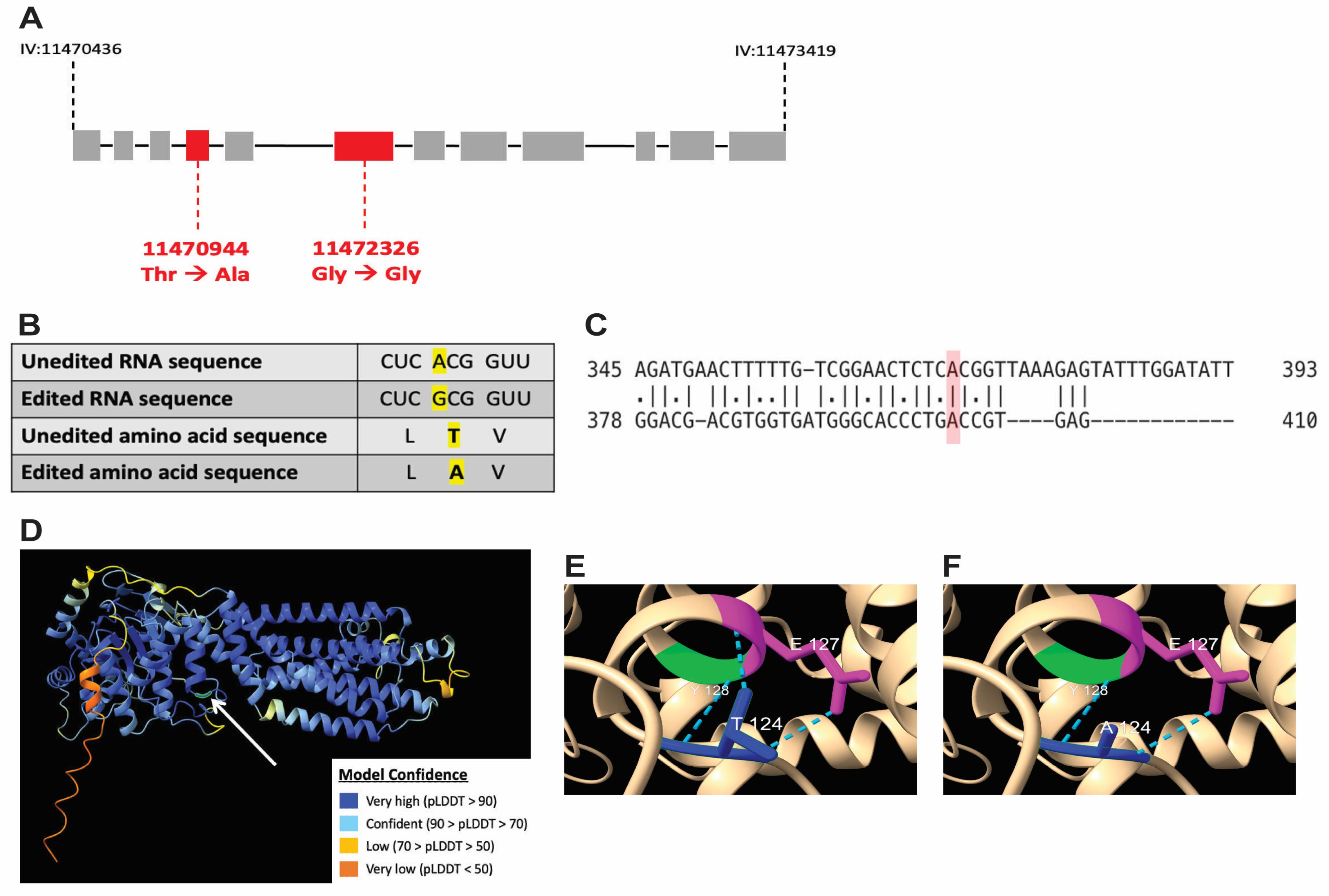
2.8. In Silico Stuctural Modeling of the WHT-2 Protein as Predicted for Unedited vs. Edited wht-2
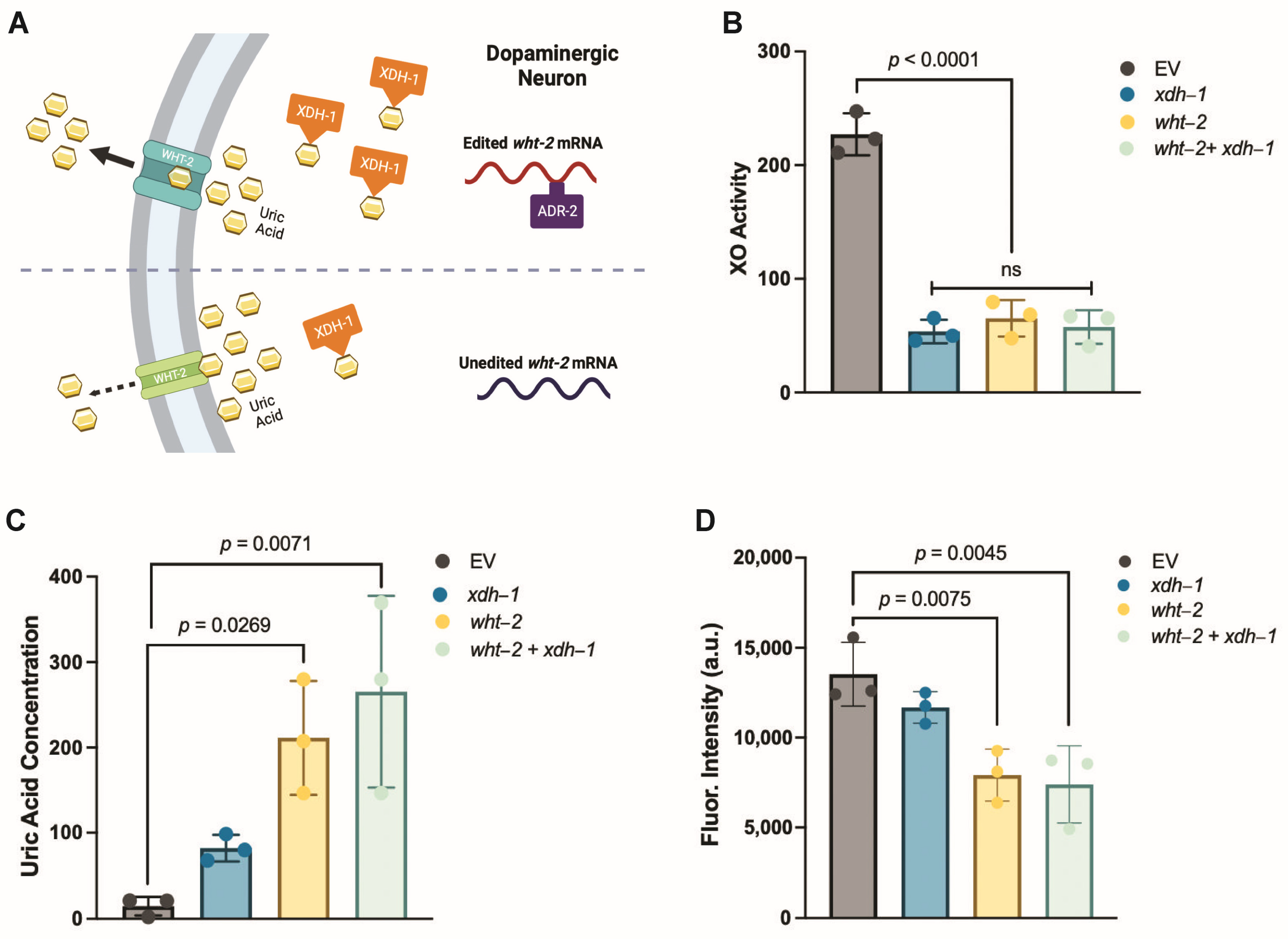
2.9. Xanthine Oxidase Activity and Uric Acid Assays
2.10. Statistical Analysis
3. Results
3.1. Genes Regulated by ADR-2 Alter α-syn Misfolding
3.2. Modifiers of Protein Misfolding Are Associated with FAD and Iron Binding
3.3. Select ADR-2-Regulated Modifiers of Protein Misfolding Impact Neurodegeneration When Knocked Down in Dopamine Neurons
3.4. Loss of XDH Leads to Neuroprotection through the Reduction in ROS
3.5. A Target of ADR-2 Editing, wht-2, Interacts in a Network with xdh-1 to Mediate PD-Associated Pathologies
3.6. A-to-I RNA Editing of wht-2 Is Predicted to Alter WHT-2 Protein Structure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Mythri, R.B.; Jagatha, B.; Pradhan, N.; Andersen, J.; Bharath, M.M.S. Mitochondrial Complex I Inhibition in Parkinson’s Disease: How Can Curcumin Protect Mitochondria? Antioxid. Redox Signal. 2007, 9, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, N.; Tanaka, M.; Ozawa, T.; Mizuno, Y. Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann. Neurol. 1991, 30, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Hattingen, E.; Magerkurth, J.; Pilatus, U.; Mozer, A.; Seifried, C.; Steinmetz, H.; Zanella, F.; Hilker, R. Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson’s disease. Brain 2009, 132, 3285–3297. [Google Scholar] [CrossRef] [Green Version]
- Cook, C.; Stetler, C.; Petrucelli, L. Disruption of Protein Quality Control in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009423. [Google Scholar] [CrossRef] [Green Version]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Jia, C.; Li, T.; Le, W. Hot Topics in Recent Parkinson’s Disease Research: Where We are and Where We Should Go. Neurosci. Bull. 2021, 37, 1735–1744. [Google Scholar] [CrossRef]
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s Disease. Subcell. Biochem. 2012, 65, 389–455. [Google Scholar] [CrossRef] [Green Version]
- Wang, I.X.; So, E.; Devlin, J.L.; Zhao, Y.; Wu, M.; Cheung, V.G. ADAR Regulates RNA Editing, Transcript Stability, and Gene Expression. Cell Rep. 2013, 5, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Reich, D.P.; Tyc, K.M.; Bass, B.L. C. elegans ADARs Antagonize Silencing of Cellular dsRNAs by the Antiviral RNAi Pathway. Genes Dev. 2018, 32, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, D.P.; Aruscavage, P.J.; Bass, B.L. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc. Natl. Acad. Sci. USA 2002, 99, 7906–7911. