3.1. Result of Protein–Protein Docking
To investigate the interactions of FXa and FIXa with the exosite of AT not binding a heparinoid ligand, we have followed two different approaches. First, we built model structures using protein–protein docking with HADDOCK, starting from a conformation of AT with a closed hinge region. Second, we have studied model systems that were constructed directly using the X-ray structures, but with the pentasaccharide deleted.
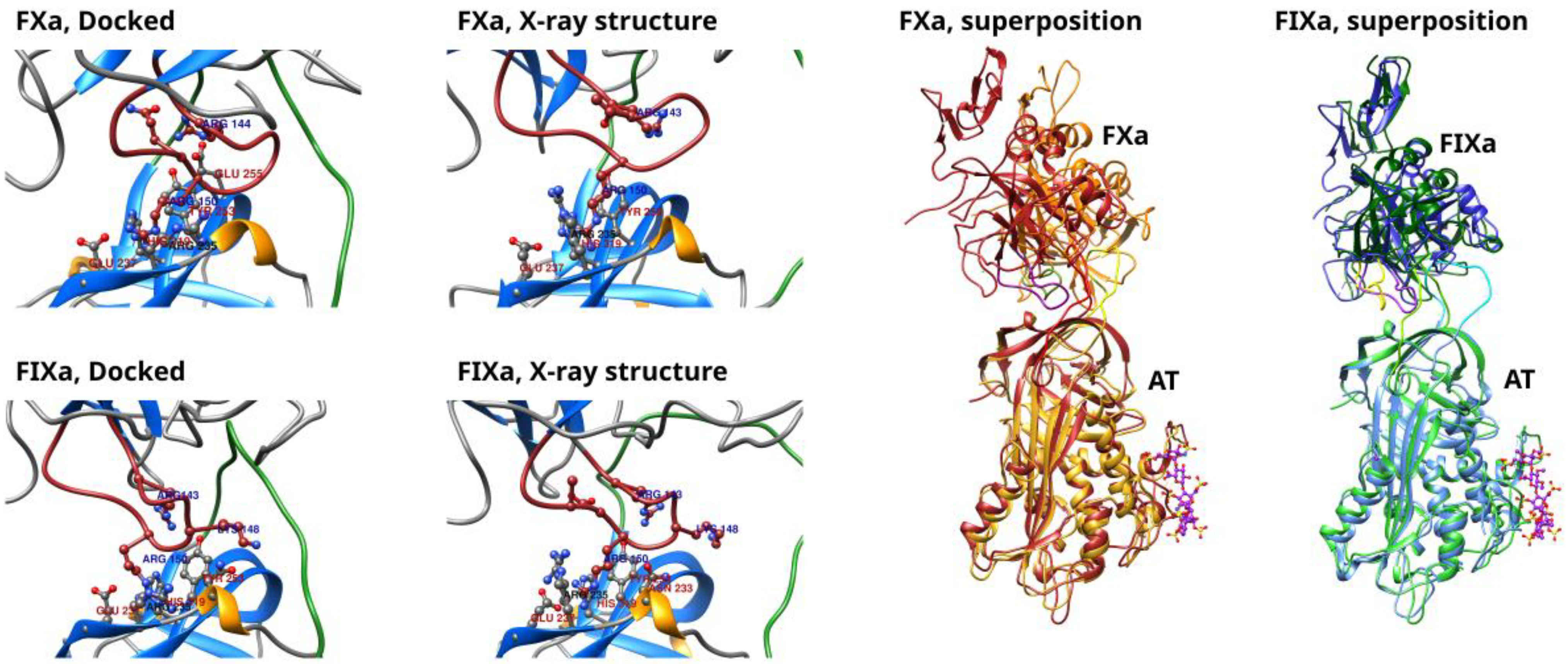
HADDOCK was able to build structures for both factors with the “hinge” region in its closed state. In the structures selected for molecular dynamics simulations—the structure with the highest score in the highest-ranked conformational cluster—all three amino acid contacts defined as “unambiguous restraints” were present (see the Methods section for further details.).That is, the Arg393–Ser394 peptide bond and the Arg393 side chain were in the proper position for the enzyme reaction and the critical Arg150 side chain [
9] was also interacting with the surface of the exosite. The closed “hinge” conformation has an effect on the conformation of the entire RCL. For optimal interactions with both the RCL and the exosite of AT at the same time, the autolysis (or 148-) loop in the factors must undergo conformational adaptation. The position of this loop, as well as the exosite interactions in the docked and the corresponding X-ray structures are compared in
Figure 1 for both factors. In the two structures obtained by docking, the Arg150 side chain interacted with the same region on the surface of AT, close to residues Tyr253 and His317; this is expected as this interaction was defined as a restraint in docking. In both factors, the autolysis loop contained two additional positively charged residues, Arg143 and Lys148, which were also involved in the interaction with the exosite [
12]. In the X-ray structure of the AT–FXa complex, the Arg143 residue interacted with Glu255 in AT. Arg143 was oriented towards Gln255 in the corresponding docked structure also, but the distance between the residues was higher. The Lys148 side chain was missing in the X-ray structure, and this made the comparison difficult for this amino acid. In the HADDOCK-generated structure, the same residue was oriented towards the RCL of AT. As for the FIXa complex, both residues interacted with the Glu255 of AT in the experimental structure, but in the selected conformation form of the docking, Arg143 was not close enough to the Glu due to the different position of the 148-loop.
