1. Introduction
The ribosome is a remarkable and extremely efficient macromolecular RNA–protein machine that synthesizes all proteins in the cytoplasm of the eukaryotic cell and is composed of a small 40S and a large 60S subunit. With the help of several hundred different assembly factors, the two subunits are assembled from ribosomal RNAs (rRNAs) and ribosomal proteins (r-proteins) in a highly conserved and complex maturation pathway called ribosome biogenesis, which is best studied in the yeast
Saccharomyces cerevisiae. This assembly and maturation process starts in the nucleolus, a non-membrane-enclosed sub-compartment of the nucleus, progresses in the nucleoplasm, and ends in the cytoplasm, where the two mature subunits form the translation-competent 80S ribosome [
1,
2,
3,
4].
R-proteins have to overcome two major obstacles before they are assembled into pre-ribosomal particles in the nucle(ol)us. First, as they contain highly basic regions as well as unstructured N- or C-terminal extensions and internal loops, which engage in interactions with the negatively charged rRNA in the ribosome, r-proteins are prone to aggregation as long as they are not assembled into (pre-)ribosomal subunits [
5]. Second, since r-proteins are synthesized in the cytoplasm but most of them are incorporated into pre-ribosomes in the nucle(ol)us, they need to be imported into the nucleus through the nuclear pore complex (NPC) before becoming available for ribosome biogenesis [
6].
Two classes of proteins are specialized in helping to overcome these obstacles and to ensure the safe delivery of r-proteins at their site of assembly with the rRNA: (1) Dedicated chaperones of r-proteins: More than ten r-proteins were found to require protection by these specialized chaperones [
5,
7,
8,
9,
10]. Most dedicated chaperones bind r-proteins already co-translationally and protect them from aggregation, presumably through shielding positively charged surfaces [
5,
7,
11]. (2) Importins of the karyopherin superfamily: Besides mediating the nuclear import of many diverse substrate proteins (cargoes) across the hydrophobic channel of the NPC [
6], selected importins have also been shown to facilitate nuclear import of r-proteins and to prevent their aggregation [
12,
13].
Importins usually recognize their cargo proteins by binding to basic nuclear localization sequences (NLSs). All importin-ßs have the ability to interact with the hydrophobic FG-repeat meshwork in the central channel of the NPC. Additionally, most importin-ßs, including Pse1 (also called Kap121), Kap123, and Kap104, can recognize the NLS of their cargoes directly; only the importin-ß Kap95 requires an adaptor, the importin-α Srp1 (also called Kap60), for binding to the NLS-containing cargo protein. Importin-α recognizes classical NLS sequences, which can be either monopartite (consensus K-K/R-X-K/R; where X can be any amino acid) or bipartite (K/R-K/R-X
10–12 K/R
3/5; with K/R
3/5 being five residues containing at least three Ks or Rs). The importin-ß Kap104 recognizes PY-NLSs containing an N-terminal basic (or hydrophobic) motif and a C-terminal R/K/H-X
2–5-P-Y/L/F motif. Additionally, Kap104 can bind to RGG regions (RG-rich NLSs). Pse1 binds to IK-NLSs with the consensus K-V/I-X-K-X
1–2-K/H/R. Importantly, however, many cargoes of the above-named importins do not harbor sequences following the so-far-described NLS consensus sequences, and, moreover, for many importins, no targeting signals have been defined at all [
6].
After nuclear import, the importins release their cargo proteins by binding to the small GTPase Ran in its GTP-bound form (RanGTP), which is highly concentrated in the nucleus, thereby controlling the directionality of transport [
6]. Nuclear import of r-proteins is believed to be mainly performed by the non-essential importin-β Kap123, with some redundant contribution of the essential Pse1 [
13].
In contrast to that notion, studies in recent years by us and others on the coordination of chaperoning and nuclear import of r-proteins revealed that several r-proteins employ importins other than Kap123. Rps3 is imported into the nucleus as a dimer, with one N-terminal domain protected by its dedicated chaperone Yar1 and the second one bound by the Srp1/Kap95 importin-α/importin-β dimer [
14,
15]. Rpl5 and Rpl11 are imported in complex with their dedicated chaperone Syo1, which functions as a transport adaptor for Kap104 [
16]. Rpl4 contains at least five different NLS sequences and is imported into the nucleus in complex with its dedicated chaperone Acl4 by importin Kap104 [
17,
18,
19]. Last but not least, Rps26 can be imported into the nucleus by Kap123, Kap104, or Pse1, and is then released from the importin in a RanGTP-independent manner by its dedicated chaperone Tsr2 [
20].
We are interested in the nuclear import of r-protein Rps2 (also called uS5 [
21]). Rps2 has an evolutionarily conserved dedicated chaperone, Tsr4 (PDCD2 in humans), which binds co-translationally to its unstructured N-terminal extension [
7,
10,
22]. In the absence of Tsr4, Rps2 accumulates in the nucleus, suggesting that Tsr4 is required for the efficient incorporation of Rps2 into pre-ribosomes [
7]. How Rps2 is imported into the nucleus has, however, remained elusive.
