1. Introduction
During the COVID-19 pandemic, whole genome sequencing (WGS) of the SARS-CoV-2 virus has played a crucial role in unraveling important biological information. Through phylogenetic analysis, it has been revealed that SARS-CoV-2 shares 50% and 79% sequence similarity with MERS-CoV and SARS-CoV, respectively, indicating their evolutionary connections [
1]. Notably, the genome sequence of SARS-CoV-2 exhibits an 85% similarity to a bat coronavirus, establishing its zoonotic origin within the Coronaviridae family and the Betacoronavirus genus [
2]. These genomic data have been instrumental in confirming the virus’s source and classification. Recognizing the significance of gathering genetic data from diverse SARS-CoV-2 sequences and variants, researchers worldwide swiftly recognized the need for comprehensive genome information [
3,
4]. The Centers for Disease Control and Prevention’s Office of Advanced Molecular Detection (AMD) released details regarding SARS-CoV-2 whole genome sequencing on various platforms, including PacBio, Illumina, and Ion Torrent. Emphasizing the importance of publicly accessible genome sequences, the World Health Organization (WHO) strongly supports their utilization in developing novel public health strategies and conducting research to combat the spread of COVID-19. A valuable resource in this endeavor is the Global Initiative on Sharing All Influenza Data (GISAID), which hosts one of the largest international databases of SARS-CoV-2 genome sequences [
5]. Leveraging GISAID, along with the open-source tools NextStrain and NextClade, researchers have made significant advancements in their investigations [
6,
7]. These resources have proven instrumental in understanding the evolution and characteristics of the virus, aiding in the development of efficient strategies to mitigate the COVID-19 infection’s spread [
8,
9,
10].
Third-generation sequencing technology has emerged as a widely used method for sequencing SARS-CoV-2 during the pandemic. These technologies, known for their ability to generate long reads, are increasingly employed in transcriptomics studies. Advancements in long-read sequencing enable the comprehensive sequencing of RNA molecules, utilizing cDNA or direct RNA protocols from Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) [
11,
12,
13]. However, the high error rates associated with long-read technologies pose challenges for accurate and efficient downstream analysis, such as genome assembly. Indels, or insertions and deletions, are the primary error types that complicate alignment processes. While various error correction tools exist, there remains a need for further development in this computational biology domain. To effectively combat the COVID-19 infection and facilitate research, an increased number of SARS-CoV-2 genome sequences are required [
14,
