
Multipurpose Iron-Chelating Ligands Inspired by Bioavailable Molecules

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Synthesis
2.3. Solution Equilibrium Studies
2.4. Cyclic Voltammetry Studies
3. Results
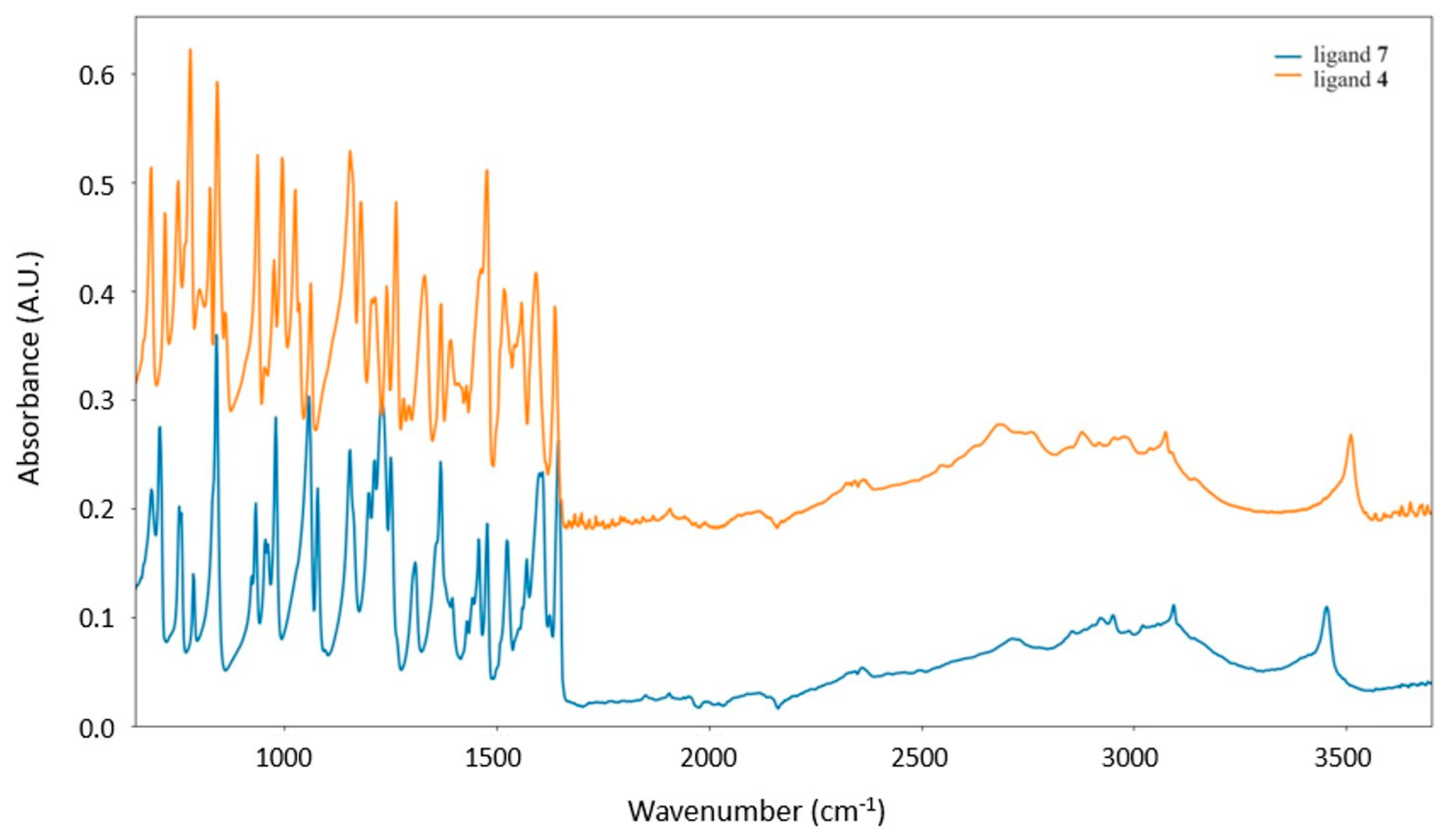
3.1. Synthesis and ATR-Characterization

3.2. Protonation Equilibria
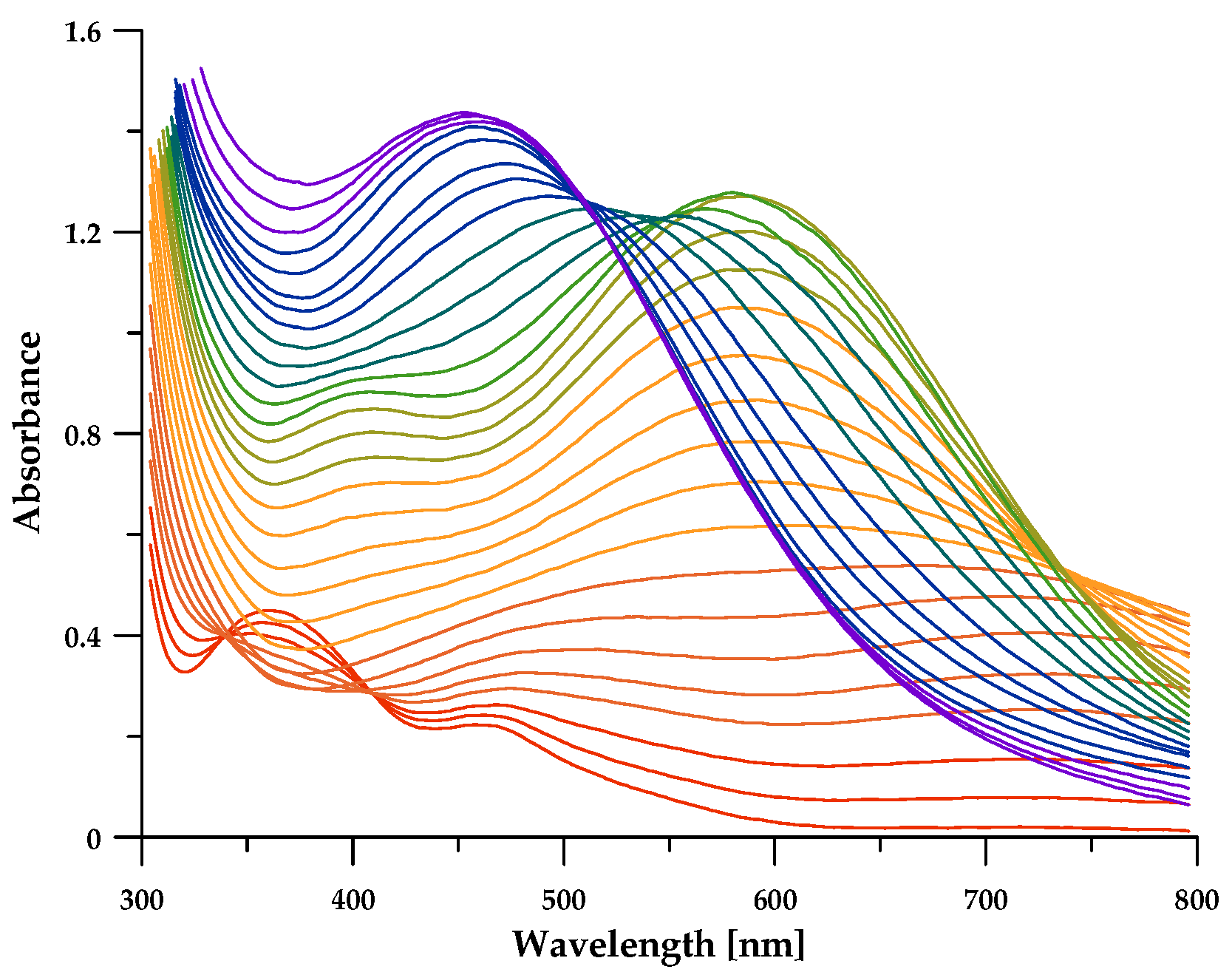
3.3. Fe3+ Complex Formation Equilibria
3.4. Cyclic Voltammetry
3.5. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Cairo, G.; Bernuzzi, F.; Recalcati, S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006, 1, 25–39. [Google Scholar] [CrossRef]
- Gozzelino, R.; Arosio, P. Iron Homeostasis in Health and Disease. Int. J. Mol. Sci. 2016, 17, 130. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J.; Pattichi, K.; Hadjigavriel, M.; Kolnagou, A. Transfusional iron overload and chelation therapy with deferoxamine and deferiprone (L1). Transfus. Sci. 2000, 23, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Nurchi, V.M.; Crisponi, G.; Lachowicz, J.I.; Medici, S.; Peana, M.; Zoroddu, M.A. Chemical features of in use and in progress chelators for iron overload. J. Trace Elements Med. Biol. 2016, 38, 10–18. [Google Scholar] [CrossRef]
- Nurchi, V.M.; de Guadalupe Jaraquemada-Pelaez, M.; Crisponi, G.; Lachowicz, J.I.; Cappai, R.; Gano, L.; Santos, M.A.; Melchior, A.; Tolazzi, M.; Peana, M.; et al. A new tripodal kojic acid derivative for iron sequestration: Synthesis, protonation, complex formation studies with Fe3+, Al3+, Cu2+ and Zn2+, and in vivo bioassays. J. Inorg. Biochem. 2019, 193, 152–165. [Google Scholar] [CrossRef]
- Cappai, R.; Chand, K.; Lachowicz, J.I.; Chaves, S.; Gano, L.; Crisponi, G.; Nurchi, V.M.; Peana, M.; Zoroddu, M.A.; Santos, M.A. A new tripodal-3-hydroxy-4-pyridinone for iron and aluminium sequestration: Synthesis, complexation and in vivo studies. N. J. Chem. 2018, 42, 8050–8061. [Google Scholar] [CrossRef]
- Nurchi, V.M.; Cappai, R.; Crisponi, G.; Sanna, G.; Alberti, G.; Biesuz, R.; Gama, S. Chelating Agents in Soil Remediation: A New Method for a Pragmatic Choice of the Right Chelator. Front. Chem. 2020, 8, 597400. [Google Scholar] [CrossRef]
- Nurchi, V.M.; Cappai, R.; Spano, N.; Sanna, G. A friendly complexing agent for spectrophotometric determination of total iron. Molecules 2021, 26, 3071. [Google Scholar] [CrossRef]
