
Regulation of Satiety by Bdnf-e2-Expressing Neurons through TrkB Activation in Ventromedial Hypothalamus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Food Consumption Test
2.3. Body Composition Analysis
2.4. Cold Tolerance Test
2.5. Western Blot
2.6. Immunohistochemistry
2.7. Real-Time qRT-PCR
2.8. Construction and Packaging of e2-BDNF-Myc
2.9. Peripheral Treatment of TrkB Agonist Antibody
2.10. Stereotactic Injection of Adeno-Associated Virus and TrkB Agonistic Antibody
2.11. Data Processing
3. Results
3.1. Body Weight and Food Intake in Promoter-Specific Bdnf Mutant Lines
3.2. Bdnf-e1−/− Mice Exhibited Hyperphagia in Isolation but Normal Food-Intake When Reared in Groups
3.3. Bdnf-e2−/− Mice Are Obese Due to Hyperphagia
3.4. Distribution of Bdnf-e2 Transcript in Brain
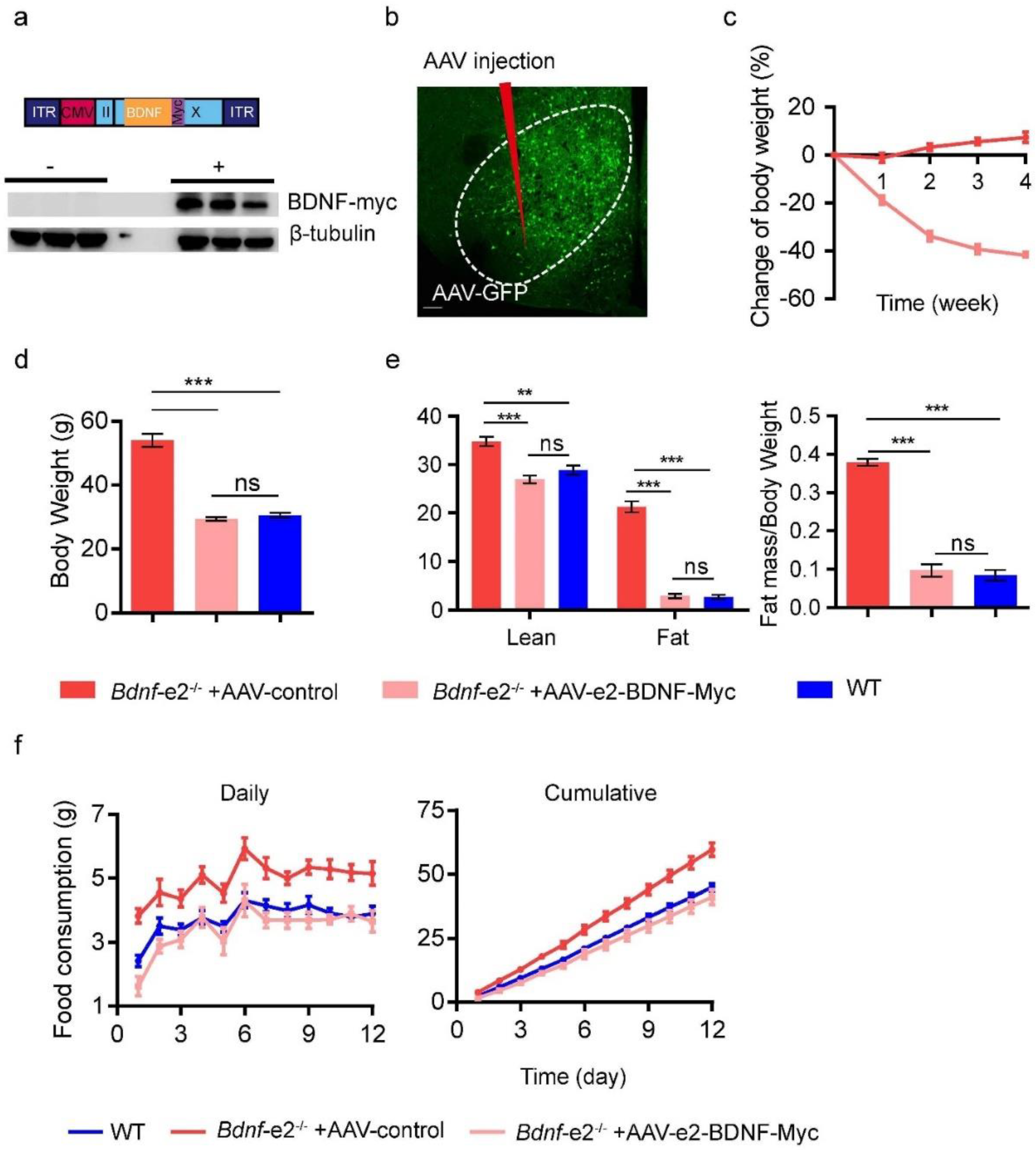
3.5. Restoration of Energy Intake through Re-Expressing Bdnf-e2 Transcript in VMH but Not DVC in Bdnf-e2−/− Mice
3.6. Rescuing Hyperphagia Deficits by Chemogenetic Activation of VMH Neurons
3.7. Rescuing Hyperphagia Deficits by Activation of VMH TrkB
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waterson, M.J.; Horvath, T.L. Neuronal Regulation of Energy Homeostasis: Beyond the Hypothalamus and Feeding. Cell Metab. 2015, 22, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-S.; Seeley, R.J.; Sandoval, D.A. Signalling from the periphery to the brain that regulates energy homeostasis. Nat. Rev. Neurosci. 2018, 19, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef]
- Thorleifsson, G.; Walters, G.B.; Gudbjartsson, D.F.; Steinthorsdottir, V.; Sulem, P.; Helgadottir, A.; Styrkarsdottir, U.; Gretarsdottir, S.; Thorlacius, S.; Jonsdottir, I.; et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 2009, 41, 18–24. [Google Scholar] [CrossRef]
- Walley, A.J.; Asher, J.E.; Froguel, P. The genetic contribution to non-syndromic human obesity. Nat. Rev. Genet. 2009, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Ernest Lyons, W.; Mamounas, L.A.; Ricaurte, G.A.; Coppola, V.; Reid, S.W.; Bora, S.H.; Wihler, C.; Koliatsos, V.E.; Tessarollo, L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc. Natl. Acad. Sci. USA 1999, 96, 15239–15244. [Google Scholar] [CrossRef] [PubMed]
- Kernie, S.G.; Liebl, D.J.; Parada, L.F. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000, 19, 1290–1300. [Google Scholar] [CrossRef]
- Han, J.C.; Liu, Q.-R.; Jones, M.; Levinn, R.L.; Menzie, C.M.; Jefferson-George, K.S.; Adler-Wailes, D.C.; Sanford, E.L.; Lacbawan, F.L.; Uhl, G.R.; et al. Brain-Derived Neurotrophic Factor and Obesity in the WAGR Syndrome. N. Engl. J. Med. 2008, 359, 918–927. [Google Scholar] [CrossRef]
- Ernst, C.; Marshall, C.R.; Shen, Y.; Metcalfe, K.; Rosenfeld, J.; Hodge, J.C.; Torres, A.; Blumenthal, I.; Chiang, C.; Pillalamarri, V.; et al. Highly Penetrant Alterations of a Critical Region Including BDNF in Human Psychopathology and Obesity. Arch. Gen. Psychiatry 2012, 69, 1238–1246. [Google Scholar] [CrossRef]