15]. Researchers worldwide rely on third-generation sequencing technologies to sequence the virus. Cutting-edge technology heavily relies on SARS-CoV-2 genomic sequences for virus tracking. To analyze genomic data effectively, scientists employ machine learning (ML) and deep learning (DL) algorithms along with embedding methods for classification purposes [
16,
17,
18,
19]. ML and DL algorithms have become valuable tools even for novice bioinformatics practitioners and core data analysts who may lack prior knowledge of sequencing technologies and associated challenges. These algorithms enable comprehensive analysis of SARS-CoV-2 sequencing data, contributing to advancements in classification techniques and aiding in our understanding of the virus’s genetic characteristics and behavior. Therefore, it is crucial to establish a robust benchmark report on SARS-CoV-2 genome sequences generated using third-generation sequencing technology, which will serve as a guide for future genomic research involving long-read sequencing.
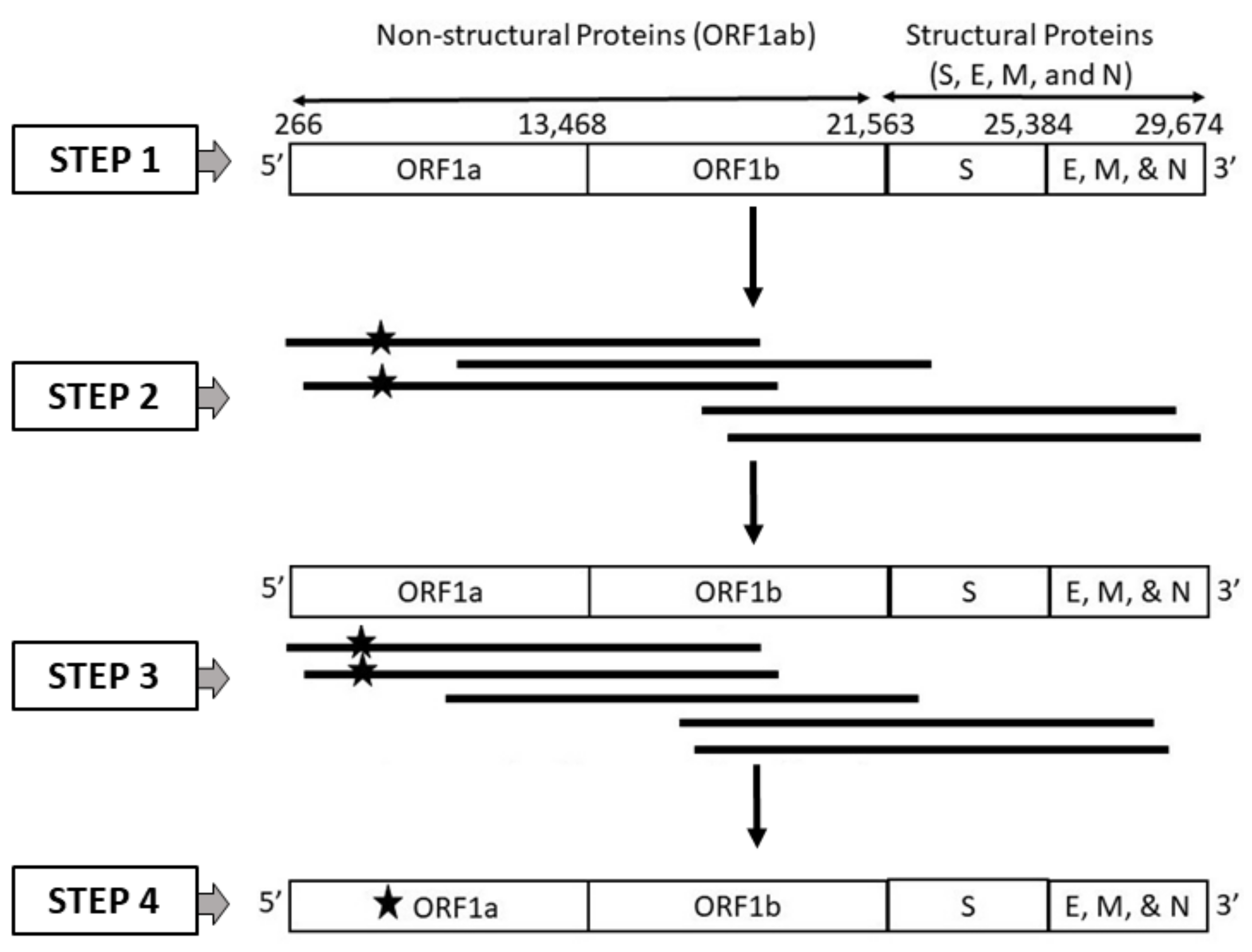
The current study aims to evaluate the performance of current classification models in handling third-generation sequencer-specific errors present in SARS-CoV-2 genome sequences. Specifically, the study investigates the effectiveness of various embedding methods under specified levels of disturbance. The evaluation of machine learning models on SARS-CoV-2 genomic sequences remains limited, with only a few existing studies in this area. For instance, a previous study [
20] conducted a benchmark of ML and DL models using different embedding methods for classifying SARS-CoV-2 genome sequences that included sequencer-specific errors. However, this study did not identify the best ML model for SARS-CoV-2 genome sequence classification. In line with a similar approach, our current study focuses exclusively on SARS-CoV-2 genomes generated using long reads obtained from third-generation sequencing (TGS) technologies, such as PacBio and Nanopore, while also considering the possibility of random errors occurring by chance. To assess the effectiveness of machine learning algorithms on SARS-CoV-2 genome sequences, we conducted simulations that accounted for various error types. Our simulations employed two primary approaches: one involved generating SARS-CoV-2 genome sequences with platform-specific errors (PacBio or ONT), while the other introduced random errors. The workflow for these simulations is depicted in
Figure 1. To analyze the SARS-CoV-2 sequences, we employed six distinct embedding methods, including one-hot encoding (OHE), Wasserstein-distance-guided representation learning (WDGRL), string kernel, spaced
k -mers, weighted
k-mers, and weighted position weight matrix (PWM). Leveraging these embedding methods, we performed supervised analyses using a variety of linear and non-linear classifiers considering both clean and error-incorporated SARS-CoV-2 sequences. This comprehensive methodology enabled us to evaluate the effectiveness of these methods in detecting errors and classifying sequences.