3.2. Interaction between the Arg150 Residue and the Exosite of AT in the Docking-Based Simulations
The critical role of the autolysis loop (or 148-loop) of both FXa and FIXa in the interaction with AT is supported by both X-ray diffraction structures [
9,
10] and mutagenesis studies [
11,
12,
39]. The Arg150 amino acid in this loop is especially important as it forms direct contacts with the surface of the serpin near the Tyr253 amino acid. To study the binding of this critical residue to AT in the non-pentasaccharide binding state, we calculated the distance between the side chain of Arg150 (CZ atom) and the β-carbon of Tyr253 in AT from the simulations that started from our docked structures. The distance values are shown as a function of the simulation time in
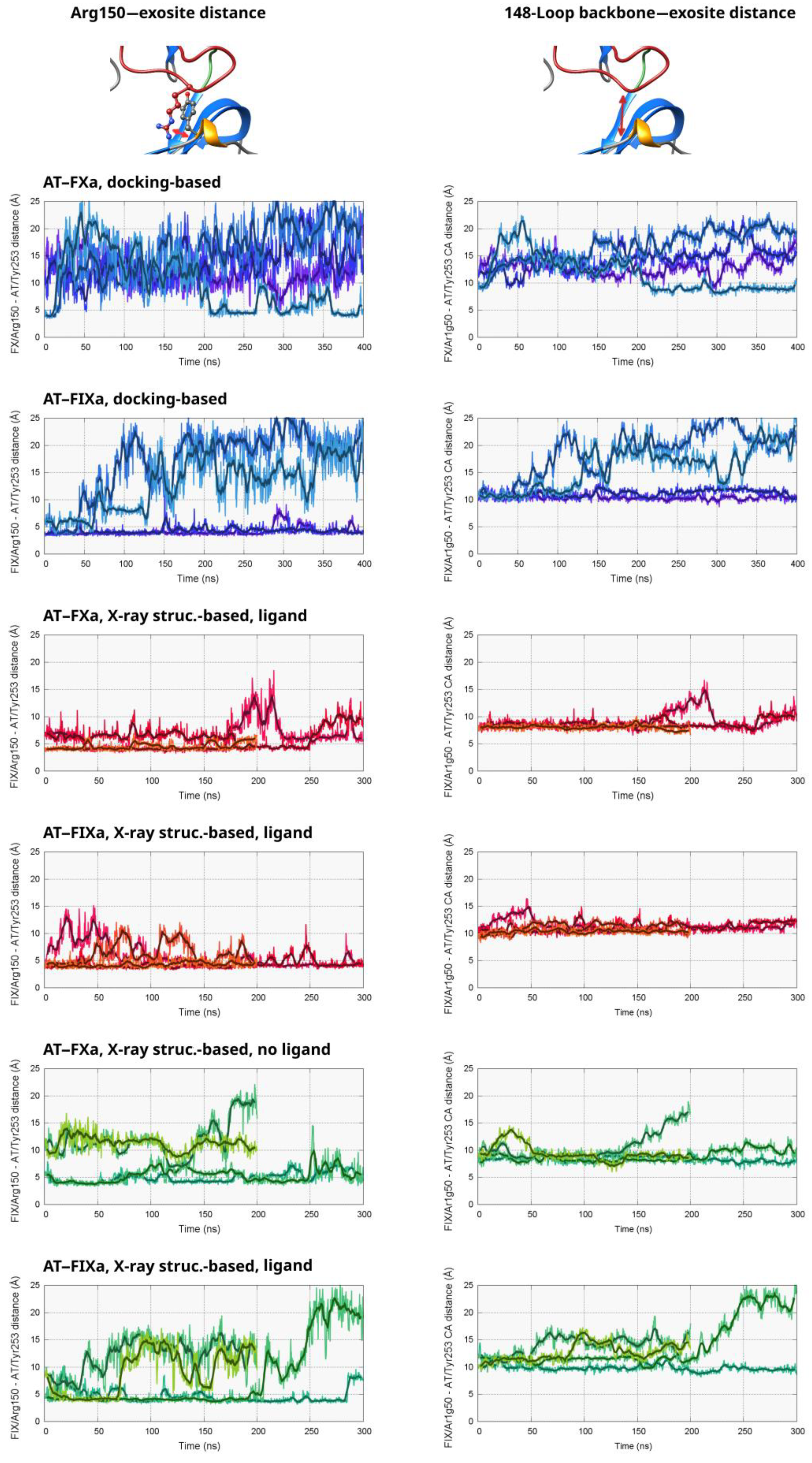
Figure 2 and in
Figures S1 and S2.
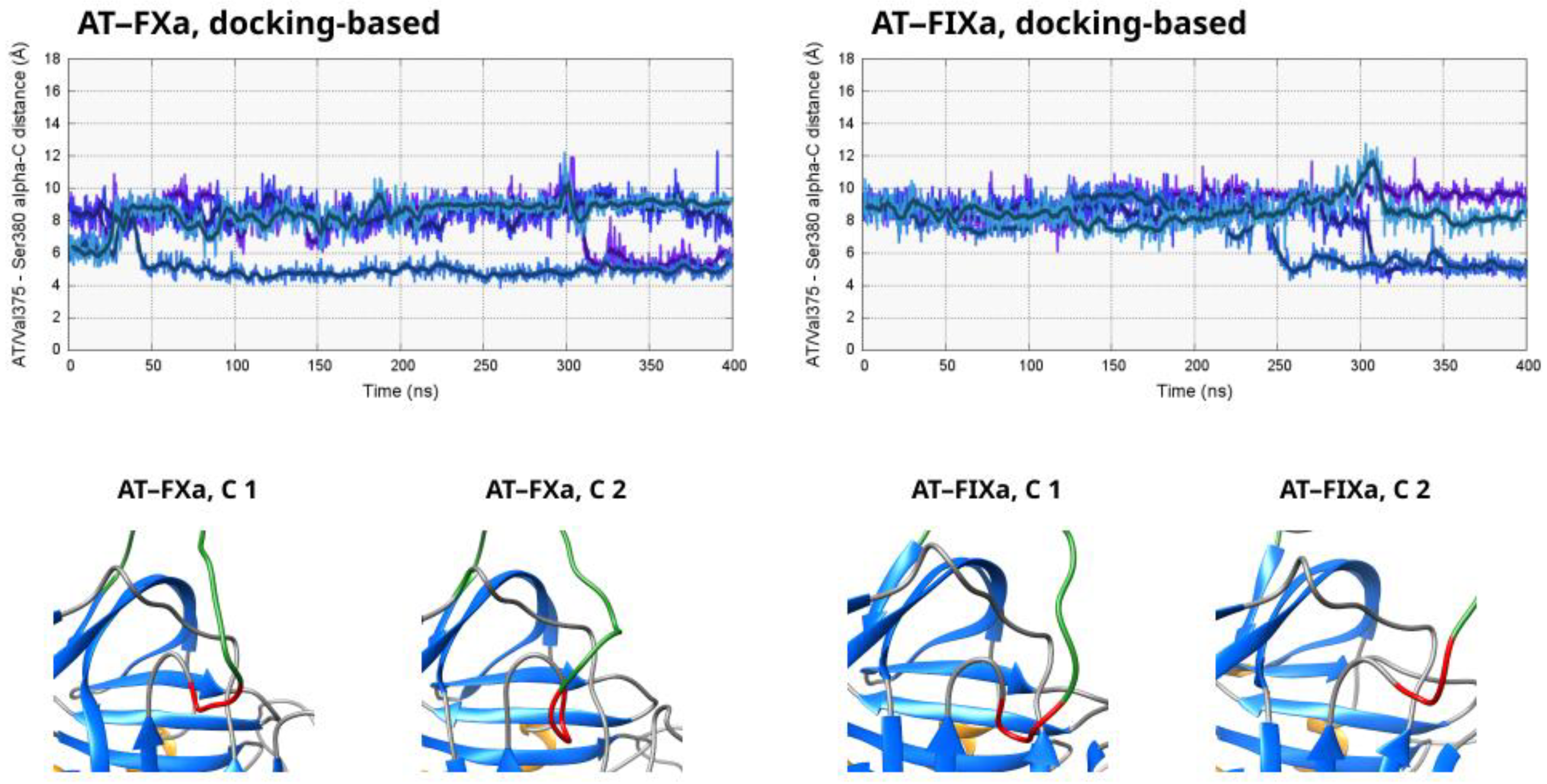
The results for the four simulations started from the docked AT–FXa complex are shown in the top left part of
Figure 2. It is evident from the distance values that the Arg150 did not bind very tightly to its binding site found in the X-ray structure. The “combined” conformational space explored by the four copies of the system was very large, with distances as low as 5 Å and as high as 22 Å, as well as many “intermediate” conformations. In one of the simulations (Number 4 in
Figure S1), the system exhibited a different behavior compared to the other three. The Arg150 residue started unbinding from its binding site on AT very soon and reached distances above 20 ns after about ~80 ns. The sidechain bound again into its pocket in the AT exosite at ~210 ns, and the distance value remained below 10 Å in most of the remaining part of the trajectory. We were not able to capture any similar event in the other simulations of the system. However, this observation at least indicates that the transition between states exhibiting a “strong” or “weak” Arg150 interaction were reversible and that the energy barrier was low.
The conformational behavior was very different in the four simulations based on the docked FIXa structure. In two trajectories, very limited movement of the Arg150 residue can be observed out of its binding “pocket”, demonstrated by the distance values below 5 Å. In the remaining two simulations, this amino acid unbound from the serpin surface between ca. 80–150 ns. With the exception of relatively short periods, the distance of Arg150 remained high from the exosite in the rest of the two trajectories.
To investigate the position of the autolysis loop without the conformational fluctuations of the Arg150 sidechain, we also analyzed the distance between the peptide backbone near Arg150 in the two factors and the AT exosite (right column of
Figure 2,
Figures S3 and S4). In AT, the α-carbon atom of Tyr253 was selected for the analysis. Typically, there is a strong correlation between this “backbone” distance and the value for the Arg150 side chain. However, the lowest and the highest observed values are much closer to each other in the former case than in the latter, indicating that the 148-loop itself moved less than did the Arg150 side chain.