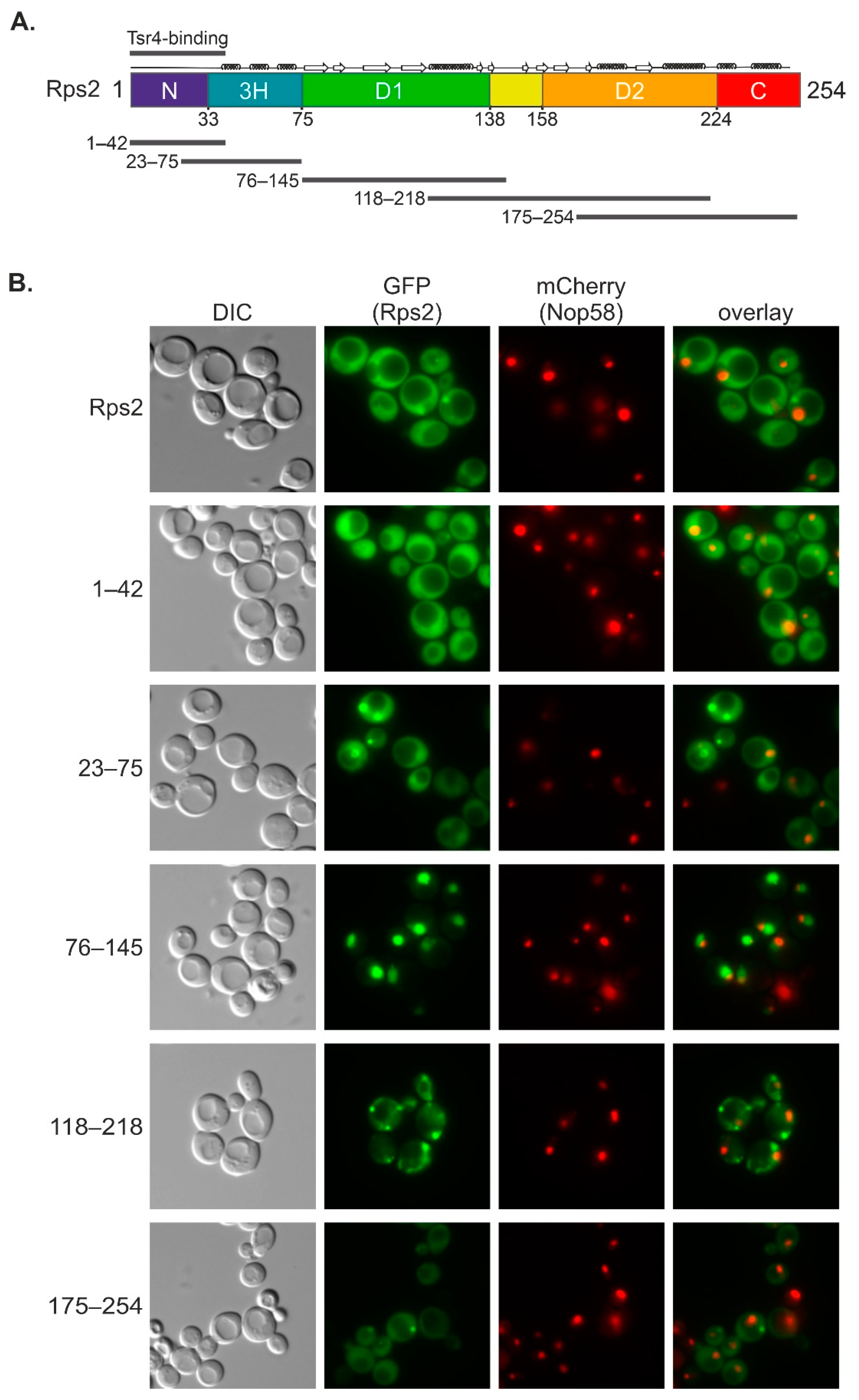
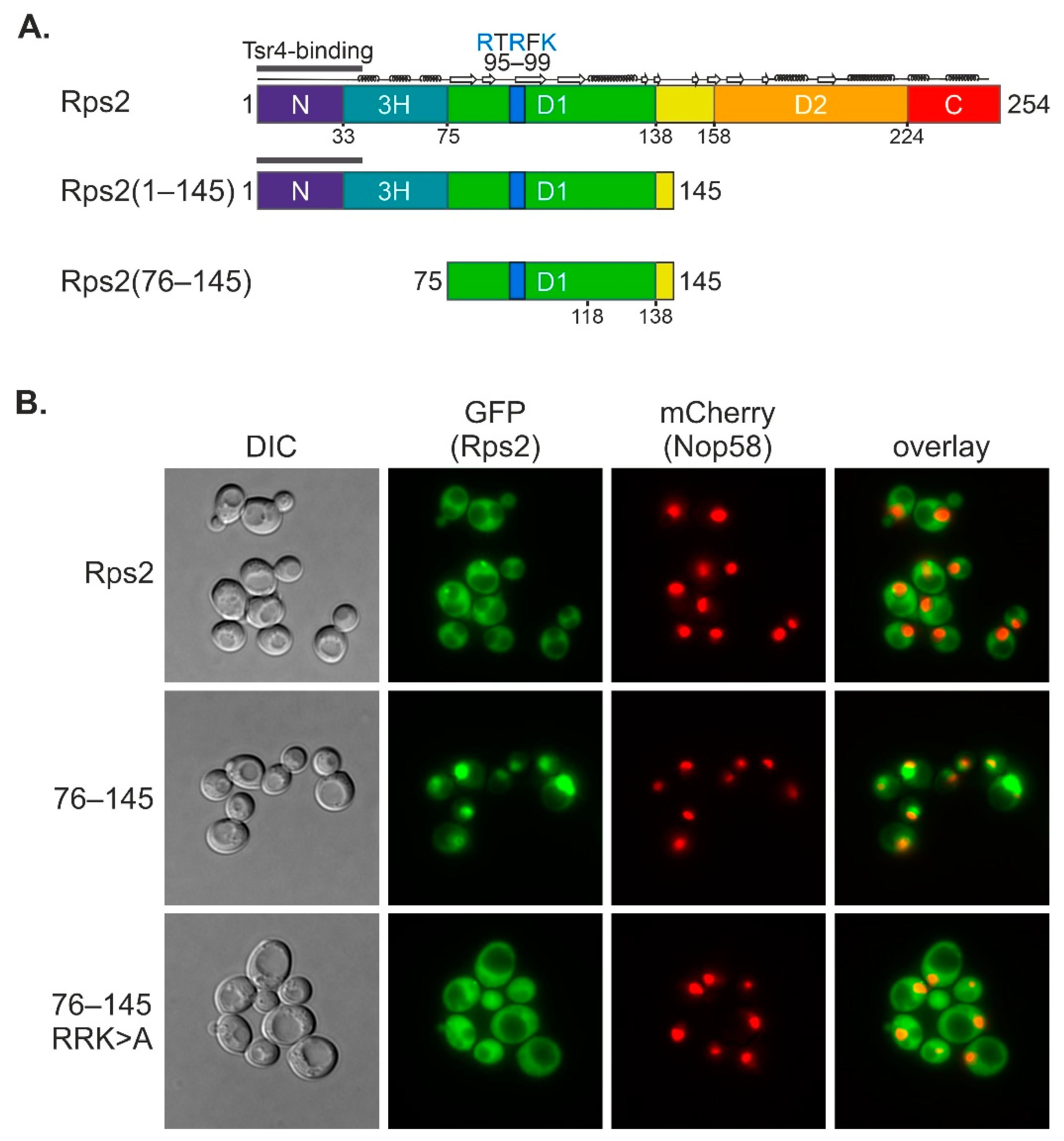
In this study, we uncovered that an internal fragment of Rps2, Rps2(76–145), interacts with the importin-β Pse1 and is sufficient to target a 3xyEGFP reporter into the nucleus, indicating that it contains a functional NLS. Nuclear import of Rps2(76–145)-3xyEGFP was blocked in a pse1-1 mutant or upon changing three basic residues in Rps2, arginine 95 (R95), R97, and lysine 99 (K99), to alanines (As), suggesting that these residues are part of the NLS. Surprisingly, when fusing a larger N-terminal Rps2 fragment, Rps2(1–145), to the 3xyEGFP reporter, nuclear targeting was no longer disturbed by the R95A, R97A, and K99A exchanges. Moreover, we identified a sequence in the N-terminal part of Rps2 (amino acids 10–28), whose basic residues are essential for the nuclear targeting of the Rps2(1–145)-3xyEGFP fusion protein bearing the R95A, R97A, and K99A mutations, strongly suggesting the presence of a second NLS in the eukaryote-specific N-terminal extension of Rps2. Our results moreover revealed that import, both via the internal and the N-terminal nuclear targeting region, also occurs in the absence of Tsr4.