The subsequent sections of the current study are described in an arranged manner as follows.
Section 2 comprises comprehensive details of the dataset statistics, dataset generation methodology, and various embedding techniques considered to convert SARS-CoV-2 genome sequences to fixed-length numerical representations. Our results for accuracy and robustness are reported in
Section 3. Finally, the current study concludes in
Section 4.
3. Results and Discussion
This section provides an overview of the outcomes achieved by our methods on the datasets employed in this study. The first subsection, labeled
Section 3.1, discusses the accuracy evaluation of machine learning classification algorithms that utilized various embedding methods. The second subsection, labeled
Section 3.2, covers the robustness evaluation of machine learning classification algorithms that used different embedding methods. The third subsection, labeled
Section 3.3, focuses on the comparison of predictive performance of machine learning models on SARS-CoV-2 sequences with errors obtained from PacBio and ONT sequencers. Lastly,
Section 3.4 explores the analysis of coronavirus variants using various embedding vector generation methods with the aid of t-SNE visualization.
3.1. Accuracy Evaluation of Machine Learning Classification Algorithms Using Different Embedding Methods
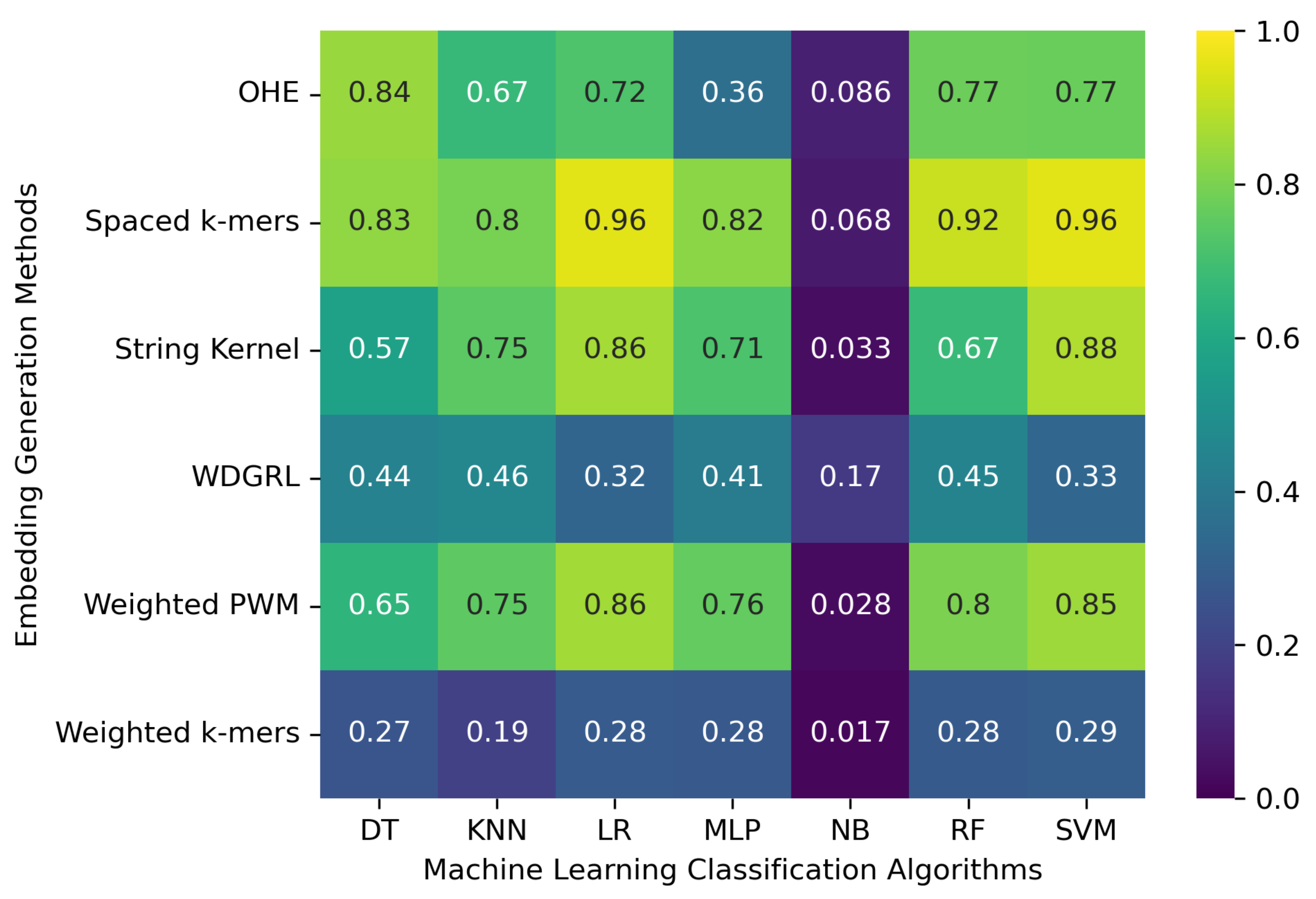
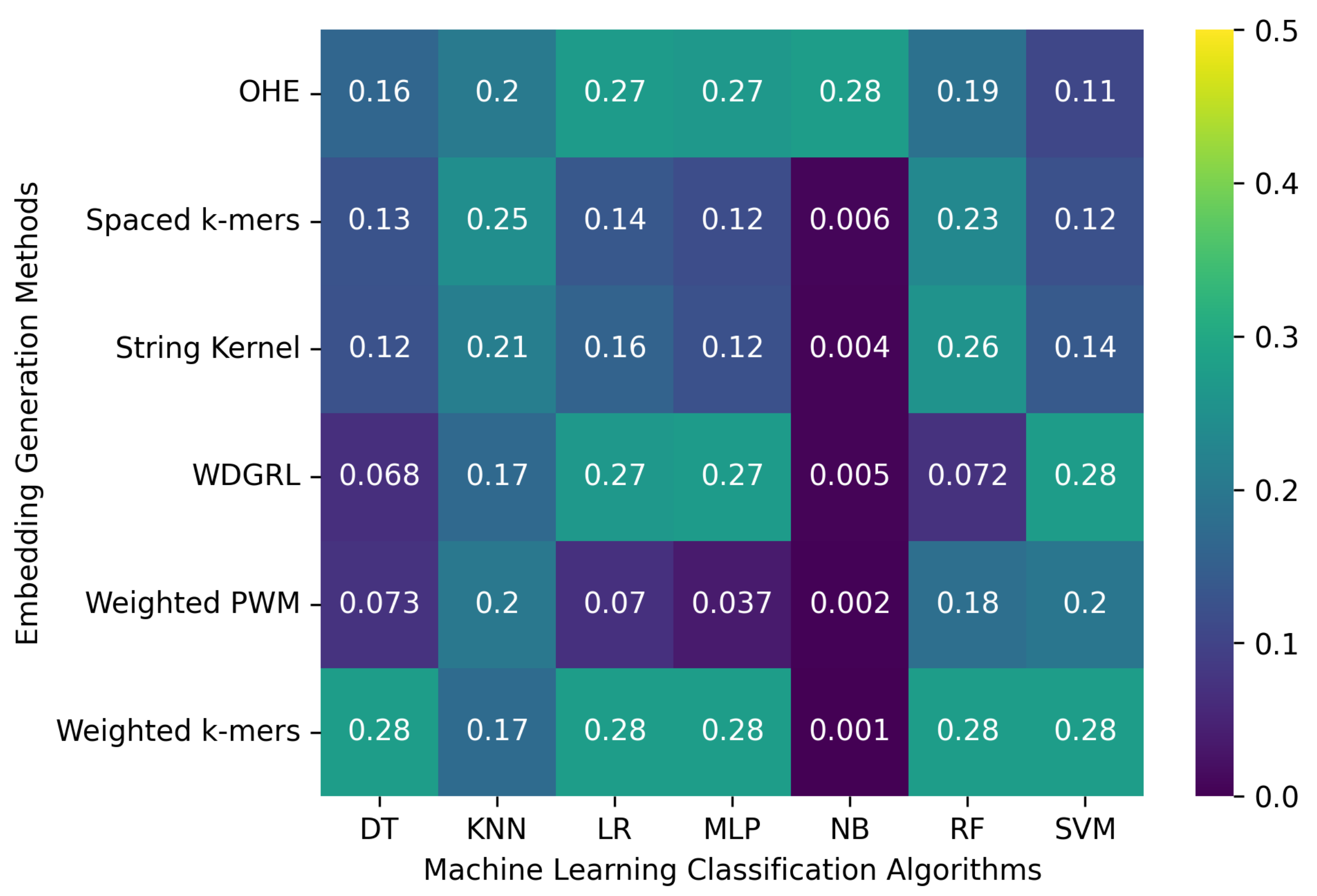
We considered 8172 clean (error-free) full-length SARS-CoV-2 nucleotide sequences from the GISAID database. These sequences were used to evaluate the machine learning models with embedding methods. In order to do that, we split the sequences into training and test sets with a 70/30% ratio. After that, we executed each analysis five times and considered the average results, reported in
Table 2 and
Figure 2. The results show that the machine learning classification algorithms’ performance significantly varies depending on the embedding method employed. Specifically, the one-hot embedding method leads to an accuracy of 0.773 for the SVM algorithm, whereas the WDGRL embedding method only results in an accuracy of 0.327. The spaced
k-mers embedding method with the SVM, RF, LR, and DT classification algorithms achieves an accuracy of up to 0.956. This method employs
g-mers and
k-mers to decrease the sparsity and size of
k-mers in the genome sequence. As a result, it generates fixed-length vectors that capture the occurrences of all possible
k-mers, which are then used to construct frequency vectors representing the frequency of each
k-mer in the sequence. This method performs well with the error-free set of SARS-CoV-2 genome sequences. However, the NB algorithm yields the worst results, with an accuracy of only 0.017 when the weighted
k-mers embedding method is used. Additionally, some algorithms, such as SVM and LR, have significantly longer training times compared to others. Thus, while selecting an algorithm and embedding method, one should consider both performance and training time.