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Okada, S.; Sakurai, M. Adenosine-to-inosine RNA editing in neurological development and disease. RNA Biol. 2021, 18, 999–1013. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Khan, A.; Paro, S.; McGurk, L.; Sambrani, N.; Hogg, M.C.; Brindle, J.; Pennetta, G.; Keegan, L.P.; O’Connell, M.A. Membrane and synaptic defects leading to neurodegeneration in Adar mutant Drosophila are rescued by increased autophagy. BMC Biol. 2020, 18, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozdyshev, D.V.; Zharikova, A.A.; Medvedeva, M.V.; Muronetz, V.I. Differential Analysis of A-to-I mRNA Edited Sites in Parkinson’s Disease. Genes 2021, 13, 14. [Google Scholar] [CrossRef]
- Calahorro, F.; Ruiz-Rubio, M. Caenorhabditis elegans as an experimental tool for the study of complex neurological diseases: Parkinson’s disease, Alzheimer’s disease and autism spectrum disorder. Invertebr. Neurosci. 2011, 11, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, K.A.; Willicott, C.W.; Caldwell, G.A. Modeling neurodegeneration in Caenorhabditis elegans. Dis. Model. Mech. 2020, 13, dmm046110. [Google Scholar] [CrossRef]
- Towlson, E.K.; Vértes, P.E.; Ahnert, S.E.; Schafer, W.R.; Bullmore, E.T. The Rich Club of the C. elegans Neuronal Connectome. J. Neurosci. 2013, 33, 6380–6387. [Google Scholar] [CrossRef] [Green Version]
- Tonkin, L.A.; Saccomanno, L.; Morse, D.P.; Brodigan, T.; Krause, M.; Bass, B.L. RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J. 2002, 21, 6025–6035. [Google Scholar] [CrossRef] [Green Version]
- Washburn, M.C.; Kakaradov, B.; Sundararaman, B.; Wheeler, E.; Hoon, S.; Yeo, G.W.; Hundley, H.A. The dsRBP and Inactive Editor ADR-1 Utilizes dsRNA Binding to Regulate A-to-I RNA Editing across the C. elegans Transcriptome. Cell Rep. 2014, 6, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Rajendren, S.; Manning, A.C.; Al-Awadi, H.; Yamada, K.; Takagi, Y.; Hundley, H.A. A protein–protein interaction underlies the molecular basis for substrate recognition by an adenosine-to-inosine RNA-editing enzyme. Nucleic Acids Res. 2018, 46, 9647–9659. [Google Scholar] [CrossRef] [Green Version]
- Arribere, J.A.; Kuroyanagi, H.; Hundley, H.A. mRNA Editing, Processing and Quality Control in Caenorhabditis elegans. Genetics 2021, 215, 531–568. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine dehydrogenase/xanthine oxidase and oxidative stress. Age 1997, 20, 127–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine oxidoreductase: One enzyme for multiple physiological tasks. Redox Biol. 2021, 41, 101882. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol.-Endocrinol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef]