2. Methods
2.1. Yeast Strains and Genetic Methods
All
S. cerevisiae strains used in this study are listed in
Supplementary Table S1. Yeast plasmids were constructed using standard recombinant DNA techniques and are listed in
Supplementary Table S2. All DNA fragments amplified by PCR were verified by sequencing.
The KAP104 shuffle strain was transformed with the YCplac22-KAP104 or the pRS314-kap104-16 plasmid (TRP1), respectively, and transformed cells were streaked on 5-FOA (Thermo Scientific) plates to counter-select against the pRS316-KAP104 (URA3) shuffle plasmid. After plasmid shuffling, cells were grown on plates lacking tryptophan (SDC-trp) and transformed with the LEU2 plasmid expressing Rps2(76–145)-3xyEGFP.
2.2. Fluorescence Microscopy
Yeast strains were grown at 30 °C in SDC media lacking leucine (SDC-leu) to an OD600 of ~0.5 (logarithmic growth phase). Cells were imaged by fluorescence microscopy using a Leica DM6 B microscope, equipped with a DFC 9000 GT camera, using the PLAN APO 100× objective, narrow-band GFP or RHOD ET filters, and LasX software. Full-length Rps2 as well as fragments and variants thereof were expressed with a C-terminal 3xyEGFP tag under the transcriptional control of the ADH1 promoter from a centromeric LEU2 plasmid. Plasmids expressing these 3xyEGFP reporter proteins were transformed into a Nop58-yEmCherry expressing strain, the C303 wild-type strain, rps2∆ and tsr4∆ rps2∆ strains containing a centromeric URA3-RPS2 plasmid, or the indicated importin mutant strains. Since Rps2 is an essential protein, we investigated the localization of the different Rps2-3xyEGFP fusion proteins in strains harboring the wild-type RPS2 gene either at the chromosomal locus or on a plasmid.
2.3. Yeast Two-Hybrid (Y2H) Assays
Protein–protein interactions between Rps2 (and fragments/variants thereof) and Pse1/Pse1.302C or Tsr4 were analyzed by yeast two-hybrid (Y2H) assays using the reporter strain PJ69-4A. This Y2H strain allows for the detection of both weak (HIS3 reporter) and strong interactions (ADE2 reporter). Two plasmids were co-transformed into PJ69-4A, whereby one plasmid was expressing fusions to the Gal4 DNA-binding domain (G4BD, BD, TRP1 marker) and the other fusions to the Gal4 transcription activation domain (G4AD, AD, LEU2 marker). For the Rps2-Tsr4 Y2H interaction assays, Rps2 variants, C-terminally fused to the G4BD, and full-length Tsr4, C-terminally fused to the G4AD, were expressed from centromeric (CEN, low-copy) plasmids (pG4BDC22 and pG4ADC111, respectively). For the Rps2-Pse1 Y2H interaction assays, Rps2 variants, either N- or C-terminally fused to the G4BD, and Pse1 or Pse1.302C, C-terminally fused to the G4AD, were expressed from episomal (2µ, high-copy) plasmids (pG4BDN112, pGAG4BDC112, and pGAG4ADC181, respectively).
After the selection of transformants on plates lacking leucine and tryptophan (SDC-leu-trp, -LT), cells were spotted onto SDC-leu-trp plates as well as onto plates lacking histidine, leucine, and tryptophan (SDC-his-leu-trp, -HLT), and lacking adenine, leucine, and tryptophan (SDC-ade-leu-trp, -ALT), respectively. Plates were incubated for 3 days at 30 °C.
2.4. Tandem Affinity Purification (TAP)
For TAP purifications, plasmids expressing Rps2(76–145) or Rps2(76–145).R95R97K99>A, C-terminally fused to the TAP tag, or a plasmid expressing the TAP tag alone were transformed into a haploid W303-derived wild-type strain. Cells were grown in 4 l yeast extract peptone dextrose medium (YPD) to an optical density (OD600) of 2 at 30 °C.
TAP purifications were performed in a buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1.5 mM MgCl2, 0.1% NP-40, 1 mM dithiothreitol (DTT), and 1x Protease Inhibitor Mix FY (Serva). Cells were lysed by mechanical disruption using glass beads and the cell lysate was incubated with 300 µL IgG SepharoseTM 6 Fast Flow (GE Healthcare, Chicago, IL, USA) for 60 min at 4 °C. After incubation, the IgG SepharoseTM beads were transferred into Mobicol columns (MoBiTec, Göttingen, Germany) and washed with 10 mL buffer. Then, TEV protease was added and elution from the beads was performed under rotation for 90 min at room temperature. After the addition of 2 mM CaCl2, TEV eluates were incubated with 300 µL Calmodulin SepharoseTM 4B beads (GE Healthcare) for 60 min at 4 °C. After washing with 5 mL buffer containing 2 mM CaCl2, proteins were eluted from Calmodulin SepharoseTM with elution buffer consisting of 10 mM Tris-HCl (pH 8.0), 5 mM EGTA, and 50 mM NaCl under rotation for 20 min at room temperature. The protein samples were separated on NuPAGETM 4–12% Bis-Tris gels (Invitrogen, Carlsbad, CA, USA) followed by Western blotting.
2.5. Western Blotting
Western blot analysis was performed using the following antibodies: α-CBP antibody (1:5000; Merck-Millipore, Burlington, MA, USA, cat. no. 07-482), α-Pse1 antibody (1:500; Matthias Seedorf [
22]), secondary α-rabbit horseradish peroxidase-conjugated antibody (1:15,000; Sigma-Aldrich, St. Louis, MO, USA, cat. no. A0545). Protein signals were visualized using the Clarity
TM Western ECL Substrate Kit (Bio-Rad, Hercules, CA, USA) and captured by the ChemiDoc
TM Touch Imaging System (Bio-Rad).