3.2. Robustness Evaluation of Machine Learning Classification Algorithms Using Different Embedding Methods
We considered 8172 clean SARS-CoV-2 sequences and incorporated errors specific to PacBio, ONT, and the random protocol, as described in the methods section. This approach helped to generate three different types of datasets: genome sequences with typical PacBio sequencing errors, ONT sequencing errors, and random errors. To evaluate the robustness of the machine learning models with embedding methods on the three different datasets, we train the models with clean SARS-CoV-2 sequences and test them on error-incorporated sequences.
3.2.1. The Robustness Results for PacBio Sequencing Error-Incorporated Datasets
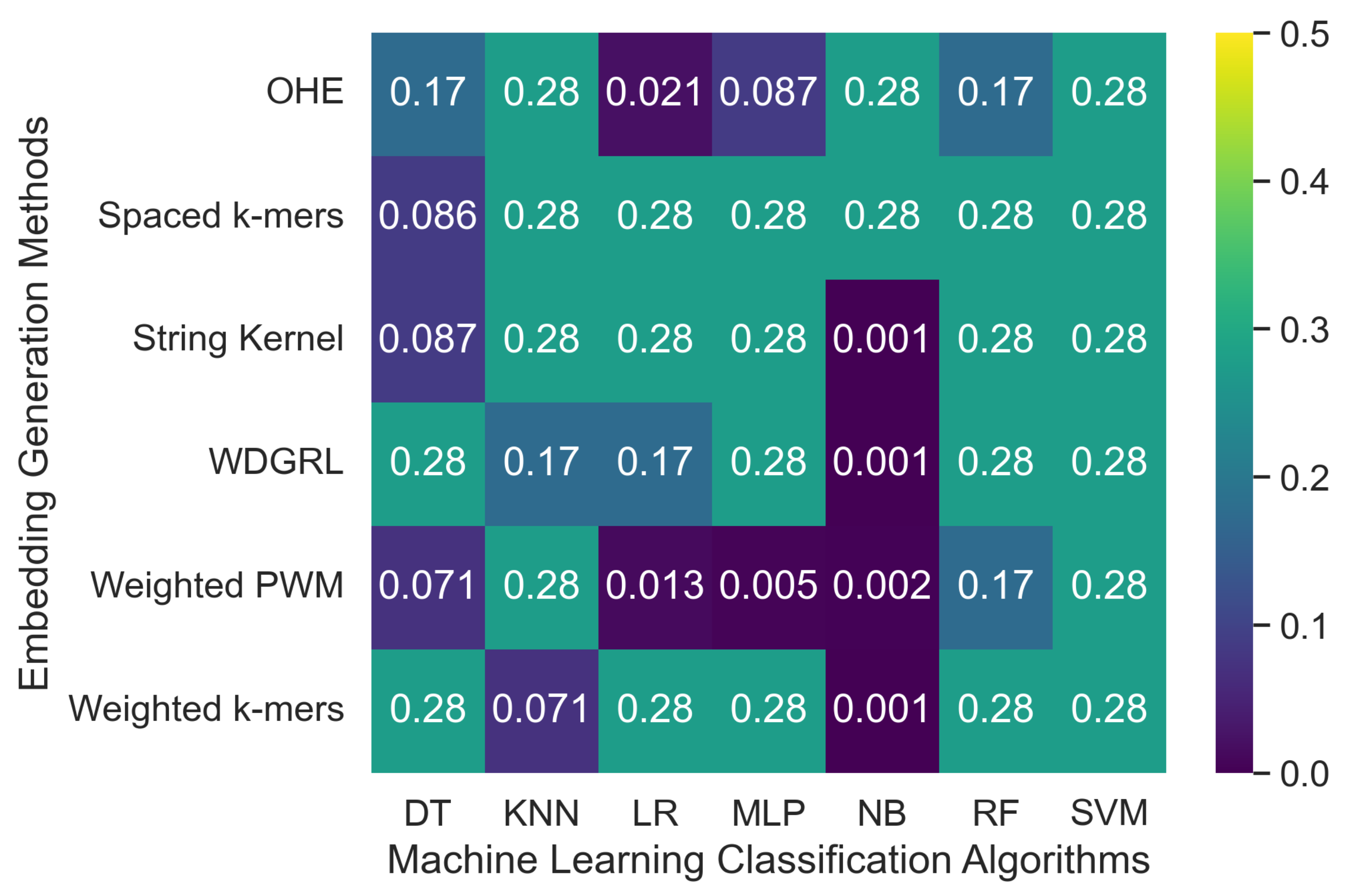
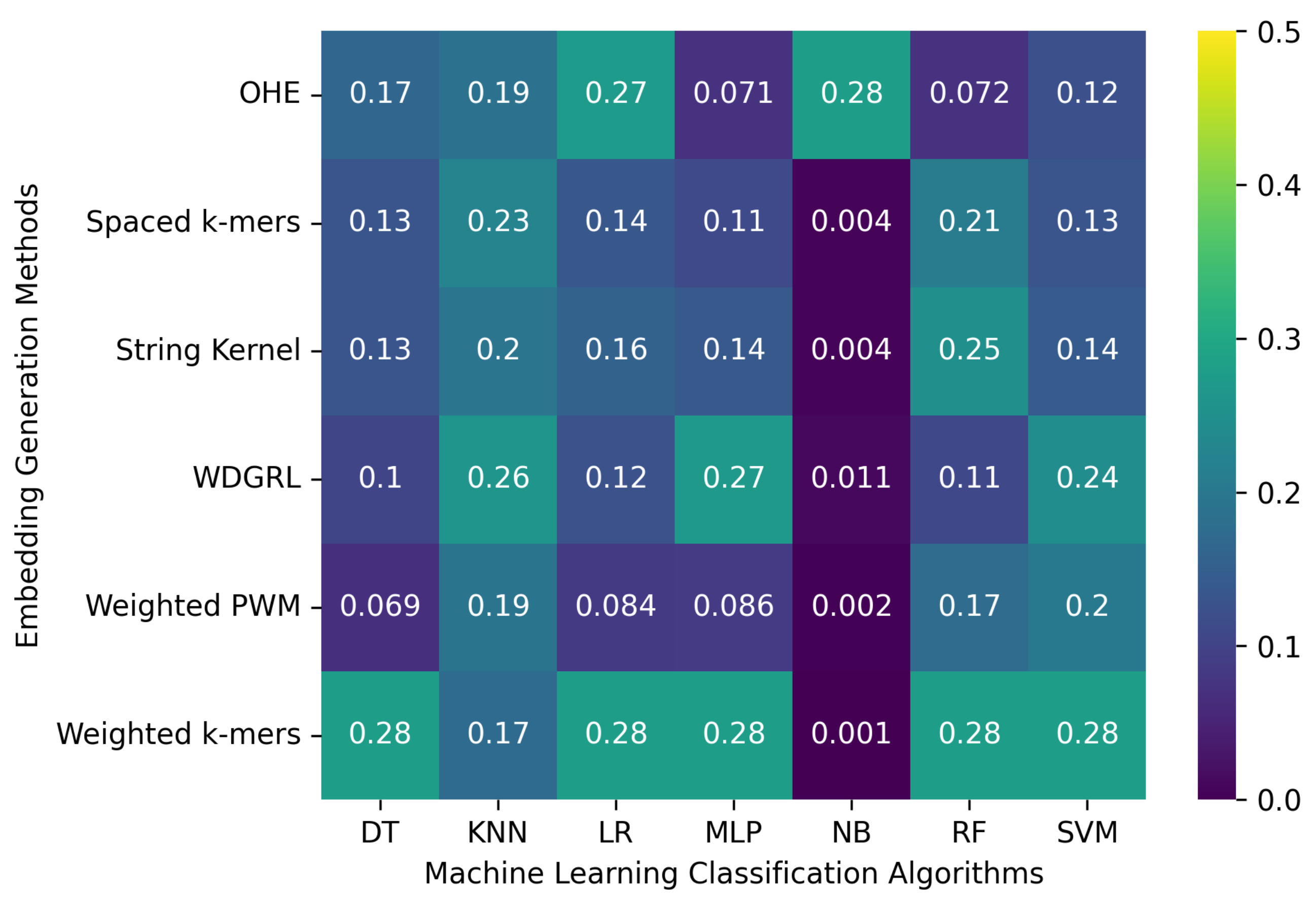
Table 3 displays the accuracy values of various machine learning classification algorithms that used different embedding methods on SARS-CoV-2 genome sequence datasets simulated at two different depths, 5 and 10, with PacBio sequencer-specific errors incorporated. Furthermore,
Figure 3 reveals that the accuracy values for machine learning algorithms ranged from 0.001 to 0.276 across all embedding methods. The spaced