To analyze the distribution of the “exosite” distance, we also plotted the “backbone distance” parameter in histograms (
Figures S4 and S5). In the two AT–FIXa simulations showing large conformational changes, a region around 12–15 Å was visible with low frequencies, suggesting that these conformations are less favorable.
3.3. The Allosteric Effects of Pentasaccharide Binding on the Exosite Interactions in the X-ray Structure-Based Simulations
Several papers have been published in which the allosteric effects of ligand binding to AT were studied based on MD trajectories for the protein [
18,
20,
40,
41]. In contrast, simulating the Michaelis complexes of AT with FXa and FIXa allowed us to study these effects directly. Therefore, we performed simulations based on the X-ray structure of the Michaelis complexes with or without a pentasaccharide to gain insights into the allosteric activation mechanism. We performed four independent simulations for all four systems (FXa vs. FIXa complex, and pentasaccharide vs. no pentasaccharide), with two 300 ns and two 200 ns runs in each case.
To investigate the interaction of Arg150 with AT, we analyzed the same atom distances as those in the docking-based simulations. As expected, conformations with a larger Arg150 distance from the exosite were observed to have a much lower frequency than that in the docking-based trajectories. However, it was still possible to draw conclusions on the effects of the pentasaccharide binding from the results. The results are shown in
Figure 2 (the “red” and the “green” plots) and in
Figures S1–S4.
In two simulations of the FXa system with a pentasaccharide (3 and 4 in
Figure S1), the Arg150 side chain of FXa was in a position very close to its binding site in the X-ray structure with only very small fluctuations. In the other two trajectories, we could observe somewhat larger variations in this parameter over the simulation time, but the distance was still well below 10 Å in a large majority of the sampled states. In the simulations not containing a pentasaccharide, the results were not significantly different from the ligand-containing ones. However, in runs 3 and 4, the interaction between the Arg150 sidechain and its binding site was less tight. The distance was above 10 Å by more than half in both trajectories, and values close to 20 Å could be observed close to the end of Simulation 3.
Regarding the simulations based on the AT–FIXa X-ray structure, the tendencies were largely similar. In the simulations with a pentasaccharide, the Arg150 remained close to Tyr253 in the case of most conformations, with some level of flexibility in its position in Simulation 1 and 3. In contrast, a significant number of states with distance values close to or above 15 Å were present in three of the four trajectories lacking the AT ligand, and such states were almost completely absent from the FIXa simulations with the heparinoid.