- Lachowicz, J.I.; Nurchi, V.M.; Fanni, D.; Gerosa, C.; Peana, M.; Zoroddu, M.A. Nutritional Iron Deficiency: The Role of Oral Iron Supplementation. Curr. Med. Chem. 2014, 21, 3775–3784. [Google Scholar] [CrossRef]
- Raymond, K.N.; Dertz, E.A.; Kim, S.S. Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. USA 2003, 100, 3584–3588. [Google Scholar] [CrossRef] [PubMed]
- Neilands, J.B. Siderophores: Structure and Function of Microbial Iron Transport Compounds. J. Biol. Chem. 1995, 270, 26723–26726. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Mohd-Nor, A.R.; Silver, J.; Morrison, I.E.G.; Rees, L.V.C. Model compounds for microbial iron-transport compounds. Part 1. Solution chemistry and Mössbauer study of iron(II) and iron(III) complexes from phenolic and catecholic systems. J. Chem. Soc. Dalton Trans. 1981, 609–622. [Google Scholar] [CrossRef]
- Kontoghiorghes, G. Deferiprone and Iron–Maltol: Forty Years since Their Discovery and Insights into Their Drug Design, Development, Clinical Use and Future Prospects. Int. J. Mol. Sci. 2023, 24, 4970. [Google Scholar] [CrossRef]
- Kandioller, W.; Kurzwernhart, A.; Hanif, M.; Meier, S.M.; Henke, H.; Keppler, B.K.; Hartinger, C.G. Pyrone derivatives and metals: From natural products to metal-based drugs. J. Organomet. Chem. 2011, 696, 999–1010. [Google Scholar] [CrossRef]
- Thompson, K.H.; Barta, C.A.; Orving, C. Metal complexes of maltol and close analogues in medicinal inorganic chemistry. Chem. Soc. Rev. 2006, 35, 545–556. [Google Scholar] [CrossRef]
- Lai, D.; Brotz-Oesterhelt, H.; Muller, W.; Wray, V.; Proksch, P. Bioactive polyketides and alkaloids from Penicillium citrinum, a fungal endophyte isolated from Ocimum tenuiflorum. Fitoterapia 2013, 91, 100–106. [Google Scholar] [CrossRef]
- Fusi, S.; Di Florio, G.; Margiotta, N.; Barbanente, A.; Cini, E.; Finetti, F.; Paradisi, L.; Trabalzini, L.; Fabrizi de Biani, F.; Corsini, M. Synthesis, characterization, electrochemistry and in vitro cytotoxicity of a new “Triazole-Maltol” ligand and its platinum(II) complex. Inorganica Chim. Acta 2022, 532, 120756. [Google Scholar] [CrossRef]
- Ehrlich, M.; Carell, T. Total Syntheses and Biological Evaluation of 3-O-Methylfunicone and Its Derivatives Prepared by TMPZnCl·LiCl-Mediated Halogenation and Carbonylative Stille Cross-Coupling. Eur. J. Org. Chem. 2013, 2013, 77–83. [Google Scholar] [CrossRef]