k-mers embedding method, in general, performed better than other embedding methods, achieving the highest accuracy value of 0.276 for the maximum number of algorithms for the depth-5 sequencing dataset, and a similar trend was observed for the depth-10 dataset. The reason for this is that the spaced
k-mers method employs
g-mers and
k-mers to decrease the sparsity and size of
k-mers in the genome sequence. As a result, it generates fixed-length vectors that capture the occurrences of all possible
k-mers, which are then used to construct frequency vectors representing the frequency of each
k-mer in the sequence. The accuracy results confirm that as the depth decreases, the error rate increases, resulting in a decrease in the performance of machine learning models. The model’s performance did not improve significantly by increasing sequencing depth from 5 between the two SARS-CoV-2 genome sequence datasets.
3.2.2. The Robustness Results for Oxford Nanopore Technologies (ONT) Sequencing Error-Incorporated Datasets
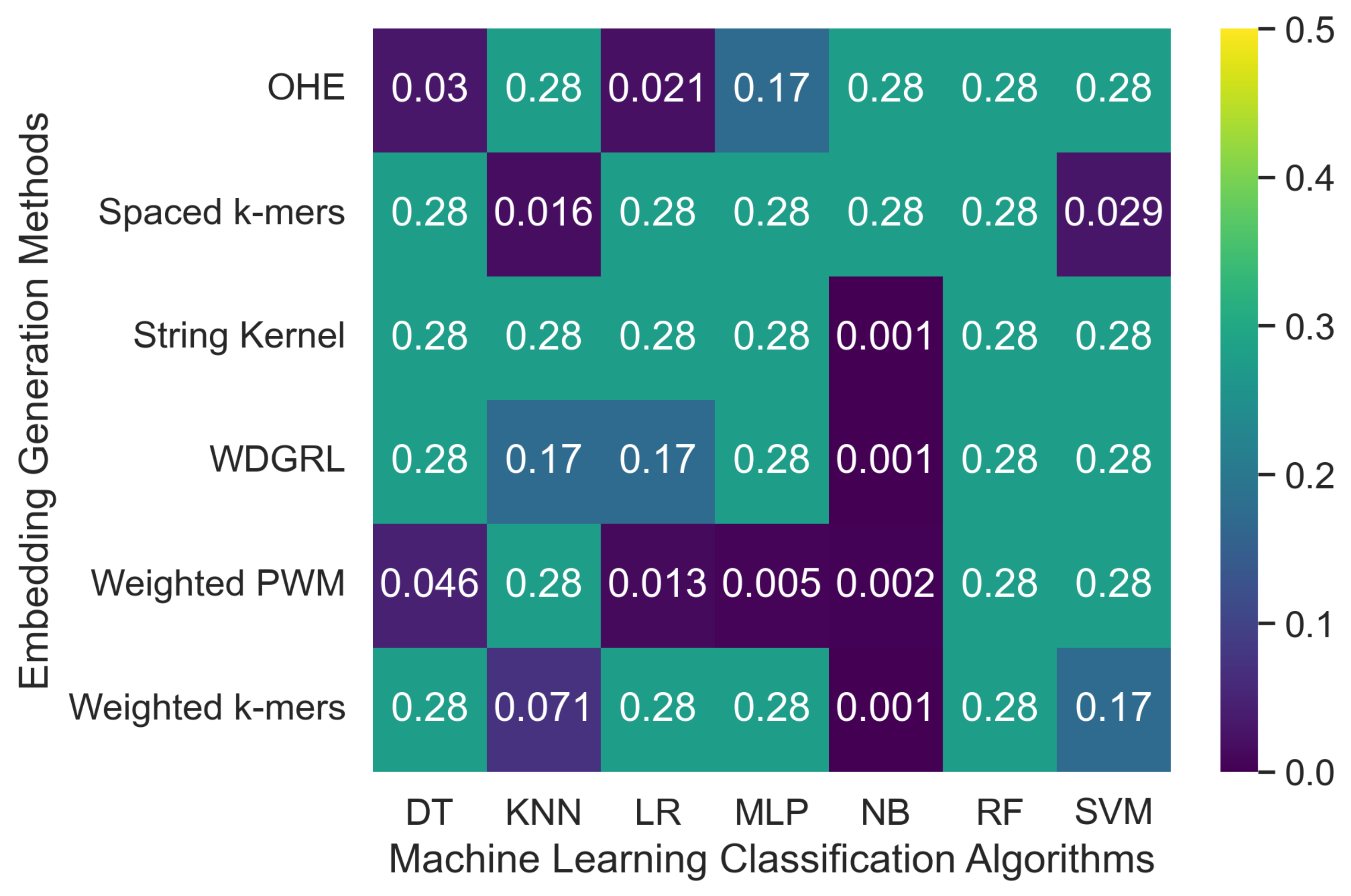
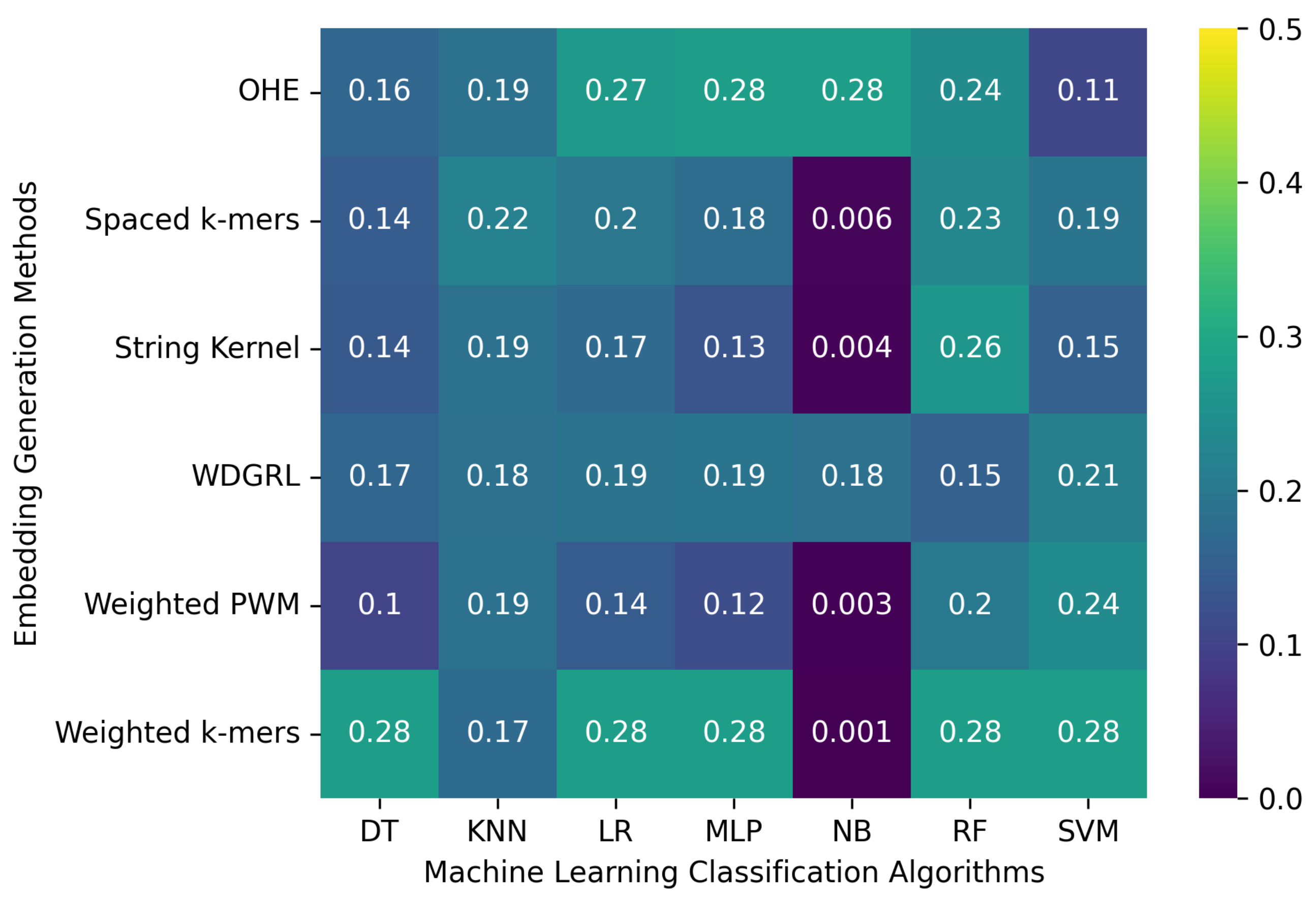
Table 4 displays the accuracy values obtained from different machine learning algorithms using various embedding methods on two SARS-CoV-2 genome sequence datasets with depths of 5 and 10, respectively, which were generated from long-reads containing Oxford Nanopore Technology (ONT) sequencer-specific errors. Moreover,
Figure 4 presents a heatmap that visualizes the accuracy values, which ranged from 0.001 to 0.276. The weighted
k-mers embedding method resulted in the highest accuracy values for the majority of the machine learning algorithms on both datasets, i.e., depths of 5 and 10. Because each
k-mer is given a weight depending on its inverse document frequency under the weighted
k-mers technique, this method generates fixed-length vectors that capture the existence of all potential
k-mers. These vectors are then used to create frequency vectors that indicate the frequency of each
k-mer in the sequence. However, due to the lower sequencing depth with ONT sequencer-specific errors, poor-quality SARS-CoV-2 genome sequences were generated, leading to a significant decrease in the predictive performance of machine learning algorithms.