Similarly to the docking-based simulations, we also investigated the distance between the peptide backbones of the interacting protein pairs at amino acid 150 in the factor and Tyr253 in AT (right column of
Figure 2). As expected, the “backbone distance” fluctuated much less than did the values for the Arg150 side chain. In agreement with the results from the docking-based MD, significant movements of the Arg150 side chain were observed in those simulations which also showed an increase in the backbone distance. Due to the relatively short simulation times, we could not observe larger conformational changes in AT towards the non-activated state after the deletion of the pentasaccharide. However, in the GaMD simulations based on the X-ray structures, we were able to observe signs that the presence (or the absence) of the pentasaccharide had an allosteric effect on the interaction of Arg150 with the exosite of AT.
We also analyzed the “backbone distance” parameter through histograms (
Figures S5 and S6). In the FIXa complex system, conformations with a distance value above 10 Å occurred in the simulations not containing a ligand. The tendencies were similar in the AT–FXa simulations; however, the difference was smaller due to the somewhat higher conformational variability in the pentasaccharide-containing state compared to the AT–FIXa complex.
However, the conformational changes described above were not observed in all simulations of the same type of system. Therefore, more conformational sampling would be needed to better understand these processes.
From the simulations containing a pentasaccharide, we have also assessed the binding of the ligand using the same “RMSD” metric as that used in our previous papers [
20,
22] (
Figure S7). The ligand binding to AT in the pentasaccharide-containing systems was stable, but there was some level of conformational variability according to the RMSD analysis. Apparently, these small movements of the pentasaccharide did not correlate with the changes in the interaction with the exosite.
3.4. Analysis of Interactions between Amino Acid Pairs
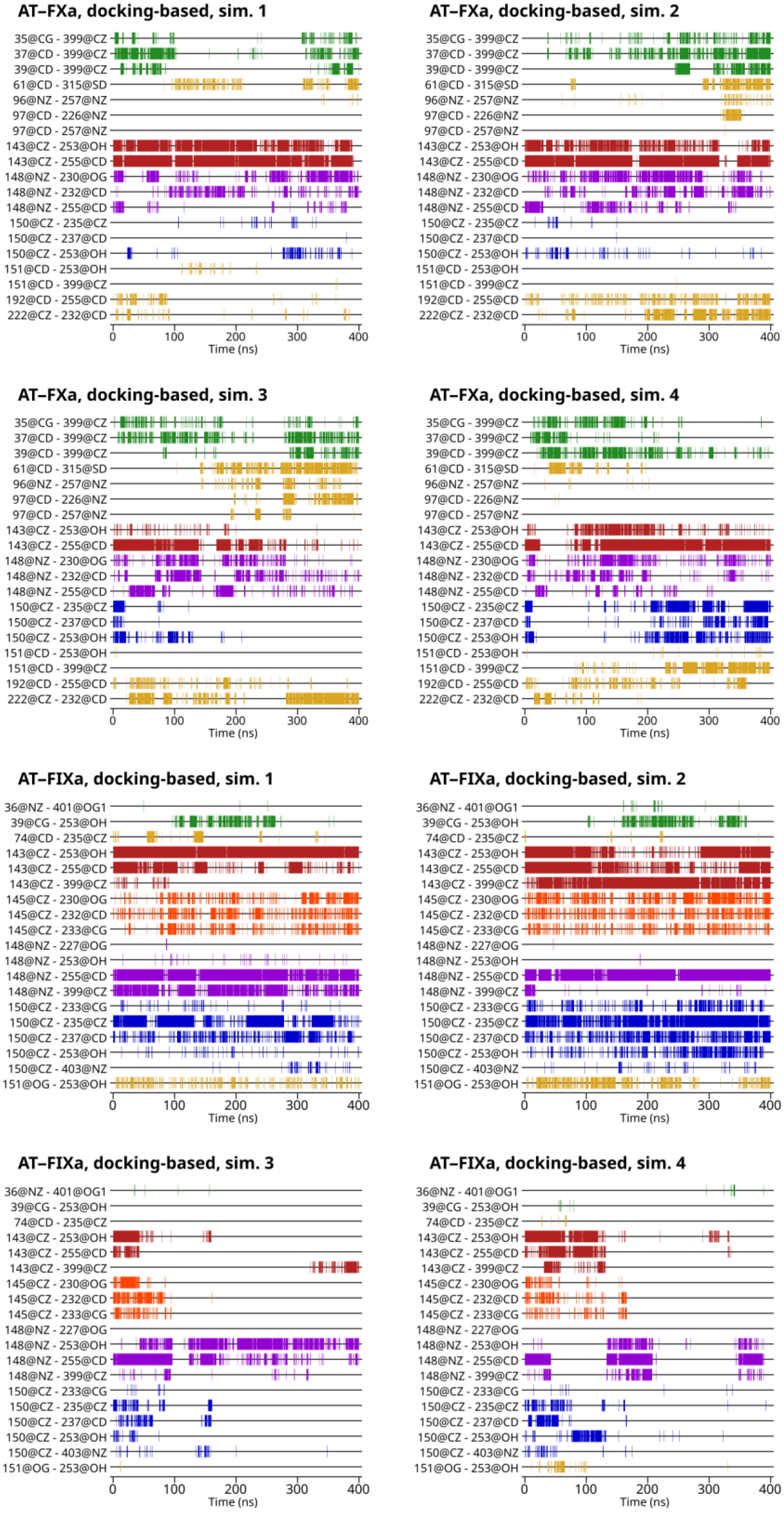
For understanding the differences in the exosite interaction between the simulated systems—that is, the two factors and the different activation states of AT—it is essential to know which amino acid pairs were involved in the interaction. The interacting pairs included in the analysis were selected using the representative conformations from the cluster analysis of all six groups of simulations. The results are shown in
Figure 3 for the docking-based trajectories and on
Figures S8 and S9 for the X-ray diffraction-based trajectories. We defined a residue pair as “interacting” if the distance was below a cut-off value of 6 Å.