- Xu, B.; Xie, X. Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 2016, 17, 282–292. [Google Scholar] [CrossRef]
- Faraguna, U.; Vyazovskiy, V.V.; Nelson, A.B.; Tononi, G.; Cirelli, C. A causal role for brain-derived neurotrophic factor in the homeostatic regulation of sleep. J. Neurosci. 2008, 28, 4088–4095. [Google Scholar] [CrossRef]
- Martinowich, K.; Lu, B. Interaction between BDNF and Serotonin: Role in Mood Disorders. Neuropsychopharmacology 2007, 33, 73–83. [Google Scholar] [CrossRef]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef]
- You, H.; Lu, B. Diverse Functions of Multiple Bdnf Transcripts Driven by Distinct Bdnf Promoters. Biomolecules 2023, 13, 655. [Google Scholar] [CrossRef]
- Maynard, K.R.; Hill, J.L.; Calcaterra, N.E.; Palko, M.E.; Kardian, A.; Paredes, D.; Sukumar, M.; Adler, B.D.; Jimenez, D.V.; Schloesser, R.J.; et al. Functional Role of BDNF Production from Unique Promoters in Aggression and Serotonin Signaling. Neuropsychopharmacology 2016, 41, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Sakata, K.; Martinowich, K.; Woo, N.H.; Schloesser, R.J.; Jimenez, D.V.; Ji, Y.; Shen, L.; Lu, B. Role of activity-dependent BDNF expression in hippocampal-prefrontal cortical regulation of behavioral perseverance. Proc. Natl. Acad. Sci. USA 2013, 110, 15103–15108. [Google Scholar] [CrossRef]
- Hill, J.L.; Hardy, N.F.; Jimenez, D.V.; Maynard, K.R.; Kardian, A.S.; Pollock, C.J.; Schloesser, R.J.; Martinowich, K. Loss of promoter IV-driven BDNF expression impacts oscillatory activity during sleep, sensory information processing and fear regulation. Transl. Psychiatry 2016, 6, e873. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Chu, P.; Guo, W.; Lu, B. A subpopulation of Bdnf-e1-expressing glutamatergic neurons in the lateral hypothalamus critical for thermogenesis control. Mol. Metab. 2020, 31, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Pandit, M.; Behl, T.; Sachdeva, M.; Arora, S. Role of brain derived neurotropic factor in obesity. Obes. Med. 2020, 17, 100189. [Google Scholar] [CrossRef]
- Martinowich, K.; Manji, H.; Lu, B. New insights into BDNF function in depression and anxiety. Nat. Neurosci. 2007, 10, 1089–1093. [Google Scholar] [CrossRef]
- McAllan, L.; Maynard, K.R.; Kardian, A.S.; Stayton, A.S.; Fox, S.L.; Stephenson, E.J.; Kinney, C.E.; Alshibli, N.K.; Gomes, C.K.; Pierre, J.F.; et al. Disruption of brain-derived neurotrophic factor production from individual promoters generates distinct body composition phenotypes in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E1168–E1184. [Google Scholar] [CrossRef] [PubMed]
- Võikar, V.; Polus, A.; Vasar, E.; Rauvala, H. Long-term individual housing in C57BL/6J and DBA/2 mice: Assessment of behavioral consequences. Genes Brain Behav. 2005, 4, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Pais, A.B.; Pais, A.C.; Elmisurati, G.; Park, S.H.; Miles, M.F.; Wolstenholme, J.T. A Novel Neighbor Housing Environment Enhances Social Interaction and Rescues Cognitive Deficits from Social Isolation in Adolescence. Brain Sci. 2019, 9, 336. [Google Scholar] [CrossRef]