- Kim, W.S.; Weickert, C.S.; Garner, B. Role of ATP-Binding Cassette Transporters in Brain Lipid Transport and Neurological Disease. J. Neurochem. 2008, 104, 1145–1166. [Google Scholar] [CrossRef]
- Kobuchi, H.; Moriya, K.; Ogino, T.; Fujita, H.; Inoue, K.; Shuin, T.; Yasuda, T.; Utsumi, K.; Utsumi, T. Mitochondrial Localization of ABC Transporter ABCG2 and Its Function in 5-Aminolevulinic Acid-Mediated Protoporphyrin IX Accumulation. PLoS ONE 2012, 7, e50082. [Google Scholar] [CrossRef] [Green Version]
- Kukal, S.; Guin, D.; Rawat, C.; Bora, S.; Mishra, M.K.; Sharma, P.; Paul, P.R.; Kanojia, N.; Grewal, G.K.; Kukreti, S.; et al. Multidrug efflux transporter ABCG2: Expression and regulation. Cell. Mol. Life Sci. 2021, 78, 6887–6939. [Google Scholar] [CrossRef]
- Stiernagle, T. Maintenance of C. elegans. In WormBook; The C. elegans Research Community, Oxford University Press: New York, NY, USA, 2006. [Google Scholar] [CrossRef] [Green Version]
- Shih, J.D.; Hunter, C.P. SID-1 is a dsRNA-selective dsRNA-gated channel. RNA 2011, 17, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Hunter, C.P. SID-1 Functions in Multiple Roles to Support Parental RNAi in Caenorhabditis elegans. Genetics 2017, 207, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Angeles-Albores, D.; Lee, R.Y.N.; Chan, J.; Sternberg, P.W. Tissue enrichment analysis for C. elegans genomics. BMC Bioinform. 2016, 17, 366. [Google Scholar] [CrossRef] [Green Version]
- Timmons, L.; Court, D.L.; Fire, A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 2001, 263, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.J.; Knight, A.L.; Caldwell, G.A.; Caldwell, K.A. Caenorhabditis elegans as a model system for identifying effectors of α-synuclein misfolding and dopaminergic cell death associated with Parkinson’s disease. Methods 2011, 53, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Tournayre, J.; Reichstadt, M.; Parry, L.; Fafournoux, P.; Jousse, C. “Do my qPCR calculation”, a web tool. Bioinformation 2019, 15, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, D.; Asaduzzaman, M.; Young, F. Real time monitoring and quantification of reactive oxygen species in breast cancer cell line MCF-7 by 2′,7′–dichlorofluorescin diacetate (DCFDA) assay. J. Pharmacol. Toxicol. Methods 2018, 94, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, B.; Agranat-Tamir, L.; Light, D.; Zgayer, O.B.-N.; Fishman, A.; Lamm, A.T. A-to-I RNA editing promotes developmental stage–specific gene and lncRNA expression. Genome Res. 2017, 27, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Deffit, S.N.; Yee, B.A.; Manning, A.C.; Rajendren, S.; Vadlamani, P.; Wheeler, E.C.; Domissy, A.; Washburn, M.C.; Yeo, G.W.; Hundley, H.A. The C. elegans neural editome reveals an ADAR target mRNA required for proper chemotaxis. eLife 2017, 6, e28625. [Google Scholar] [CrossRef]