In
Figure 3, a conformational change in two simulations based on the FIXa-docked structure is visible. After ~100 ns in Simulation 3 and ~180 ns in Simulation 4, very few interactions remained between the two proteins. A particularly interesting example is Arg143. This residue interacted with Glu255 in AT in most simulations discussed in this study (FXa, FIXa, docking and X-ray structure-based). The only exceptions are the two simulations of the FIXa-docked structure that showed significant unbinding of Arg150 from its pocket. The interaction was present at the beginning of both simulations, but the distance increased between the two sidechains in parallel with the partial dissociation of the Arg150 residue, as discussed before. The notable exception is the interaction between Lys148 of FIXa autolysis loop and Tyr253 and Glu255 in AT, as well as a few other contacts in short time intervals in Simulation 4. Some of the very few remaining contacts were even repulsive, being found between positively charged Arg residues. The behavior of the docking-based FXa simulations was markedly different. Even in the conformations with larger distances between the autolysis loop and the AT exosite, the interactions were still present between several amino acid pairs.
According to the available experimental data, the interaction of FIXa with non-activated AT was weak, even relative to the FXa binding to AT in this state. This raises the question of how this can be explained with our MD data. The difference in interaction’s free energy may be due to the combination of two factors with opposite effects. In the docking-based FXa complex simulations, the binding of the Arg150 residue appeared to be less stable to the AT exosite, but this may have been compensated by several other, less specific interactions between the two proteins. In contrast, the simulations of the AT–FIXa systems suggested the existence of a state with favorable binding that could exist for longer time periods, but there was very limited interaction in other conformations. Additionally, the transition was observed in one direction, and this may suggest that the opposite process (from a large distance to close contact) may not have been kinetically favorable.
The data obtained from our simulations are certainly not sufficient for an accurate comparison of the stability of the states discussed above. However, we still expect that the conformations observed in our simulations—not previously reported—provide insights into the biochemical background of the weaker interaction of non-activated AT with the factors.
3.4.1. Contribution of the Arg143 and Lys148 Amino Acids
As mentioned before, the autolysis loop of both factors contains two further amino acids with a positive charge, Arg143 and Lys148. Izagurre et al. have published mutagenesis studies involving these two amino acids in factor Xa [
12]. However, to our knowledge, no such data are available for factor IXa.
The Arg143 amino acid of FXa and FIXa mainly interacted with one AT residue in the AT–FXa complex simulations, Glu255. Additionally, it could be in close proximity to the ‘binding pocket-forming’ Tyr253 amino acid. Arg143–Glu255 was one of the most stable interactions in these simulations as it was present in the majority of the sampled states in all 12 simulations. The role of Arg143 was largely similar in the AT–FIXa model systems: its most important interaction was with Glu255 and could be positioned close to Tyr253. However, it could also be found close to Arg399 in some of the states in the trajectory. The Arg143–Glu255 interaction was “stable” except in the two trajectories where a conformational change occurred.
In the AT–FXa complex simulations, the Lys148 in FXa appeared to play a more important role in the docking-based simulations than it did in the X-ray diffraction-based trajectories. This residue could interact with two positively charged residues in AT, Glu232 and Glu255. In the docking-based simulations, both interactions were present in a large number of sampled conformations, while at least one of the two was absent or very weak in the X-ray-based simulations. In the AT–FIXa complex trajectories, Lys148 couldonly interact with Glu255, but not Glu232. The Lys148–Glu255 distance varied significantly between each trajectory and with the simulation time. Notably, Lys148–Glu255 was one of the very few interactions that remained in Simulation 3 after the conformational change to a state with a weaker interaction.
The Gln151 residue of the autolysis loop of FXa could make contact with the Arg399 amino acid of AT. This interaction was almost entirely absent in the docking-based simulations, but was found in a significant number of conformations in the X-ray diffraction-based simulations. Residue 151 was substituted with a serine in Factor IX. This residue was close to the binding site in most of the simulation time, except in the two trajectories where the aforementioned conformational change occurred.
In the autolysis loop of factor IXa, one further amino acid made close contact with AT, namely Phe145. Its side chain was positioned close to Glu232 and Asn233 in AT. This interaction was stable in all simulations of FIXa except the two where the previously discussed conformational change occurred. In those simulations, the interaction disappeared in parallel with the unbinding of Arg150, Arg143 and other residues.
In FXa, two further residues could be found in contact with the surface of AT, Gln192 and Arg222. Such conformations were mainly found in the docking-based trajectories and were almost completely absent from the simulations containing a pentasaccharide ligand.
3.4.2. Involvement of Regions Outside the Autolysis Loop in the Exosite Interactions
Yang et al. have suggested that the sequence differences in the 36-loop (or 39-loop) between FXa and FIXa may be partially responsible for the slower reactivity of FIXa with non-activated AT [
42]. Notably, this structural element contains three negatively charged Glu residues in FXa (residues 36, 37 and 39), while in FIXa, residue 37 is missing and the other two are substituted with a Lys and an Asp, respectively.
As expected, the main interaction of all three Glu residues was with the Arg399 residue in AT (
Figure 3). Such interactions were present in all three types of systems investigated (docking-based and X-ray diffraction-based, with and without a pentasaccharide). In some trajectories, however, such binding was limited to specific parts of the trajectory. This suggests that this interaction depends on the binding mode of FX. From our results, it is clear that the 36-loop of FIXa could not interact with Arg399 in the AT in any of the three states simulated. Instead, in two of the four docking-based simulations, the Asp39 residue was frequently found in proximity to the Tyr253 residue of the binding site in AT. This was not observed in any of the simulations based on the X-ray diffraction structure. Residues Gln61, Lys96 and Glu97 in factor X were also observed to make contact with AT. Although Gln61 was located in a different loop than the other two, it seems reasonable to discuss them as a single group. These amino acids were located far from the surface of AT in the X-ray structure. However, there were some interactions between these amino acids and AT in all four docking-based simulations at least in some part of the trajectory. This interaction was absent from the X-ray diffraction-based simulations, with the exception of one simulation without a pentasaccharide.