2.6. TurboID-Based Proximity Labeling
Plasmids expressing C-terminal TurboID-tagged bait proteins under the control of the copper-inducible CUP1 promoter were transformed into the wild-type strain YDK11-5A. Transformed cells were grown at 30 °C in 100 mL SDC-leu medium, prepared with copper-free yeast nitrogen base (FORMEDIUM), to an OD600 between 0.4 and 0.5. Then, copper sulfate, to induce expression from the CUP1 promoter, and freshly prepared biotin (Sigma-Aldrich, St. Louis, MO, USA) were added to a final concentration of 500 μM, and cells were grown for an additional hour, typically reaching a final OD600 between 0.6 and 0.8, and harvested by centrifugation at 4000 rpm for 5 min at 4 °C. Then, cells were washed with 50 mL ice-cold H2O, resuspended in 1 mL ice-cold lysis buffer (LB: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1.5 mM MgCl2, 0.1% SDS, and 1% Triton X-100) containing 1 mM PMSF, transferred to 2 mL safe-lock tubes, pelleted by centrifugation, frozen in liquid nitrogen, and stored at −80 °C. Extracts were prepared, upon the resuspension of cells in 400 μL lysis buffer containing 0.5% sodium deoxycholate and 1 mM PMSF (LB-P/D), by glass bead lysis with a Precellys 24 homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France) set at 5000 rpm using a 3 × 30 s lysis cycle with 30 s breaks in between at 4 °C. Lysates were transferred to 1.5 mL tubes. For complete extract recovery, 200 μL LB-P/D was added to the glass beads and, after brief vortexing, combined with the already transferred lysate. Cell lysates were clarified by centrifugation for 10 min at 13,500 rpm at 4 °C, transferred to a new 1.5 mL tube. Total protein concentration in the clarified cell extracts was determined with the Pierce™ BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA) using a microplate reader (BioTek 800 TS). To reduce non-specific binding, 100 μL of Pierce™ High Capacity Streptavidin Agarose Resin (Thermo Scientific) slurry, corresponding to 50 μL of settled beads, were transferred to a 1.5 mL safe-lock tube, blocked by incubation with 1 mL LB containing 3% BSA for 1 h at RT, and then washed four times with 1 mL LB. For the affinity purification of biotinylated proteins, 2 mg of total protein in an adjusted volume of 800 μL LB-P/D was added to the blocked and washed streptavidin beads, and binding was carried out for 1 h at RT on a rotating wheel. Beads were then washed once for 5 min with 1 mL of wash buffer (50 mM Tris-HCl (pH 7.5), 2% SDS), five times with 1 mL LB, and finally five times with 1 mL ABC buffer (100 mM ammonium bicarbonate (pH 8.2)). Bound proteins were eluted by two consecutive incubations with 30 μL 3× SDS sample buffer, containing 10 mM biotin and 20 mM DTT, for 10 min at 75 °C. The eluates were combined in one 1.5 mL safe-lock tube and stored at –20 °C. Upon reduction with DTT and alkylation with iodoacetamide, samples were separated on NuPAGE 4–12% Bis-Tris gels (Invitrogen, Carlsbad, CA, USA), run in NuPAGE 1× MES SDS running buffer (Novex) at 200 V for a total of 12 min. The gels were incubated with Brilliant Blue G Colloidal Coomassie (Sigma-Aldrich) until the staining of proteins was visible. Each lane was cut, from slot to the migration front, into three gel pieces that were, upon their fragmentation into smaller pieces, transferred into separate 1.5 mL low-binding tubes.
Gel pieces were covered with 100–150 μL of ABC buffer, prepared in HPLC-grade H
2O, and incubated for 10 min at RT in a thermoshaker set to 1000 rpm. Then, gel pieces were covered with 100–150 μL of HPLC-grade absolute EtOH and incubated for 10 min at RT in a thermoshaker set to 1000 rpm. These two wash steps were repeated two more times. For the in-gel digestion of proteins, gel pieces were covered with 120 μL of ABC buffer containing 1 μg sequencing-grade modified trypsin (Promega Madison, WI, USA) and incubated overnight at 37 °C with shaking at 1000 rpm. To stop the digestion and recover the peptides, 50 μL of a 2% trifluoroacetic acid (TFA) solution was added, and, after a 10 min incubation at RT with shaking at 1000 rpm, the supernatant was transferred to a new 1.5 mL low-binding tube. The gel pieces were then incubated, again for 10 min at RT with shaking at 1000 rpm, another two times with 100–150 μL EtOH, and these two supernatants were combined with the first supernatant. Finally, using a SpeedVac, the organic solvents were evaporated and the volume was reduced to around 50 μL. Then, 200 μL of buffer A (0.5% acetic acid) were added, and the samples were applied to C18 StageTips [
23], equilibrated with 50 μL of buffer B (80% acetonitrile, 0.3% TFA) and washed twice with 50 μL of buffer A, for desalting and peptide purification. StageTips were washed once with 100 μL of buffer A, and the peptides were eluted with 50 μL of buffer B. The solvents were completely evaporated using a SpeedVac. Peptides were resuspended by first adding 3 µL buffer A* (3% acetonitrile, 0.3% TFA) and then 17 µL buffer A*/A (30% buffer A*/70% buffer A), with each solvent addition being followed by vortexing for 10 s. Samples were stored at –80 °C.
LC-MS/MS measurements were performed on a Q Exactive HF-X (Thermo Scientific) coupled to an EASY-nLC 1200 nanoflow-HPLC (Thermo Scientific). HPLC-column tips (fused silica) with 75 μm inner diameter were self-packed with ReproSil-Pur 120 C18-AQ, 1.9 μm particle size (Dr. Maisch GmbH, Ammerbuch, Germany) to a length of 20 cm. Samples were directly applied onto the column without a pre-column. A gradient of A (0.1% formic acid in H2O) and B (0.1% formic acid in 80% acetonitrile in H2O) with increasing organic proportion was used for peptide separation (loading of sample with 0% B; separation ramp: from 5–30% B within 85 min). The flow rate was 250 nL/min and for sample application, it was 600 nL/min. The mass spectrometer was operated in the data-dependent mode and switched automatically between MS (max. of 1 × 106 ions) and MS/MS. Each MS scan was followed by a maximum of ten MS/MS scans using a normalized collision energy of 25% and a target value of 1000. Parent ions with a charge state form z = 1 and unassigned charge states were excluded for fragmentation. The mass range for MS was m/z = 370–1750. The resolution for MS was set to 70,000 and for MS/MS to 17,500. MS parameters were as follows: spray voltage 2.3 kV, no sheath and auxiliary gas flow, ion-transfer tube temperature 250 °C.
