

A New Generation of IMiDs as Treatments for Neuroinflammatory and Neurodegenerative Disorders
 , , , , and
, , , , and
Abstract
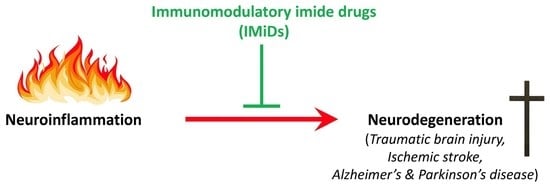
:
1. Introduction
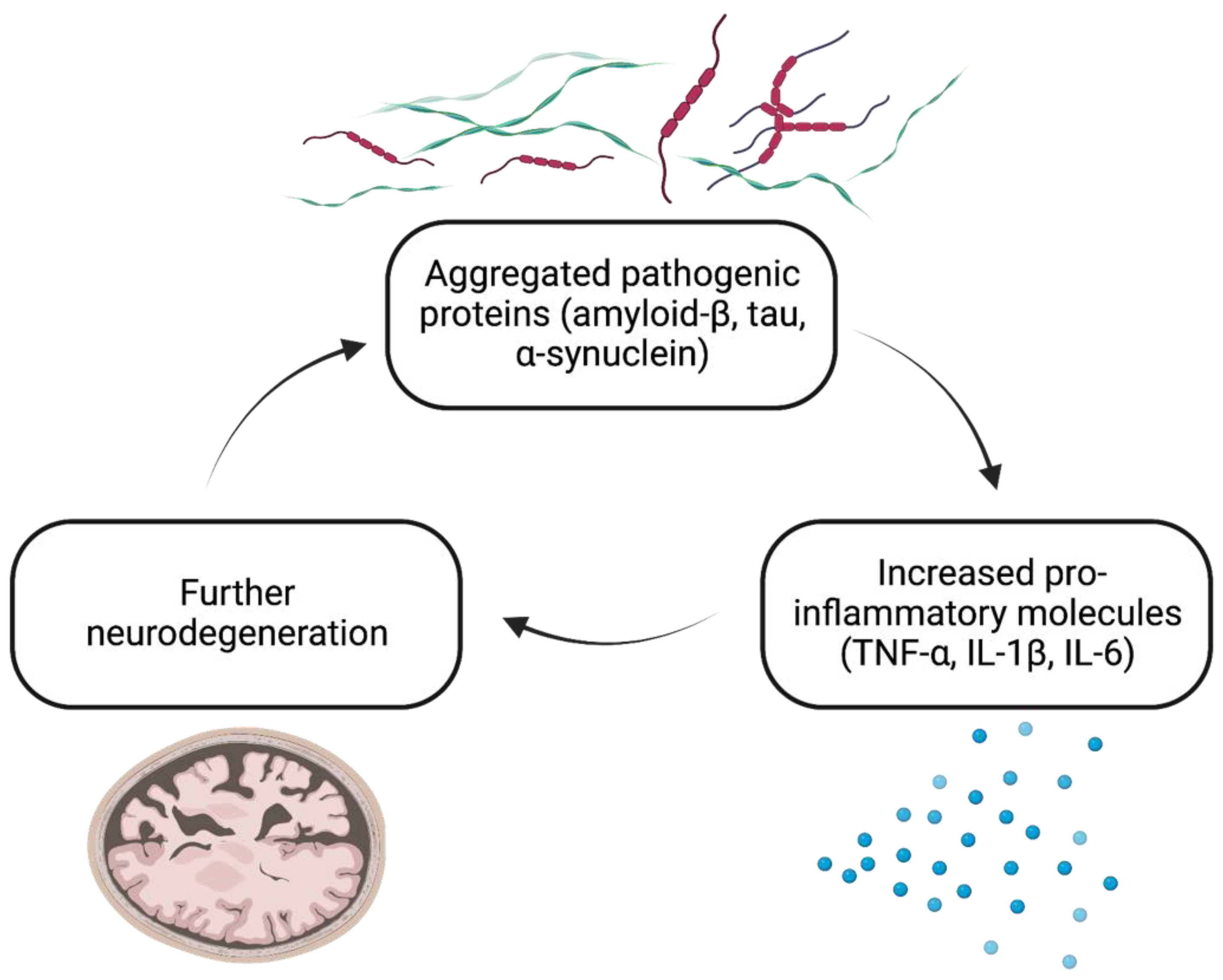
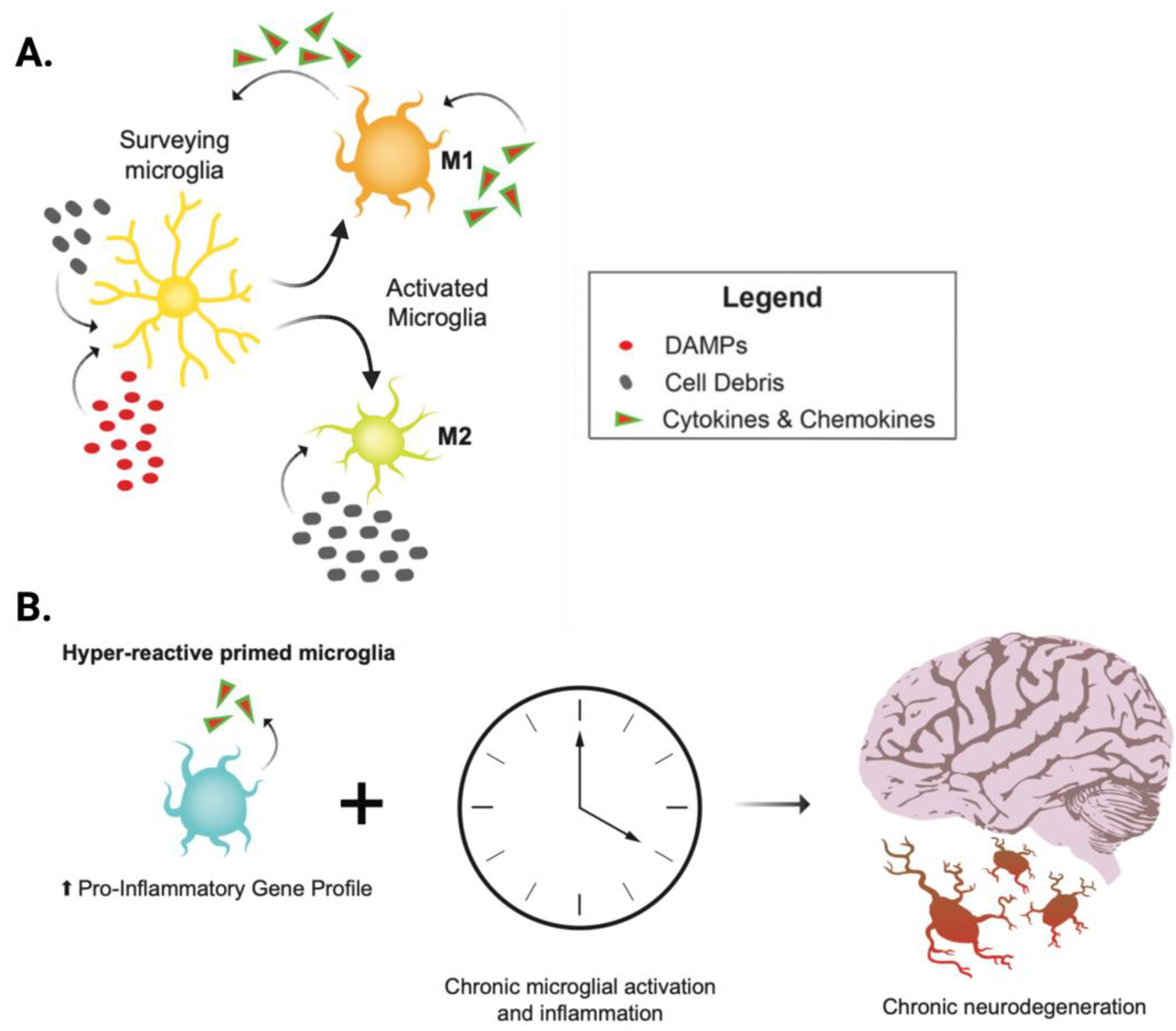