- Liu, N.; Wang, Y.; An, A.Y.; Banker, C.; Qian, Y.H.; O’Donnell, J.M. Single housing-induced effects on cognitive impairment and depression-like behavior in male and female mice involve neuroplasticity-related signaling. Eur. J. Neurosci. 2020, 52, 2694–2704. [Google Scholar] [CrossRef] [PubMed]
- Zelikowsky, M.; Hui, M.; Karigo, T.; Choe, A.; Yang, B.; Blanco, M.R.; Beadle, K.; Gradinaru, V.; Deverman, B.E.; Anderson, D.J. The Neuropeptide Tac2 Controls a Distributed Brain State Induced by Chronic Social Isolation Stress. Cell 2018, 173, 1265–1279.e19. [Google Scholar] [CrossRef]
- Schipper, L.; Harvey, L.; van der Beek, E.M.; van Dijk, G. Home alone: A systematic review and meta-analysis on the effects of individual housing on body weight, food intake and visceral fat mass in rodents. Obes. Rev. 2018, 19, 614–637. [Google Scholar] [CrossRef]
- Liu, Y.; Lv, L.; Wang, L.; Zhong, Y. Social Isolation Induces Rac1-Dependent Forgetting of Social Memory. Cell Rep. 2018, 25, 288–295.e3. [Google Scholar] [CrossRef]
- Morrison, S.F.; Madden, C.J.; Tupone, D. Central neural regulation of brown adipose tissue thermogenesis and energy expenditure. Cell Metab. 2014, 19, 741–756. [Google Scholar] [CrossRef]
- Castren, E.; Thoenen, H.; Lindholm, D. Brain-derived neurotrophic factor messenger RNA is expressed in the septum, hypothalamus and in adrenergic brain stem nuclei of adult rat brain and is increased by osmotic stimulation in the paraventricular nucleus. Neuroscience 1995, 64, 71–80. [Google Scholar] [CrossRef]
- Dietrich, M.O.; Horvath, T.L. Hypothalamic control of energy balance: Insights into the role of synaptic plasticity. Trends Neurosci. 2013, 36, 65–73. [Google Scholar] [CrossRef]
- Liao, G.Y.; Kinney, C.E.; An, J.J.; Xu, B. TrkB-expressing neurons in the dorsomedial hypothalamus are necessary and sufficient to suppress homeostatic feeding. Proc. Natl. Acad. Sci. USA 2019, 116, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Bariohay, B.; Lebrun, B.; Moyse, E.; Jean, A. Brain-Derived Neurotrophic Factor Plays a Role as an Anorexigenic Factor in the Dorsal Vagal Complex. Endocrinology 2005, 146, 5612–5620. [Google Scholar] [CrossRef] [PubMed]
- Klöckener, T.; Hess, S.; Belgardt, B.F.; Paeger, L.; Verhagen, L.A.W.; Husch, A.; Sohn, J.W.; Hampel, B.; Dhillon, H.; Zigman, J.M.; et al. High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nat. Neurosci. 2011, 14, 911–918. [Google Scholar] [CrossRef]
- Dhillon, H.; Zigman, J.M.; Ye, C.; Lee, C.E.; McGovern, R.A.; Tang, V.; Kenny, C.D.; Christiansen, L.M.; White, R.D.; Edelstein, E.A.; et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 2006, 49, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Sternson, S.M.; Shepherd, G.M.G.; Friedman, J.M. Topographic mapping of VMH → arcuate nucleus microcircuits and their reorganization by fasting. Nat. Neurosci. 2005, 8, 1356–1363. [Google Scholar] [CrossRef]