The MS raw data files were analyzed with the MaxQuant software package version 1.6.2.10 [
24] for peak detection, generation of peak lists of mass-error-corrected peptides, and database searches. The UniProt
Saccharomyces cerevisiae database (version March 2016), additionally including common contaminants, trypsin, TurboID, and GFP, was used as reference. Carbamidomethylcysteine was set as fixed modification and protein amino-terminal acetylation, oxidation of methionine, and biotin were set as variable modifications. Four missed cleavages were allowed, enzyme specificity was Trypsin/P, and the MS/MS tolerance was set to 20 ppm. Peptide lists were further used by MaxQuant to identify and relatively quantify proteins using the following parameters: peptide and protein false discovery rates, based on a forward–reverse database, were set to 0.01, minimum peptide length was set to seven, and minimum number of unique peptides for identification and quantification of proteins was set to one. The ‘match-between-run’ option (0.7 min) was used.
For quantification, missing iBAQ (intensity-based absolute quantification) values in the two control purifications from cells expressing either the GFP-TurboID or the NLS-GFP-TurboID bait were imputed in Perseus [
25]. For normalization of intensities in each independent purification, iBAQ values were divided by the median iBAQ value, derived from all nonzero values, of the respective purification. To calculate the enrichment of a given protein compared to its average abundance in the two control purifications, the normalized iBAQ values were log2-transformed and those of the control purifications were subtracted from the ones of each respective bait purification. For graphical presentation, the normalized iBAQ value (log10 scale) of each protein detected in a given bait purification was plotted against its relative abundance (log2-transformed enrichment compared to the control purifications). To visualize the effects of the RRK>A mutations on the proximal protein neighborhood of Rps2, the normalized iBAQ value (log10 scale) of each protein detected in the purification from cells expressing a wild-type Rps2 bait protein (full-length Rps2, Rps2(1–145), or Rps2(76–145)) was plotted against its relative abundance (log2-transformed enrichment) compared to the purification from cells expressing the respective RRK>A mutant protein (with prior imputation of missing iBAQ values).