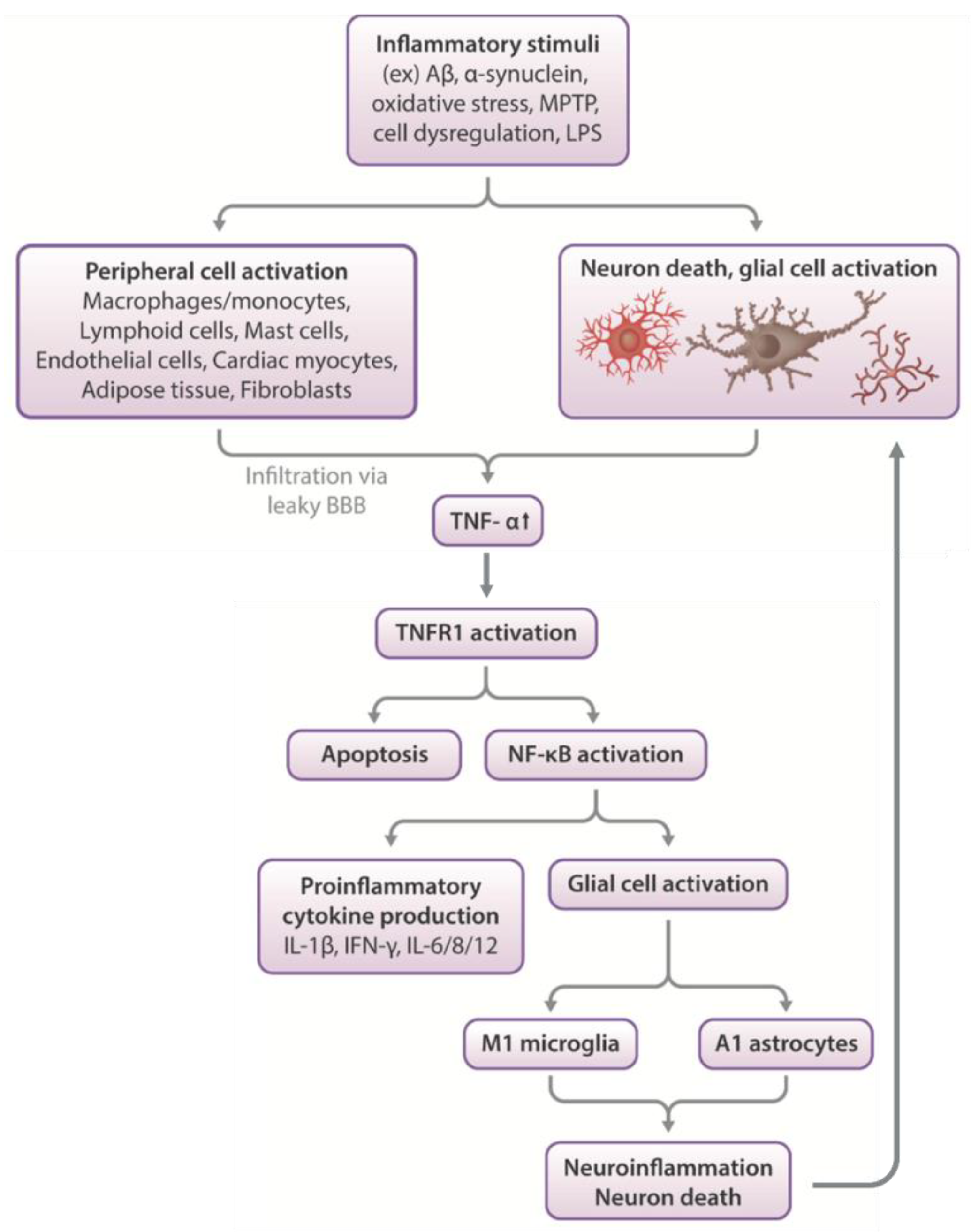
2. Neuroinflammation and Neurodegeneration: A Feed-Forward Cycle