- Hamamichi, S.; Rivas, R.N.; Knight, A.L.; Cao, S.; Caldwell, K.A.; Caldwell, G.A. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA 2008, 105, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Corte, E.; Stirpe, F. The regulation of rat liver xanthine oxidase. Involvement of thiol groups in the conversion of the enzyme activity from dehydrogenase (type D) into oxidase (type O) and purification of the enzyme. Biochem. J. 1972, 126, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaye, D.D.; Greenwald, I. OrthoList: A Compendium of C. elegans Genes with Human Orthologs. PLoS ONE 2011, 6, e20085. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Davis, P.; Paulini, M.; Tuli, M.A.; Williams, G.; Yook, K.; Durbin, R.; Kersey, P.; Sternberg, P.W. WormBase. Worm 2012, 1, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Sternberg, P.W. Genome-Wide Prediction of C. elegans Genetic Interactions. Science 2006, 311, 1481–1484. [Google Scholar] [CrossRef] [Green Version]
- Dalbeth, N.; Gosling, A.L.; Gaffo, A.; Abhishek, A. Gout. Lancet 2021, 397, 1843–1855. [Google Scholar] [CrossRef]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [Green Version]
- Podoly, E.; Hanin, G.; Soreq, H. Alanine-to-threonine substitutions and amyloid diseases: Butyrylcholinesterase as a case study. Chem. Interact. 2010, 187, 64–71. [Google Scholar] [CrossRef]
- Larosa, V.; Remacle, C. Insights into the respiratory chain and oxidative stress. Biosci. Rep. 2018, 38, BSR20171492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mash, D.C.; Pablo, J.; Buck, B.E.; Sanchez-Ramos, J.; Weiner, W.J. Distribution and number of transferrin receptors in Parkinson’s disease and in MPTP-treated mice. Exp. Neurol. 1991, 114, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Wang, J.; Jiang, H.; Xie, J. Ferroportin 1 but not hephaestin contributes to iron accumulation in a cell model of Parkinson’s disease. Free. Radic. Biol. Med. 2010, 48, 332–341. [Google Scholar] [CrossRef]
- Patel, D.; Xu, C.; Nagarajan, S.; Liu, Z.; Hemphill, W.O.; Shi, R.; Uversky, V.N.; Caldwell, G.A.; Caldwell, K.A.; Witt, S.N. Alpha-synuclein inhibits Snx3–retromer-mediated retrograde recycling of iron transporters in S. cerevisiae and C. elegans models of Parkinson’s disease. Hum. Mol. Genet. 2018, 27, 1514–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-H.; Lee, J.; Mok, K.H.; Lee, J.H.; Han, K.-H. Salient Features of Monomeric Alpha-Synuclein Revealed by NMR Spectroscopy. Biomolecules 2020, 10, 428. [Google Scholar] [CrossRef] [Green Version]