4. Discussion
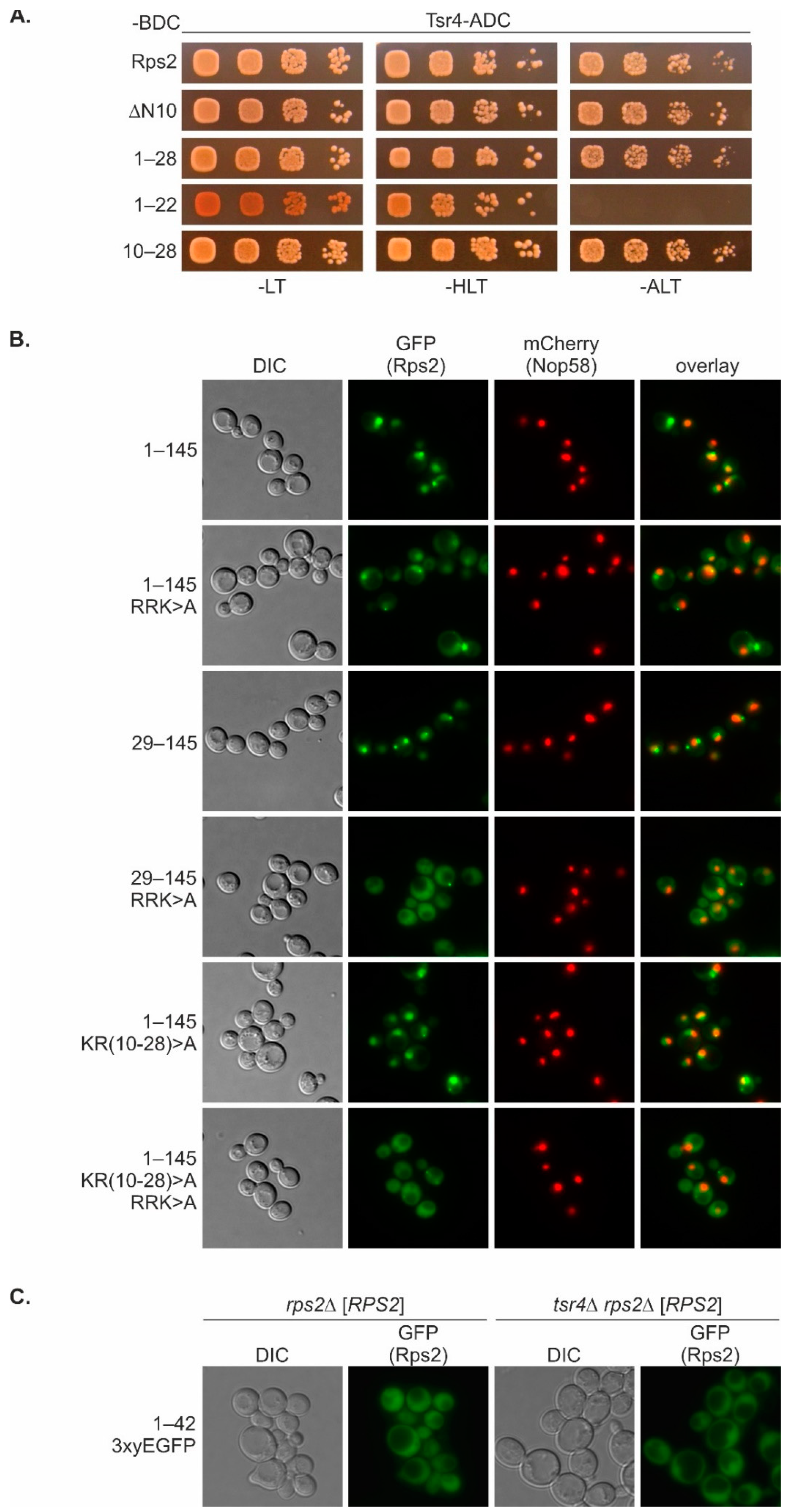
With this study, we have provided insights into the intricate mechanisms underlying nuclear import of the r-protein Rps2. We found that amino acids 76 to 145 are sufficient to target the protein to the nucleus, with residues R95, R97, and K99 being essential for the nuclear localization of this fragment. The main importin responsible for import via Rps2(76–145) is Pse1. Hence, the preference of Rps2 for Pse1 deviates from the common preference of r-proteins for Kap123, with Pse1 stepping in place mainly in the absence of Kap123 [
13]. Our data moreover demonstrate that the mutation of R95, R97, and K99 in the Rps2(76–145) fragment greatly reduces the interaction with Pse1, suggesting that these residues are critical determinants for Pse1 binding. Previous structural analyses of Pse1 in complex with NLS sequences of three different Pse1 cargoes have led to the definition of the IK-NLS with the consensus K-V/I-X-K-X
1–2-K/H/R [
27,
28]. The segment ranging from residues 95 to 99 of Rps2 (RTRFK), however, does not match this consensus. Moreover, it is positioned within a beta-sheet (
Supplementary Figure S1B), while IK-NLSs are unstructured [
27,
28]. Hence, Rps2 likely uses a binding mode that differs from the one reported for the interaction of Pse1 with IK-NLSs, and seems to involve structured elements. In our tandem affinity purification experiment, where we expressed either Rps2(76–145)-TAP or Rps2(76–145).R
95R
97K
99>A-TAP from plasmids in a wild-type strain, we observed higher levels of the Rps2 fragment carrying the substitutions in cell lysates compared to the wild-type fragment (
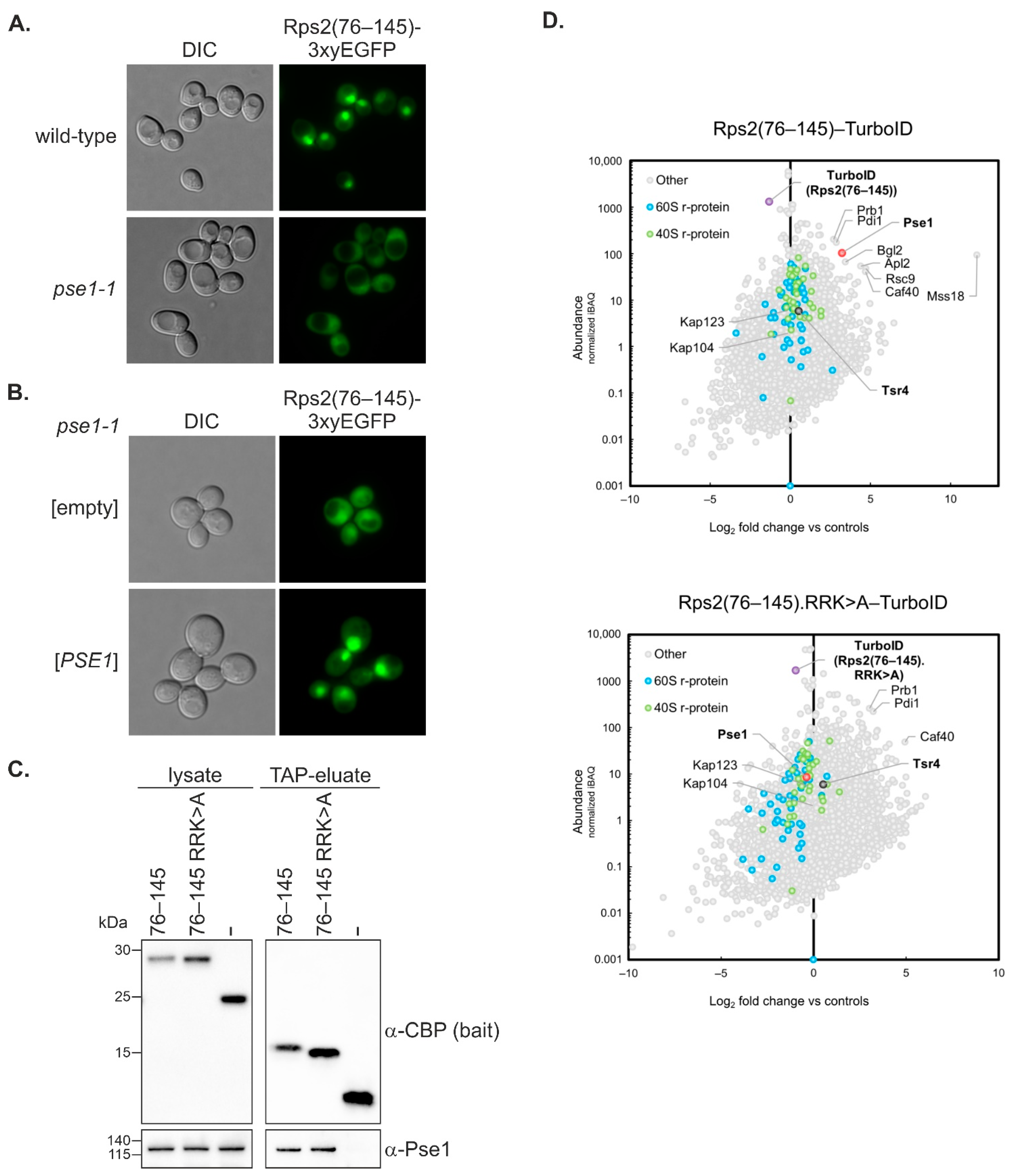
Figure 3C). This observation suggests that the substitutions of R95, R97, and K99 to A might induce structural changes that enhance the stability of the Rps2(76–145) fragment. These altered structural features might impede the efficient binding of Pse1, despite promoting protein stability.