- Cordeira, J.W.; Felsted, J.A.; Teillon, S.; Daftary, S.; Panessiti, M.; Wirth, J.; Sena-Esteves, M.; Rios, M. Hypothalamic dysfunction of the thrombospondin receptor alpha2delta-1 underlies the overeating and obesity triggered by brain-derived neurotrophic factor deficiency. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 554–565. [Google Scholar] [CrossRef]
- Unger, T.J.; Calderon, G.A.; Bradley, L.C.; Sena-Esteves, M.; Rios, M. Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J. Neurosci. 2007, 27, 14265–14274. [Google Scholar] [CrossRef]
- An, J.J.; Kinney, C.E.; Tan, J.W.; Liao, G.Y.; Kremer, E.J.; Xu, B. TrkB-expressing paraventricular hypothalamic neurons suppress appetite through multiple neurocircuits. Nat. Commun. 2020, 11, 1729. [Google Scholar] [CrossRef]
- Houtz, J.; Liao, G.Y.; An, J.J.; Xu, B. Discrete TrkB-expressing neurons of the dorsomedial hypothalamus regulate feeding and thermogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2017218118. [Google Scholar] [CrossRef]
- Wang, S.; Yao, H.; Xu, Y.; Hao, R.; Zhang, W.; Liu, H.; Huang, Y.; Guo, W.; Lu, B. Therapeutic potential of a TrkB agonistic antibody for Alzheimer’s disease. Theranostics 2020, 10, 6854–6874. [Google Scholar] [CrossRef]
- Wang, P.; Loh, K.H.; Wu, M.; Morgan, D.A.; Schneeberger, M.; Yu, X.; Chi, J.; Kosse, C.; Kim, D.; Rahmouni, K.; et al. A leptin-BDNF pathway regulating sympathetic innervation of adipose tissue. Nature 2020, 583, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Tsao, D.; Thomsen, H.K.; Chou, J.; Stratton, J.; Hagen, M.; Loo, C.; Garcia, C.; Sloane, D.L.; Rosenthal, A.; Lin, J.C. TrkB agonists ameliorate obesity and associated metabolic conditions in mice. Endocrinology 2008, 149, 1038–1048. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Allen, H.L.; Lindgren, C.M.; Luan, J.a.; Mägi, R.; et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef]
- Mou, Z.; Hyde, T.M.; Lipska, B.K.; Martinowich, K.; Wei, P.; Ong, C.J.; Hunter, L.A.; Palaguachi, G.I.; Morgun, E.; Teng, R.; et al. Human Obesity Associated with an Intronic SNP in the Brain-Derived Neurotrophic Factor Locus. Cell Rep. 2015, 13, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Voisin, S.; Almén, M.S.; Zheleznyakova, G.Y.; Lundberg, L.; Zarei, S.; Castillo, S.; Eriksson, F.E.; Nilsson, E.K.; Blüher, M.; Böttcher, Y.; et al. Many obesity-associated SNPs strongly associate with DNA methylation changes at proximal promoters and enhancers. Genome Med. 2015, 7, 103. [Google Scholar] [CrossRef]
- Tuvikene, J.; Esvald, E.-E.; Rähni, A.; Uustalu, K.; Zhuravskaya, A.; Avarlaid, A.; Makeyev, E.V.; Timmusk, T. Intronic enhancer region governs transcript-specific Bdnf expression in rodent neurons. eLife 2021, 10, e65161. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.V.; Akana, S.F.; Malkovska, I.; Dallman, M.F.; Parada, L.F.; Ingraham, H.A. Diminished hypothalamic bdnf expression and impaired VMH function are associated with reduced SF-1 gene dosage. J. Comp. Neurol. 2006, 498, 637–648. [Google Scholar] [CrossRef]