3.2.3. The Robustness Results for Random-Error-Incorporated Datasets
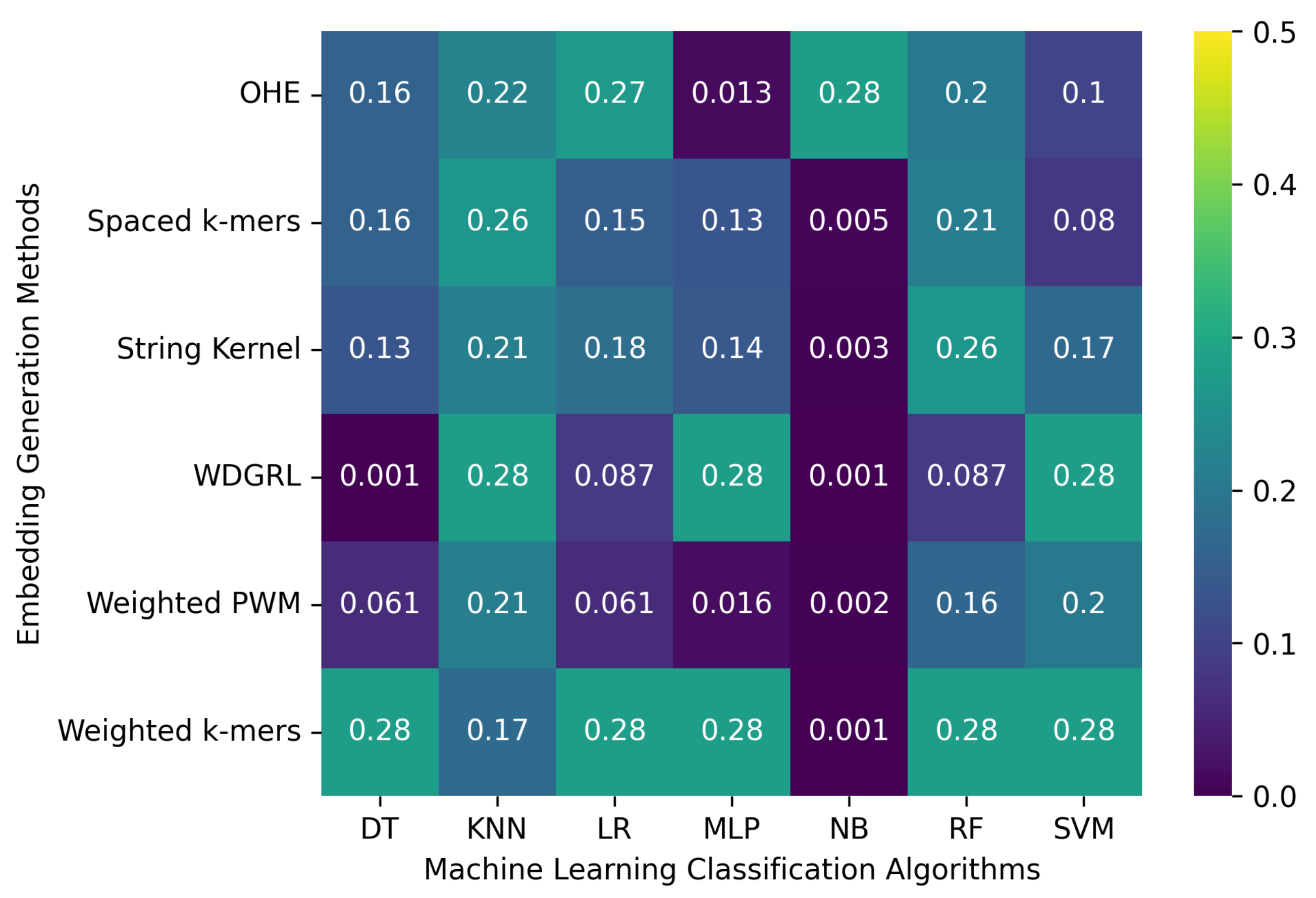
In this section, we evaluated the accuracy of various machine learning algorithms using different embedding methods on two SARS-CoV-2 genome sequence datasets. These datasets were generated by incorporating random errors into long-reads at depths of 5 and 10. The results, presented in
Table 5 and
Figure 5, indicate that the weighted
k-mers method achieved the highest accuracy of 0.276 across the majority of machine learning classification algorithms for both datasets. The main objective of incorporating random errors into the SARS-CoV-2 datasets was to compare the performance of machine learning models on datasets generated by different types of errors, including sequencer-specific errors and random errors. Interestingly, we found that there was not much difference in accuracy between these two types of errors.
3.3. Comparison of Predictive Performance of Machine Learning Models on SARS-CoV-2 Sequences with Errors from PacBio and ONT Sequencers
Third-generation sequencing (TGS) technologies such as PacBio and Oxford Nanopore Technology (ONT) are widely used for generating long reads with high error rates. However, PacBio technology sequences a DNA molecule multiple times, whereas ONT sequences it only twice, making PacBio generate higher-quality data with lower error rates compared to ONT. Through our analysis, we discovered that the errors specific to the PacBio sequencer have a more significant impact on the predictive performance of machine learning (ML) models on SARS-CoV-2 sequences than errors specific to ONT. Our ML model’s predictive performance indicated that PacBio sequences have a lower error rate than ONT, but the low predictive power was due to low coverage. We also compared the predictive performance of ML models on SARS-CoV-2 sequences incorporated with random errors with other datasets and observed that the results were similar to the ONT scenario.