- Gran, G. Determination of the equivalence point in potentiometric acid-base titrations. Analyst 1952, 77, 661–671. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Gans, P.; Sabatini, A.; Vacca, A. To improve accuracy of the calculated pKa values. Ann. Chim. 1999, 89, 45–49. [Google Scholar]
- Baes, C.F.; Mesmer, R.E. The Hydrolysis of Cations; John Wiley & Sons: New York, NY, USA, 1976. [Google Scholar]
- Kostelnik, T.I.; Scheiber, H.; Cappai, R.; Choudhary, N.; Lindheimer, F.; De Guadalupe Jaraquemada-Peláez, M.; Orvig, C. Phosphonate Chelators for Medicinal Metal Ions. Inorg. Chem. 2021, 60, 5343–5361. [Google Scholar] [CrossRef]
- Migliorini, F.; Cini, E.; Dreassi, E.; Finetti, F.; Ievoli, G.; Macrì, G.; Petricci, E.; Rango, E.; Trabalzini, L.; Taddei, M. A pH-responsive crosslinker platform for antibody-drug conjugate (ADC) targeting delivery. Chem. Commun. 2022, 58, 10532–10535. [Google Scholar] [CrossRef] [PubMed]
- Fusi, S.; Frosini, M.; Biagi, M.; Zór, K.; Rindzevicius, T.; Baratto, M.C.; De Vico, L.; Corsini, M. Iron(III) complexing ability of new ligands based on natural γ-pyrone maltol. Polyhedron 2020, 187, 114650. [Google Scholar] [CrossRef]
- Zborowski, K.; Grybos, R.; Proniewicz, L.M. Structure modification of maltol (3-hydroxy-2-methyl-4H-pyran-4-one) upon cation and anion formation studied by vibrational spectroscopy and quantum-mechanical calculations. Vib. Spectrosc. 2007, 43, 344–350. [Google Scholar] [CrossRef]
- Mohammed-Ziegler, I.; Billes, F. Vibrational spectroscopic calculations on pyrogallol and gallic acid. J. Mol. Struct.-Theochem. 2002, 618, 259–265. [Google Scholar] [CrossRef]
- Williams, D.H.; Fleming, I. Spectroscopic Methods in Organic Chemistry; McGraw-Hill Book Company: Maidenhead, UK, 1966. [Google Scholar]
- Parajon-Costa, B.S.; Baran, E.J. Vibrational spectra of bis(maltolato)zinc(II), an interesting insulin mimetic agent. Spectrochim. Acta A 2013, 113, 337–339. [Google Scholar] [CrossRef]
- Ostacoli, G.; Vanni, A.; Roletto, E. Complex formation between alkyl-substituted malonic acids and bivalent metal ions in aqueous solutions. Ric. Sci. 1968, 38, 318–321. [Google Scholar]