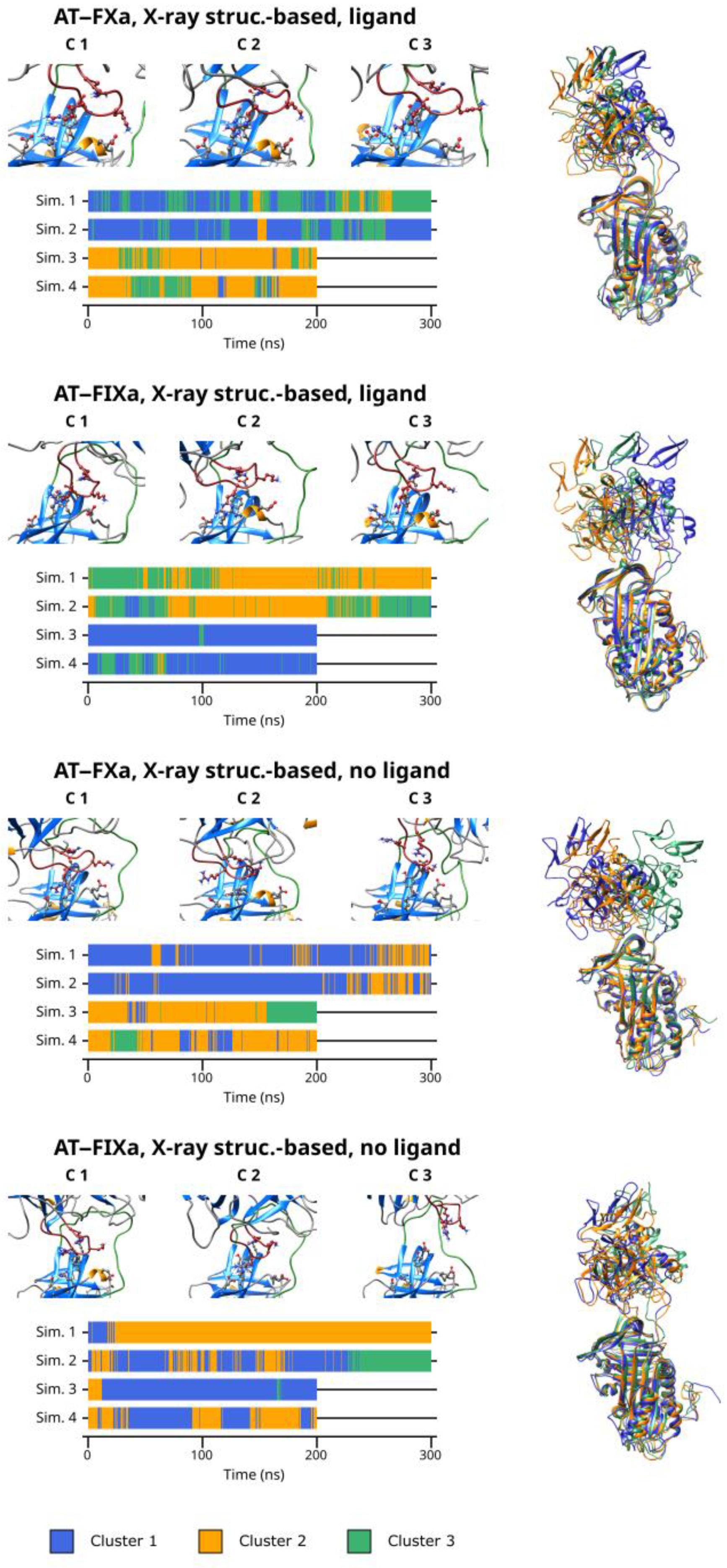
3.5. Cluster Analysis
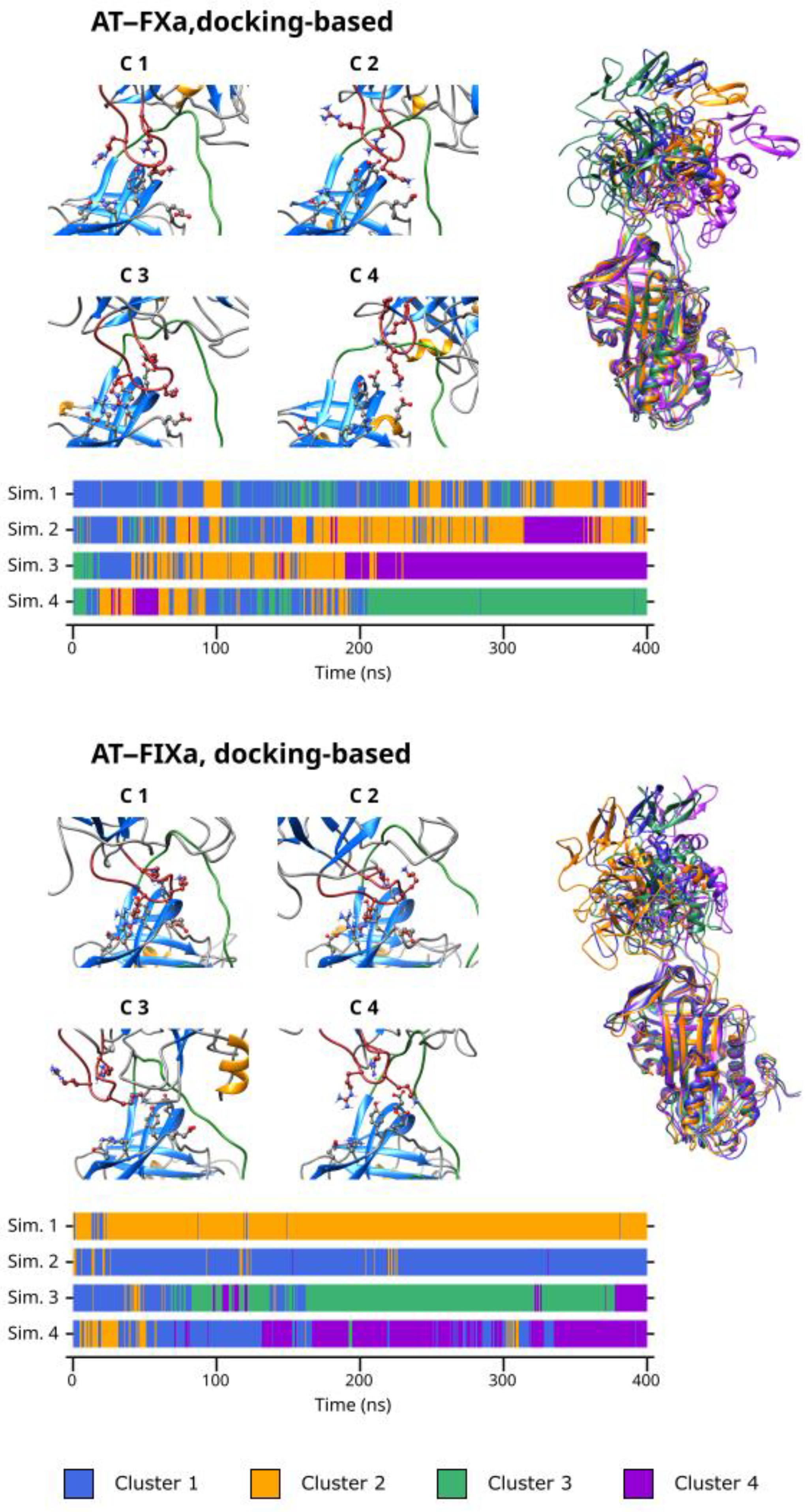
As discussed before, our simulations revealed that multiple binding modes exist in both the AT–FXa and the AT–FIXa complexes. We performed cluster analysis to identify conformational types in both the docking- and the X-ray diffraction-based simulations. The cluster number parameter for the K-means algorithm was four in the docking-based systems but only three in the X-ray diffraction-based simulations as these have smaller variability in conformation. The results obtained from cluster analysis of the docking-based simulations are presented in
Figure 4. The figure shows the representative conformation for each cluster as well as the cluster number as a function of time. The superpositions of the conformational clusters are found in the right column, with each structure colored according to the cluster number.
From the result of the cluster analysis, it is evident that the four docking-based simulations of the FXa system together explored a large conformational space. Cluster 3 and 4 represent the two extremes of the “spectrum” of conformations observed, while Cluster 1 and 2 are “intermediate” states between them. Cluster 3 contained the conformations with the most favorable binding mode of Arg150, while the distance between the autolysis loop of FX and AT was the highest in Cluster 4. Most of Simulation 1 and 2 as well as the first ~200 ns of Simulation 3 and 4 are characterized by frequent and rapid changes between the conformational types. However, in the second half of the latter two simulations, most conformations belonged to a single cluster—Cluster 4 in the former and Cluster 3 in the latter trajectory. This, of course correlates with the large (in Simulation 3) and small (in simulation 4) Arg150exosite distances observed in the simulations.
Similarly, the docking-based FIXa system had significant conformational flexibility. However, a few important differences are evident (