- Dohgu, S.; Takata, F.; Matsumoto, J.; Kimura, I.; Yamauchi, A.; Kataoka, Y. Monomeric α-synuclein induces blood–brain barrier dysfunction through activated brain pericytes releasing inflammatory mediators in vitro. Microvasc. Res. 2019, 124, 61–66. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Yu, Z.F.; Bruce-Keller, A.J.; Goodman, Y.; Mattson, M.P. Uric Acid Protects Neurons against Excitotoxic and Metabolic Insults in Cell Culture, and against Focal Ischemic Brain Injury in vivo. J. Neurosci. Res. 1998, 53. [Google Scholar] [CrossRef]
- Ghio, A.J.; Kennedy, T.P.; Stonehuerner, J.; Carter, J.D.; Skinner, K.A.; Parks, D.A.; Hoidal, J.R. Iron regulates xanthine oxidase activity in the lung. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2002, 283, L563–L572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Parkinson Study Group SURE-PD3 Investigators; Bluett, B.; Togasaki, D.M.; Mihaila, D.; Evatt, M.; Rezak, M.; Jain, S.; Schwarzschild, M.A.; Ascherio, A.; Casaceli, C.; et al. Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 2021, 326, 926–939. [Google Scholar] [CrossRef]
- Devos, D.; Cabantchik, Z.I.; Moreau, C.; Danel, V.; Mahoney-Sanchez, L.; Bouchaoui, H.; Gouel, F.; Rolland, A.-S.; Duce, J.A.; Devedijan, J.C.; et al. Conservative iron chelation for neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis. J. Neural Transm. 2020, 127, 189–203. [Google Scholar] [CrossRef]
- Gu, T.; Buaas, F.W.; Simons, A.K.; Ackert-Bicknell, C.; Braun, R.E.; Hibbs, M.A. Canonical A-to-I and C-to-U RNA Editing Is Enriched at 3′UTRs and microRNA Target Sites in Multiple Mouse Tissues. PLoS ONE 2012, 7, e33720. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Zhan, Z.; Litman, T.; Bates, S.E. Regulation of ABCG2 Expression at the 3′ Untranslated Region of Its mRNA through Modulation of Transcript Stability and Protein Translation by a Putative MicroRNA in the S1 Colon Cancer Cell Line. Mol. Cell. Biol. 2008, 28, 5147–5161. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.; Kay, M. How do miRNAs mediate translational repression? Silence 2010, 1, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Emin, D.; Zhang, Y.P.; Lobanova, E.; Miller, A.; Li, X.; Xia, Z.; Dakin, H.; Sideris, D.I.; Lam, J.Y.L.; Ranasinghe, R.T.; et al. Small soluble α-synuclein aggregates are the toxic species in Parkinson’s disease. Nat. Commun. 2022, 13, 5512. [Google Scholar] [CrossRef]
- Mougenot, A.-L.; Nicot, S.; Bencsik, A.; Morignat, E.; Verchère, J.; Lakhdar, L.; Legastelois, S.; Baron, T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 2012, 33, 2225–2228. [Google Scholar] [CrossRef]