- Liao, G.Y.; An, J.J.; Gharami, K.; Waterhouse, E.G.; Vanevski, F.; Jones, K.R.; Xu, B. Dendritically targeted Bdnf mRNA is essential for energy balance and response to leptin. Nat. Med. 2012, 18, 564–571. [Google Scholar] [CrossRef]
- Cao, L.; Lin, E.J.D.; Cahill, M.C.; Wang, C.; Liu, X.; During, M.J. Molecular therapy of obesity and diabetes by a physiological autoregulatory approach. Nat. Med. 2009, 15, 447–454. [Google Scholar] [CrossRef]
- Baj, G.; Leone, E.; Chao, M.V.; Tongiorgi, E. Spatial segregation of BDNF transcripts enables BDNF to differentially shape distinct dendritic compartments. Proc. Natl. Acad. Sci. USA 2011, 108, 16813–16818. [Google Scholar] [CrossRef]
- An, J.J.; Liao, G.Y.; Kinney, C.E.; Sahibzada, N.; Xu, B. Discrete BDNF Neurons in the Paraventricular Hypothalamus Control Feeding and Energy Expenditure. Cell Metab. 2015, 22, 175–188. [Google Scholar] [CrossRef]
- Kamitakahara, A.; Xu, B.; Simerly, R. Ventromedial hypothalamic expression of Bdnf is required to establish normal patterns of afferent GABAergic connectivity and responses to hypoglycemia. Mol. Metab. 2016, 5, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Ye, C.; McCrimmon, R.J.; Dhillon, H.; Choi, B.; Kramer, M.D.; Yu, J.; Yang, Z.; Christiansen, L.M.; Lee, C.E.; et al. Synaptic Glutamate Release by Ventromedial Hypothalamic Neurons Is Part of the Neurocircuitry that Prevents Hypoglycemia. Cell Metab. 2007, 5, 383–393. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, D.; Sweeney, P.; Yang, Y. An excitatory ventromedial hypothalamus to paraventricular thalamus circuit that suppresses food intake. Nat. Commun. 2020, 11, 6326. [Google Scholar] [CrossRef] [PubMed]
- Noble, E.E.; Billington, C.J.; Kotz, C.M.; Wang, C. The lighter side of BDNF. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1053–R1069. [Google Scholar] [CrossRef] [PubMed]
- Ozek, C.; Zimmer, D.J.; De Jonghe, B.C.; Kalb, R.G.; Bence, K.K. Ablation of intact hypothalamic and/or hindbrain TrkB signaling leads to perturbations in energy balance. Mol. Metab. 2015, 4, 867–880. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, P.; Guo, W.; You, H.; Lu, B. Regulation of Satiety by Bdnf-e2-Expressing Neurons through TrkB Activation in Ventromedial Hypothalamus. Biomolecules 2023, 13, 822. https://doi.org/10.3390/biom13050822
Chu P, Guo W, You H, Lu B. Regulation of Satiety by Bdnf-e2-Expressing Neurons through TrkB Activation in Ventromedial Hypothalamus. Biomolecules. 2023; 13(5):822. https://doi.org/10.3390/biom13050822
Chicago/Turabian StyleChu, Pengcheng, Wei Guo, He You, and Bai Lu. 2023. "Regulation of Satiety by Bdnf-e2-Expressing Neurons through TrkB Activation in Ventromedial Hypothalamus" Biomolecules 13, no. 5: 822. https://doi.org/10.3390/biom13050822