Figure 5). Here, Cluster 1 and 2 contained the conformations with a more-or-less optimal interaction between the autolysis loop and the AT exosite, while the states showing a less tight interaction were placed in the remaining two clusters. At the beginning of Simulation 3 and 4, the sampled conformations belonged to Cluster 1 and to a lesser extent, Cluster 2. This corresponds to the states in which a short distance was observed between the Arg150 residue and the exosite. The previously discussed conformational change in the other two simulations are clearly visible in the cluster analysis results. After this event, Cluster 3 became ‘dominant’ in Simulation 3 and Cluster 4 became dominant in Simulation 4.
Even in the X-ray diffraction-based simulations with a pentasaccharide, the position of the factor compared to that of the AT differed significantly between the three clusters. This demonstrates the remarkable flexibility of the system despite the stable binding of the Arg150 residue in a large majority of the conformations analyzed. Apparently, the main cause of this conformational variability was the flexibility of the RCL but the position of the 148-loop could adapt to the changes.
The cluster analysis of the X-ray structure-based simulations without the ligand revealed both significant similarities and differences between the two factors. In both cases, the first two clusters represent the movements of the proteins within the complexes in states with close exosite interaction, while the states with an apparently less favorable binding modes were placed in the third cluster. When comparing the Cluster 3 conformations obtained for the FXa and the FIXa systems, the large difference in the position of the factors is clearly visible. This further supports our conclusions about the different binding and unbinding mechanisms between the two factors.
In general, the cluster analysis revealed multiple important aspects of the interaction. First, it demonstrated the highly dynamic nature of the interaction, even in simulations where Arg150 was in stable interaction with the AT exosite. Second, it provided further insights into the differences in the conformational space available to the two factors.