- Ma, J.; Gao, J.; Wang, J.; Xie, A. Prion-Like Mechanisms in Parkinson’s Disease. Front. Neurosci. 2019, 13, 552. [Google Scholar] [CrossRef]
- Maulik, M.; Mitra, S.; Bult-Ito, A.; Taylor, B.E.; Vayndorf, E.M. Behavioral Phenotyping and Pathological Indicators of Parkinson’s Disease in C. elegans Models. Front. Genet. 2017, 8, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivard, L.; Srinivasan, J.; Stone, A.; Ochoa, S.; Sternberg, P.W.; Loer, C.M. A comparison of experience-dependent locomotory behaviors and biogenic amine neurons in nematode relatives of Caenorhabditis elegans. BMC Neurosci. 2010, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawin, E.R.; Ranganathan, R.; Horvitz, H. C. elegans Locomotory Rate Is Modulated by the Environment through a Dopaminergic Pathway and by Experience through a Serotonergic Pathway. Neuron 2000, 26, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaeta, A.L.; Caldwell, K.A.; Caldwell, G.A. Found in Translation: The Utility of C. elegans Alpha-Synuclein Models of Parkinson’s Disease. Brain Sci. 2019, 9, 73. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Wen, Q.; Ren, J.; Hendricks, M.; Gershow, M.; Qin, Y.; Greenwood, J.; Soucy, E.R.; Klein, M.; Smith-Parker, H.K.; et al. Dynamic Encoding of Perception, Memory, and Movement in a C. elegans Chemotaxis Circuit. Neuron 2014, 82, 1115–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdmann, E.A.; Mahapatra, A.; Mukherjee, P.; Yang, B.; Hundley, H.A. To protect and modify double-stranded RNA—The critical roles of ADARs in development, immunity and oncogenesis. Crit. Rev. Biochem. Mol. Biol. 2020, 56, 54–87. [Google Scholar] [CrossRef] [PubMed]
- Mew, M.; Caldwell, K.A.; Caldwell, G.A. From bugs to bedside: Functional annotation of human genetic variation for neurological disorders using invertebrate models. Hum. Mol. Genet. 2022, 31, R37–R46. [Google Scholar] [CrossRef]
- Kropp, P.A.; Bauer, R.; Zafra, I.; Graham, C.; Golden, A. Caenorhabditis elegans for rare disease modeling and drug discovery: Strategies and strengths. Dis. Model. Mech. 2021, 14, dmm049010. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Strains Crossed |
|---|---|---|
| UA455 | xdh-1(ok3134); baIn11[Pdat-1::α-syn, Pdat-1::GFP] | RB2379 × UA44 |
| UA456 | xdh-1(ok3134); vtIs7 [Pdat-1::GFP] | RB2379 × BY250 |
| UA457 | adr-2(ok735); vtIs7[Pdat-1::α-syn, Pdat-1::GFP] | RB886 × BY250 |
| Genes | |||
|---|---|---|---|
| acdh-1 | ctc-3 | hpo-29 | papl-1 |
| acox-1 | ctl-2 | hsp-12.3 | pept-1 |
| acox-1.4 | cyp-13A12 | hsp-12.6 | pho-1 |
| act-5 | cyp-25A2 | ifp-1 | srsx-33 |
| ads-1 | dhs-28 | K02F6.8 | T05C3.2 |
| asns-2 | dod-17 | K03H1.5 | T22F3.7 |
| C06G8.3 | dsc-4 | K08D8.6 | ttll-9 |