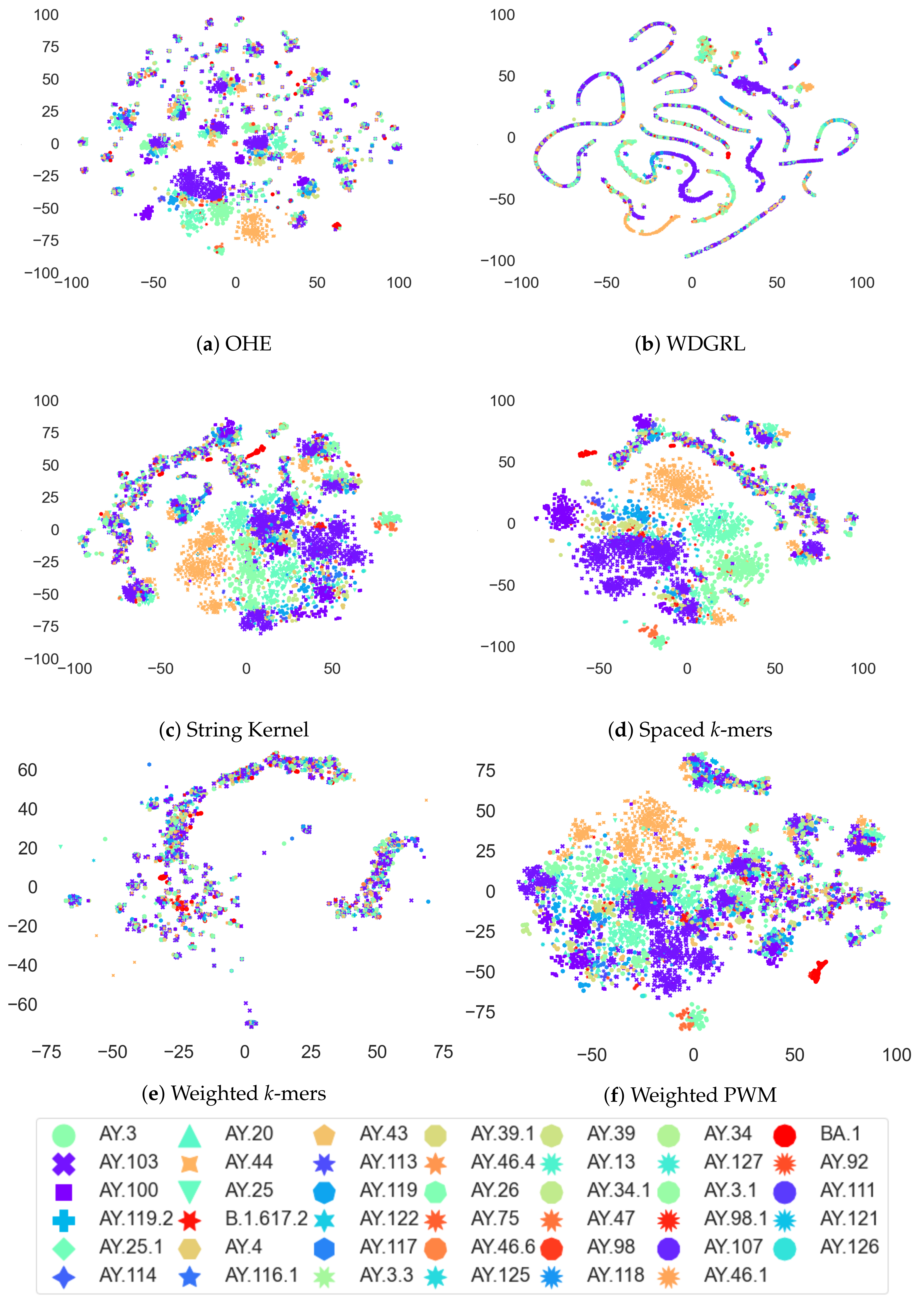
3.4. Analysis of Coronavirus Variants Based on Different Embedding Vector Generation Methods Using t-SNE Visualization
The t-distributed stochastic neighbor embedding (t-SNE) method is a widely used data visualization technique that preserves the pairwise distances between high-dimensional vectors in a lower-dimensional space. In this study, we employed t-SNE to visualize the clustering patterns of different coronavirus variants using various embedding vector generation methods, including one-hot encoding (OHE), Wasserstein-distance-guided representation learning (WDGRL), string kernel, spaced
k-mer, weighted
k-mer, and weighted position weight matrix (PWM). Our analysis, as depicted in
Figure 6, reveals the remarkable effectiveness of t-SNE in capturing the pairwise distance information and unveiling the distinct grouping patterns of coronavirus variants in a two-dimensional space. Specifically, the t-SNE plot based on the OHE vector demonstrated that AY.44 variants were more clearly grouped than the other variants, while the WDGRL vector maintained a smaller group of variants than OHE vector. Furthermore, the string-kernel-vector-based t-SNE plot exhibited clearer grouping patterns of AY.44 and other variants than the OHE vector. Additionally, the spaced
k-mer vector method showed a more distinct grouping of variants compared to other embedding vector generation methods. The weighted
k-mer vector exhibited grouping of the variants similar to the WDGRL vector, whereas the weighted PWM vector showed grouping patterns more similar to the string kernel vector.
4. Conclusions
In summary, the COVID-19 pandemic has emphasized the importance of transitioning from second-generation to third-generation sequencing technology. Long-read sequencing has emerged as a critical tool for unraveling various genomic features of the SARS-CoV-2 virus. With the ability to read longer DNA fragments, ranging from 5000 to 30,000 base pairs, long-read sequencing addresses a major challenge faced by short-read sequencing methods. This extended read length has enabled researchers to detect complex structural variations, including large insertions/deletions, inversions, repeats, duplications, and translocations. Additionally, long-read sequencing has facilitated the phasing of SNPs into haplotypes and facilitated de novo genome assembly. However, it is important to acknowledge that the high error rate associated with long-read sequencing may impact the interpretation of SARS-CoV-2’s biology.
In this study, we have demonstrated that the accuracy of machine learning classification algorithms in analyzing SARS-CoV-2 genome sequences greatly depends on the selection of appropriate embedding methods. Our analysis of simulated SARS-CoV-2 viral sequences underscores the value of employing robust embedding techniques capable of effectively managing errors and accurately categorizing genome sequences considering both long-read sequencer-specific errors and random error types. Specifically, we have identified certain embedding methods, such as WDGRL and weighted PWM, as superior in detecting errors and classifying sequences. These findings highlight the potential of machine learning in analyzing SARS-CoV-2 genomic data, contributing to a deeper understanding of the virus’s evolution and spread.
In the future, we want to explore more sequence embedding and advanced deep learning methods on SARS-CoV-2 genomic sequences generated at different long-read sequencing depths with third-generation sequence-specific errors. These experiments will help us develop robust models to improve our ability to adapt long-read sequencing technology (PacBio and ONT) to produce error-free SARS-CoV-2 genome sequences to understand and answer critical biological questions.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}