- Amico, P.; Bonomo, R.P.; Cali, R.; Cucinotta, V.; Daniele, P.G.; Ostacoli, G.; Rizzarelli, E. Ligand-ligand Interactions in Metal Complexes: Thermodynamic and Spectroscopic Investigation of Simple and Mixed Copper(II) and Zinc(II) Substituted-malonate Complexes with 2,2’-bipyridyl in Aqueous Solution. Inorg. Chem. 1989, 28, 3555–3561. [Google Scholar] [CrossRef]
- DeRobertis, A.; DeStefano, C.; Foti, C. Medium effects on the protonation of carboxylic acids at different temperatures. J. Chem. Eng. Data 1999, 44, 262–270. [Google Scholar] [CrossRef]
- Petrola, R. Spectrophotometric study on the equilibria of pyromeconic acid derivatives with proton in aqueous solution. Finn. Chem. Lett. 1985, 12, 207. [Google Scholar]
- Chiaccherini, E.; Bartušek, M. Complex of maltol with uranyl and aluminium. Collect. Czechoslov. Chem. Commun. 1969, 34, 530–536. [Google Scholar] [CrossRef]
- Herrero-Martínez, J.M.; Sanmartin, M.; Rosés, M.; Bosch, E.; Ràfols, C. Determination of dissociation constant of flavonoids by capillary electrophoresis. Electrophoresis 2005, 10, 1886–1895. [Google Scholar] [CrossRef] [PubMed]
- Sigel, H.; Huber, P.R.; Griesser, R.; Bernhard, P. Ternary Complexes in Solution. XV.1 Mixed-Ligand Copper(II) Complexes with 2,2 -Bipyridyl or 1,10-Phenanthroline and Pyrocatecholate or Derivatives Thereof. Inorg. Chem. 1973, 12, 1198–1200. [Google Scholar] [CrossRef]
- Swain, C.G.; Lupton, E.C. Field and Resonance Components of Substituent Effects. J. Am. Chem. Soc. 1968, 90, 4328–4337. [Google Scholar] [CrossRef]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): A utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Stefanović, A.; Havel, J.; Sommel, L. On the reaction of iron(III). Collect. Czechoslov. Chem. Commun. 1968, 33, 4198–4214. [Google Scholar] [CrossRef]
- Salvadó, V.; Ribas, X.; Zelano, V.; Ostacoli, G.; Valiente, M. The chemistry of iron in biosystems-III. Complex formation between FeIII and malonic acid in aqueous solutions. Polyhedron 1989, 8, 813–818. [Google Scholar] [CrossRef]
- Nurchi, V.M.; Pivetta, T.; Lachowicz, J.I.; Crisponi, G. Effect of substituents on complex stability aimed at designing new iron(III) and aluminum(III) chelators. J. Inorg. Biochem. 2009, 103, 227–236. [Google Scholar] [CrossRef]