| C29F3.7 | ets-4 | K10C2.6 | ugt-16 |
| C29F3.7 | ets-9 | K10D11.3 | ugt-37 |
| C31H5.6 | F09F7.5 | lec-10 | ugt-44 |
| ccpp-6 | F11C7.2 | lipl-1 | xdh-1 |
| chil-13 | F15E6.6 | lipl-7 | Y32F68.1 |
| clec-3 | F21D5.3 | ltah-1.2 | Y44A6D.5 |
| clec-4 | F41E7.6 | metr-1 | Y47H10A.5 |
| clec-41 | F52E1.2 | mth-1 | Y48A6B.7 |
| clec-42 | fmo-5 | nduo-1 | |
| crn-6 | folt-2 | nep-17 | |
| ctc-2 | H43E16.1 | nep-22 | |
| Gene Name | Human Ortholog(s) | Differential Expression in adr-2 Mutant | Change in Protein Misfolding | Change in Dopamine Neuron Degeneration | Description |
|---|---|---|---|---|---|
| xdh - 1 | XDH | Downregulated | +8.55% | Protective (p = 0.0002) | Catalyzes the final two steps of purine catabolism; low oxidase activity toward aldehydes; produces ROS. |
| acdh - 1 | ACADSB | Downregulated | +14.47% | Protective (p = 0.0164) | Promotes acyl-CoA dehydrogenase activity; involved in innate immune response. |
| pho - 1 | ACP2, ACPT | Downregulated | +10.92% | Protective (p = 0.0198) | Converts orthophosphoric monoesters to alcohol and phosphate via hydrolysis; encodes phosphatase activity. |
| F52E1.2 | CLEC4A, CLEC4C, CLEC4D, CLEC4E, CLEC6A, ASGR1 | Upregulated | +11.93% | Protective (p = 0.0452) | Promotes carbohydrate binding activity; involved in cell signaling, adhesion, glycoprotein degradation and production, inflammation, immune response. |
| papl - 1 | ACP7 | Downregulated | +9.195% | Enhanced (p = 0.0361) | Promotes acid phosphatase activity; enables metal ion binding. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starr, L.A.; McKay, L.E.; Peter, K.N.; Seyfarth, L.M.; Berkowitz, L.A.; Caldwell, K.A.; Caldwell, G.A. Attenuation of Dopaminergic Neurodegeneration in a C. elegans Parkinson’s Model through Regulation of Xanthine Dehydrogenase (XDH-1) Expression by the RNA Editase, ADR-2. J. Dev. Biol. 2023, 11, 20. https://doi.org/10.3390/jdb11020020
Starr LA, McKay LE, Peter KN, Seyfarth LM, Berkowitz LA, Caldwell KA, Caldwell GA. Attenuation of Dopaminergic Neurodegeneration in a C. elegans Parkinson’s Model through Regulation of Xanthine Dehydrogenase (XDH-1) Expression by the RNA Editase, ADR-2. Journal of Developmental Biology. 2023; 11(2):20. https://doi.org/10.3390/jdb11020020
Chicago/Turabian StyleStarr, Lindsey A., Luke E. McKay, Kylie N. Peter, Lena M. Seyfarth, Laura A. Berkowitz, Kim A. Caldwell, and Guy A. Caldwell. 2023. "Attenuation of Dopaminergic Neurodegeneration in a C. elegans Parkinson’s Model through Regulation of Xanthine Dehydrogenase (XDH-1) Expression by the RNA Editase, ADR-2" Journal of Developmental Biology 11, no. 2: 20. https://doi.org/10.3390/jdb11020020