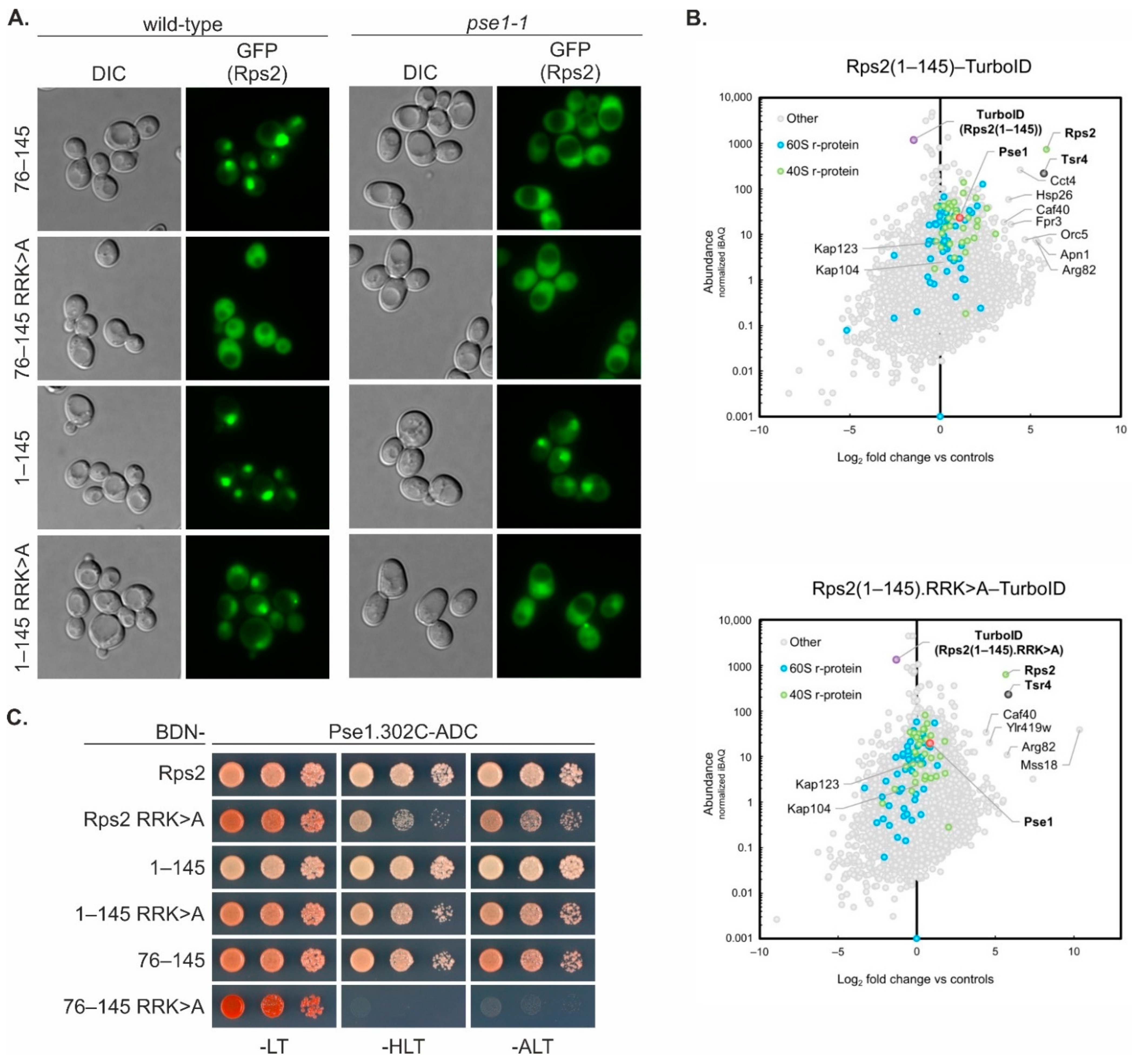
The Rps2(1–145) fragment, containing in addition to the above-discussed nuclear-targeting domain also the N-terminal part of Rps2, enters the nucleus as well, even when the residues critical for the nuclear targeting of the Rps2(76–145) fragment are mutated. Moreover, R95A, R97A, and K99A mutation reduces the Y2H interaction of the Rps2(1–145) fragment with Pse1 only slightly, while the interaction of Rps2(76–145) with Pse1 is severely reduced by these exchanges. This suggests that Pse1 may possess an additional binding site within amino acids 1–75 of Rps2. Indeed, Rps2(1–145).R
95R
97K
99>A-3xEGFP displayed an increased cytoplasmic signal in
pse1-1 mutant cells compared to wild-type cells, indicating that even if the internal NLS is not available for interaction with Pse1, Pse1 is capable of importing the Rps2(1–145) fragment. It is worth noting that none of the tested Rps2 fragment 3xEGFP fusions showed complete import inhibition in the
pse1-1 mutant, implying that, as also suggested in previous studies (see for example [
29,
30,
31]), other importins can compensate for the loss of one importin. Nevertheless, the significant defects observed in the
pse1-1 mutant strongly indicate that Pse1 is the primary importin binding to the two Rps2 NLS elements described in this study.
Nuclear import via this N-terminal nuclear-targeting region requires basic residues within the RG-rich, unstructured N-terminal part (amino acids 10–28) of Rps2. It is already known that such RGG regions can function as NLSs for Kap104 [
32,
33]; however, recognition of RG-rich NLSs by Pse1 has not been reported so far. Although our findings suggest that Rps2(1–145) can still be imported into the nucleus by Pse1 when either the N-terminal or the internal NLS is mutated, it remains unclear whether Pse1 interacts simultaneously with both binding sites in the wild-type scenario, or if it only utilizes one of them at a time. It will be interesting to further define and map the two Pse1-binding regions of Rps2 in the future, which might lead to the definition of novel NLS consensus motifs for Pse1. Notably, while the N-terminal and internal NLSs share some sequence similarities, such as positively charged amino acids with similar spacing (e.g., 95-RTRFK-99 and 17-RNRGR-21), they are embedded in entirely different structural contexts. The N-terminal NLS resides within an unstructured region, whereas the internal NLS lies within a beta-sheet (
Supplementary Figure S1B). Consequently, the two NLSs may employ distinct binding modes for Pse1 interaction.
It is important to acknowledge that the basic residues within amino acids 10–28 of Rps2, although being necessary for the nuclear targeting of a Rps2(1–145) fragment with a mutated internal NLS (
Figure 6B), are not sufficient for efficient import, as concluded from the fact that a small Rps2 fragment containing these amino acids, Rps2(1–42), does not exclusively localize to the nucleus (
Figure 1B and
Figure 6C). Hence, additional sequence elements are required for the complete functionality of the N-terminal NLS. Furthermore, it is possible that not all eight positively charged amino acids within Rps2(10–28) are essential for the function of the N-terminal NLS. It is plausible that a few specific residues within this range are crucial for the import via the N-terminal NLS, or that multiple clusters of positively charged amino acids within this sequence can fulfill this function alternatively, as recently reported for the NLS of the viral protein HIV-1 Tat [
34]. Future in-depth biochemical and structural studies might provide further insights into the binding modes and interplay between the two Rps2 NLSs.