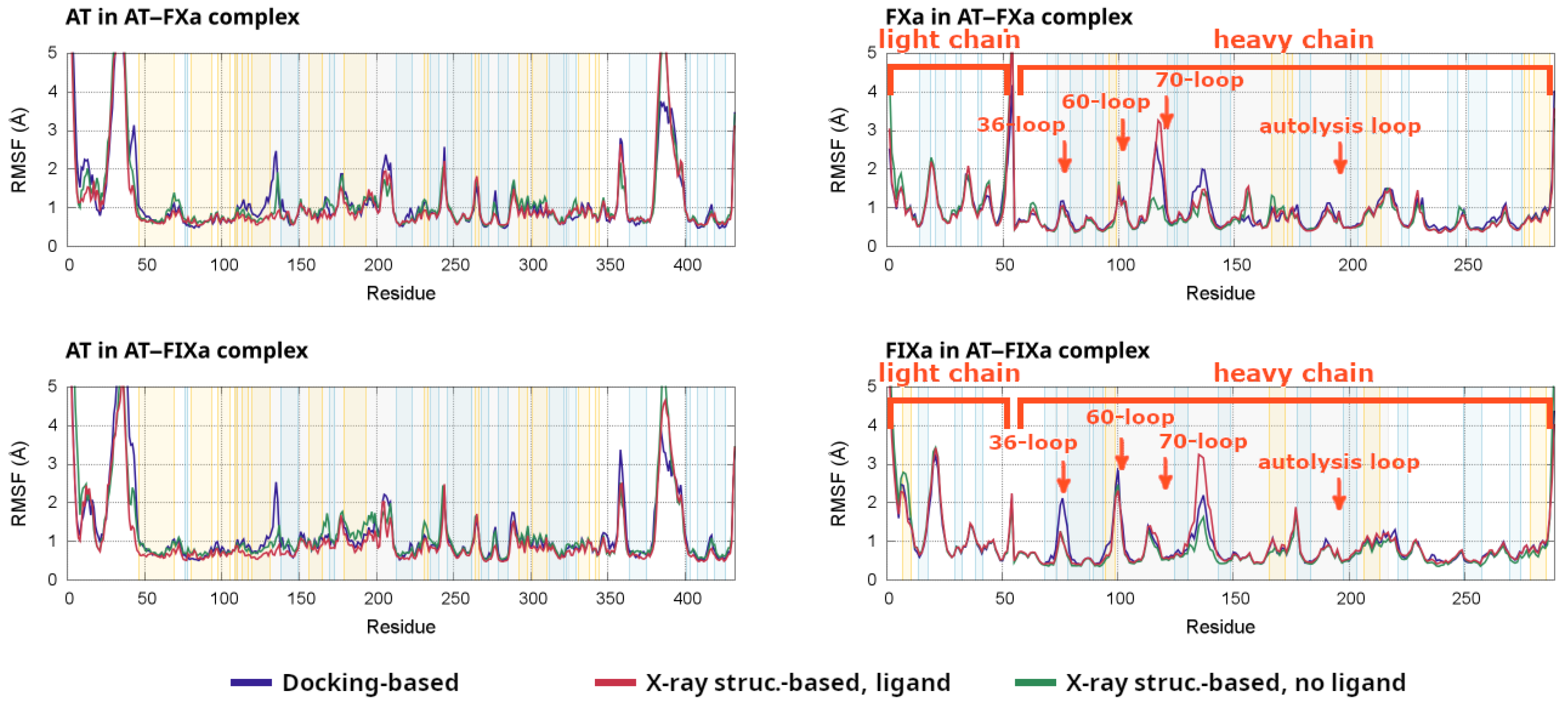
Root mean square fluctuations (RMSF) are a simple and frequently used technique for measuring the flexibility of specific parts of a protein from an MD trajectory. In the present study, we performed the calculation separately for AT and the clotting factor Xa or IXa for all simulations. The RMSF values from the individual simulations are plotted in
Figures S10–S13. For all α-carbon atoms, averages were calculated from the four simulations for all six systems (docking-based and X-ray structure-based, the latter with or without a pentasaccharide) for both factors. In each of the four plots, we compared the averaged fluctuations for the three types of model systems.
Results from the analysis are presented in
Figure 6 for both AT–factor complexes studied. In both cases, fluctuations in AT are shown on the left and the RMSF values in the clotting factor Xa or IXa are shown on the right. Due to technical reasons, the X axis for FXa and FIXa uses a different numbering than that used in the rest of the paper. Instead, we added labels for the most important loops in these two plots, showing the residue numbers used in the text.
The RMSF analysis of AT provided insights into the consequences of the pentasaccharide binding on the conformational behavior of the system. In several regions of AT, the fluctuations were the smallest in the X-ray-based simulation with the pentasaccharide. In the region close to the heparin binding site (100–140), however, significantly higher fluctuations in the values could be observed in the docking-based systems compared to the values from the X-ray diffraction-based simulations of both factors. This indicates that the conformation of several structural elements in the region were stabilized once all conformational changes triggered by the pentasaccharide had taken place. The difference was the largest close to the C-terminal end of helix D, which underwent a conformational change in the later steps of activation.
The most notable difference between the FXa and FIXa complexes is that the removal of the pentasaccharide caused much higher increases in the fluctuations of several regions in the simulations of FIXa. These increases were most visible between amino acids 80–200 in AT, and this region includes much of the heparin binding site, two strands off beta sheet A as well as helices E and F. This may indicate that the conformation of AT was more sensitive to the pentasaccharide binding in the FIXa-containing simulations, compared to that in the FXa-containing ones.
Of particular interest is the region around amino acids 230–240 in AT, which includedmultiple residues involved in the exosite interaction. The differences were not particularly significant in this region among the groups of simulations analyzed. However, the pentasaccharide-containing simulations tended to have lower values for both factors. The lower fluctuations may indicate a conformation more favorable for interaction with the factors. It should be noted, however, that this was a region that underwent only smaller visible conformational changes.
As expected, the differences between the activation states had relatively small effects on the RMSF values in the catalytic domains of the factors. However, we could observe important differences between FXa and FIXa. The 36-loop, as mentioned before, contributed to the AT–FXa interactions, but its role was less important in the AT–FIXa complex. Interestingly, the RMSF of this loop was higher in the docking-based simulations of the AT–FIXa complex than it was in any other systems. This corresponded to a conformational change that resulted in the Asp39 residue becoming better-positioned to interact with the exosite. The flexibility of the 60-loop was not significantly higher than that of other loops in FXa. In contrast, in the FIXa complex simulations, it was one of the most flexible parts of the heavy chain. The behavior of this region was similar between the docking-based and X-ray based simulations, with slightly higher RMSF values in the former group. In contrast, the 70-loop or the calcium binding loop was found to have a higher tendency to conformational changes in the FXa simulations based on the RMSF values. Interestingly, the autolysis loop did not show significantly higher fluctuations than those of several other loops in the factor, despite our observation of some level of conformational flexibility. This may indicate that the conformation of this structural element was stable to a limited extent and that this is required for interaction. The RMSF values were somewhat higher in the docking-based simulations of the two factors compared to those in the X-ray diffraction-based ones. This may correspond to the “conformational adaptation” of this loop to the changes in the relative position between the proteins.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