- Barham, A.S.; Kennedy, B.M.; Cunnane, V.J.; Daous, M.A. The Electrochemical polymerisation of 1,2 dihydroxybenzene and 2-hydroxybenzyl alcohol prepared in different solutions media. Electrochim. Acta 2014, 147, 19–24. [Google Scholar] [CrossRef]
- Marczewska, B.; Przegaliński, M. Poly(catechol) electroactive film and its electrochemical properties. Synth. Met. 2013, 182, 33–39. [Google Scholar] [CrossRef]
- Ball, V. Electrodeposition of pyrogallol versus pyrocatechol using cyclic voltammetry and chronoamperometry. J. Electroanal. Chem. 2022, 909, 116142. [Google Scholar] [CrossRef]
- Crisponi, G.; Remelli, M. Iron chelating agents for the treatment of iron overload. Coord. Chem. Rev. 2008, 252, 1225–1240. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–26. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 | 4 | 7 | |||||
|---|---|---|---|---|---|---|---|
| species | log β | log K | species | log β | log K | log β | log K |
| [LH]2− | 8.999 (1) | 8.999 | [LH]− | 11.55 (2) | 11.55 | 11.30 (6) | 11.30 |
| [LH2]− | 13.87 (4) | 4.87 | LH2 | 20.46 (2) | 8.91 | 20.25 (3) | 8.95 |
| LH3 | 16.70 (5) | 2.83 | |||||
| 3 | 7 | ||||
|---|---|---|---|---|---|
| Species | log β | pK | Species | log β | pK |
| [FeLH]+ | 20.39 (1) | [FeLH]2+ | 22.44 (1) | 3.97 | |
| [FeL2H]2− | 30.33 (1) | 5.20 | [FeL]+ | 18.47 (2) | |
| [FeL2]3− | 25.13 (1) | 9.06 | FeL2H | 37.29 (1) | 5.32 |
| [FeL2H-2]5− | 7.01 (2) | [FeL2]− | 31.97 (1) | 9.27 | |
| [FeL3]6− | 29.60 (3) | [FeL2H-2]3− | 13.43 (2) | ||
| [FeL3]3− | 39.03 (1) | ||||
| pFe3+ | 17.72 | 16.86 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cini, E.; Crisponi, G.; Fantasia, A.; Cappai, R.; Siciliano, S.; Florio, G.D.; Nurchi, V.M.; Corsini, M. Multipurpose Iron-Chelating Ligands Inspired by Bioavailable Molecules. Biomolecules 2024, 14, 92. https://doi.org/10.3390/biom14010092
Cini E, Crisponi G, Fantasia A, Cappai R, Siciliano S, Florio GD, Nurchi VM, Corsini M. Multipurpose Iron-Chelating Ligands Inspired by Bioavailable Molecules. Biomolecules. 2024; 14(1):92. https://doi.org/10.3390/biom14010092
Chicago/Turabian StyleCini, Elena, Guido Crisponi, Alessandra Fantasia, Rosita Cappai, Sofia Siciliano, Giuseppe Di Florio, Valeria M. Nurchi, and Maddalena Corsini. 2024. "Multipurpose Iron-Chelating Ligands Inspired by Bioavailable Molecules" Biomolecules 14, no. 1: 92. https://doi.org/10.3390/biom14010092