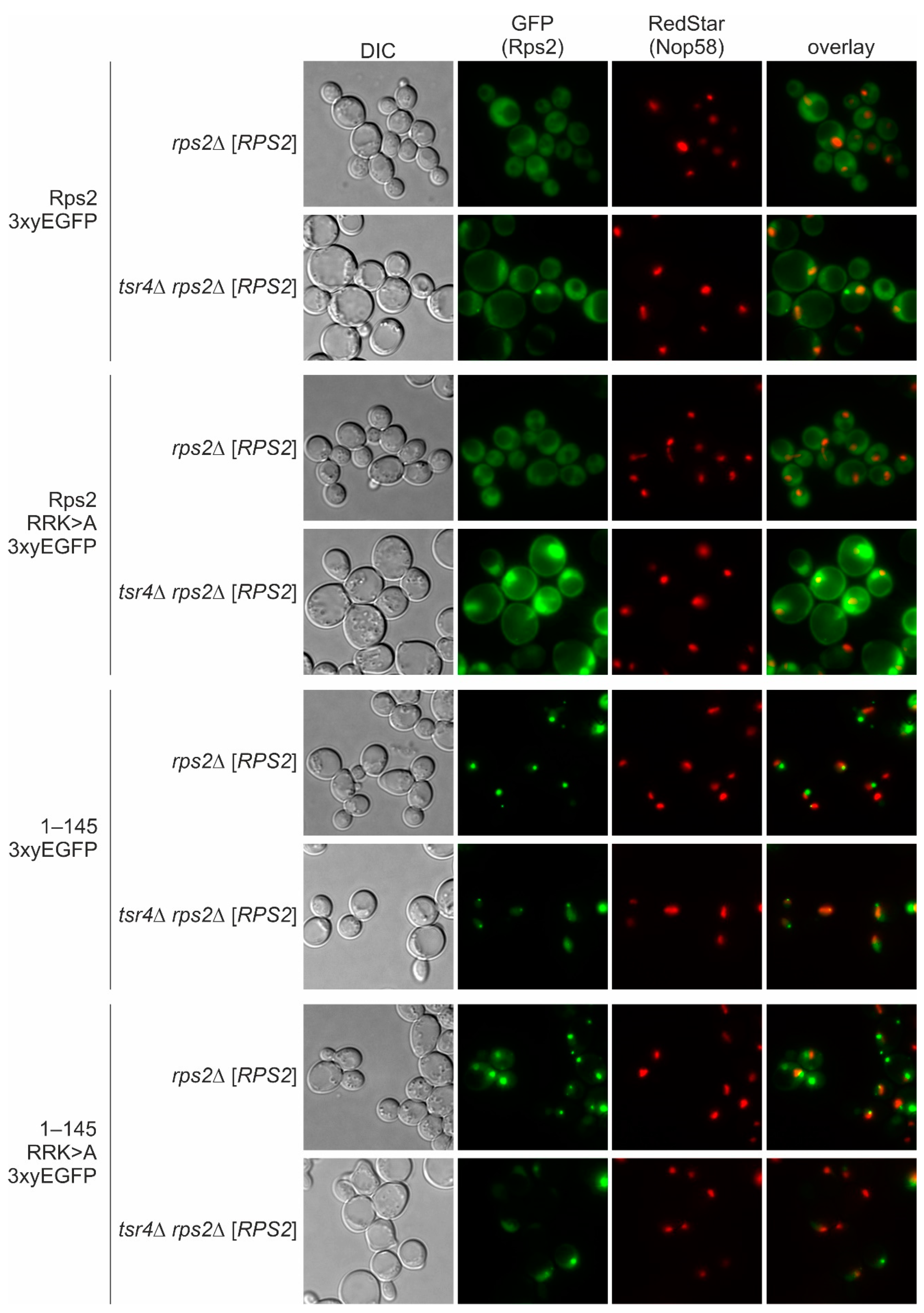
Importantly, the amino acids required for the function of the N-terminal NLS of Rps2 overlap with the binding site of its dedicated chaperone, Tsr4. Therefore, it was important to investigate whether the presence of Tsr4 affects the nuclear targeting of Rps2 via the N-terminal NLS.
We can exclude the possibility that import mediated by Rps2 amino acids 10–28 occurs via a ‘piggyback’ mechanism in which Tsr4 binds Rps2 and provides the NLS for the nuclear import of the Rps2-Tsr4 complex, as our data revealed that the presence of Tsr4 is not required for the import involving the N-terminal region of Rps2 (
Figure 5). Rps2(1–42)-3xyEGFP even exhibited a stronger nuclear signal in the absence of Tsr4 (
Figure 6C), suggesting that its import is less efficient when Tsr4 is present. One potential explanation for this effect is that Tsr4 shields the N-terminal NLS, thereby reducing the efficiency of importin binding. It is yet to be determined whether Tsr4 accompanies Rps2 into the nucleus or dissociates from Rps2 already in the cytoplasm, e.g., upon the binding of importin. Tsr4-GFP does not accumulate in the nucleus upon inhibition of the main exportin Crm1 [
10]. However, the human Tsr4 homolog PDCD2 accompanies human RPS2 into the nucleus [
35], as does the closely related PDCD2L [
36]. Further, our data demonstrate that in the absence of Tsr4, Rps2 accumulates in the nucleolus (
Figure 5 and [
7]), suggesting that Rps2’s efficient incorporation into pre-ribosomal particles is prevented. The simplest explanation for this phenotype would be that Tsr4 functions in promoting Rps2 pre-ribosome incorporation in the nucleus. On the other hand, the more efficient import of the Rps2(1–42) fragment in the absence of Tsr4 suggests that nuclear import might occur after the release of Tsr4. Alternatively, the interaction of Pse1 with the internal NLS of Rps2 may be sufficient to mediate the nuclear targeting of Rps2 bound to Tsr4, even if the N-terminal NLS is not fully accessible to the importin.
The binding of Pse1 to Rps2 could serve a second function beside nuclear import as importins have been implicated in functioning as chaperones for exposed basic domains [
12]. The richness in positive charges, together with the flexibility of the Rps2 N-terminal region, might make Rps2 particularly prone to aggregation, which could be the reason why two different, potentially redundant mechanisms for chaperoning of this region have evolved, with the main one relying on a dedicated chaperone and the second one involving an importin.
Interestingly, the N-terminal RG-rich region of Rps2 is absent in bacteria and archaea (
Supplementary Figure S1A), suggesting that it serves a eukaryote-specific function, as is the case for a targeting sequence for nuclear import. In contrast, parts of the internal positively charged NLS residues are also found in archaea and bacteria. For instance,
Pyrococcus furiosus uS5 contains all three of these residues, while
Sulfolobus and
Bacillus subtilis have two positively charged amino acids in the corresponding region (
Supplementary Figure S1A). NLS-type motifs have been observed in archaea before, suggesting that NLS sequences may have originated from sequences that originally served other functions [
37,
38].
Intriguingly, Rps2’s unstructured N-terminal region seems to be a hub for the binding of multiple interaction partners (elaborated in detail in a review article by the Bachand group within this Special Issue [
39]). Besides the binding partners investigated in this study (Tsr4 and importins), the N-terminal part of Rps2 also likely interacts (at least transiently) with Hmt1, as this enzyme methylates an arginine in the N-terminal region of Rps2 [
40,
41]. In the human system, RPS2 is bound by PDCD2 or PDCD2L, and is additionally stably bound by the arginine methyl transferase PRMT3, which competes with the zinc finger protein ZNF277 for RPS2 binding [
22,
36,
42,
43]. The investigation of the timing and coordination of these manifold interactions will be an interesting subject for future studies.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