3. Anti-Inflammatory Therapies to Treat Neurodegenerative Disease
4. IMiDs
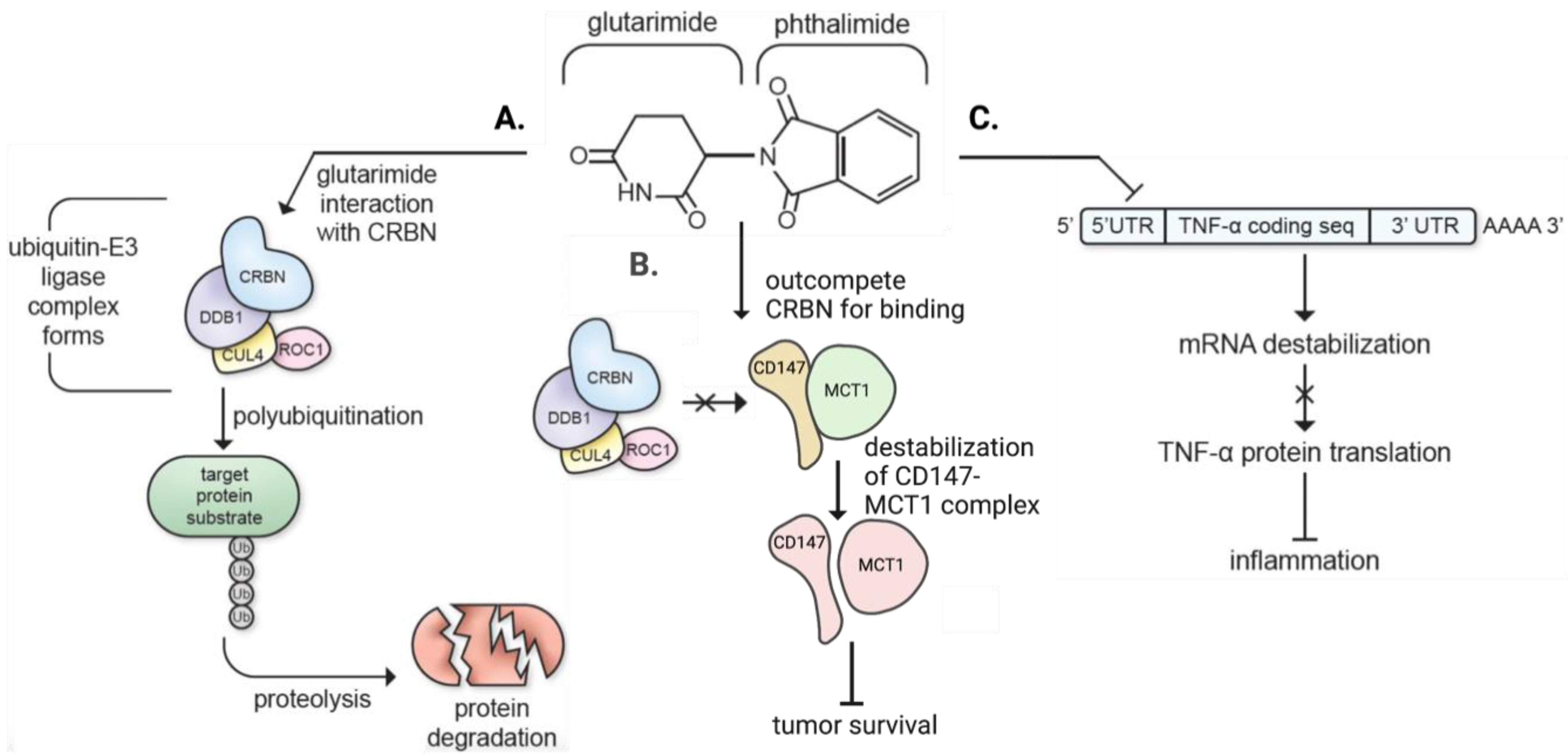
4.1. IMiD Signaling
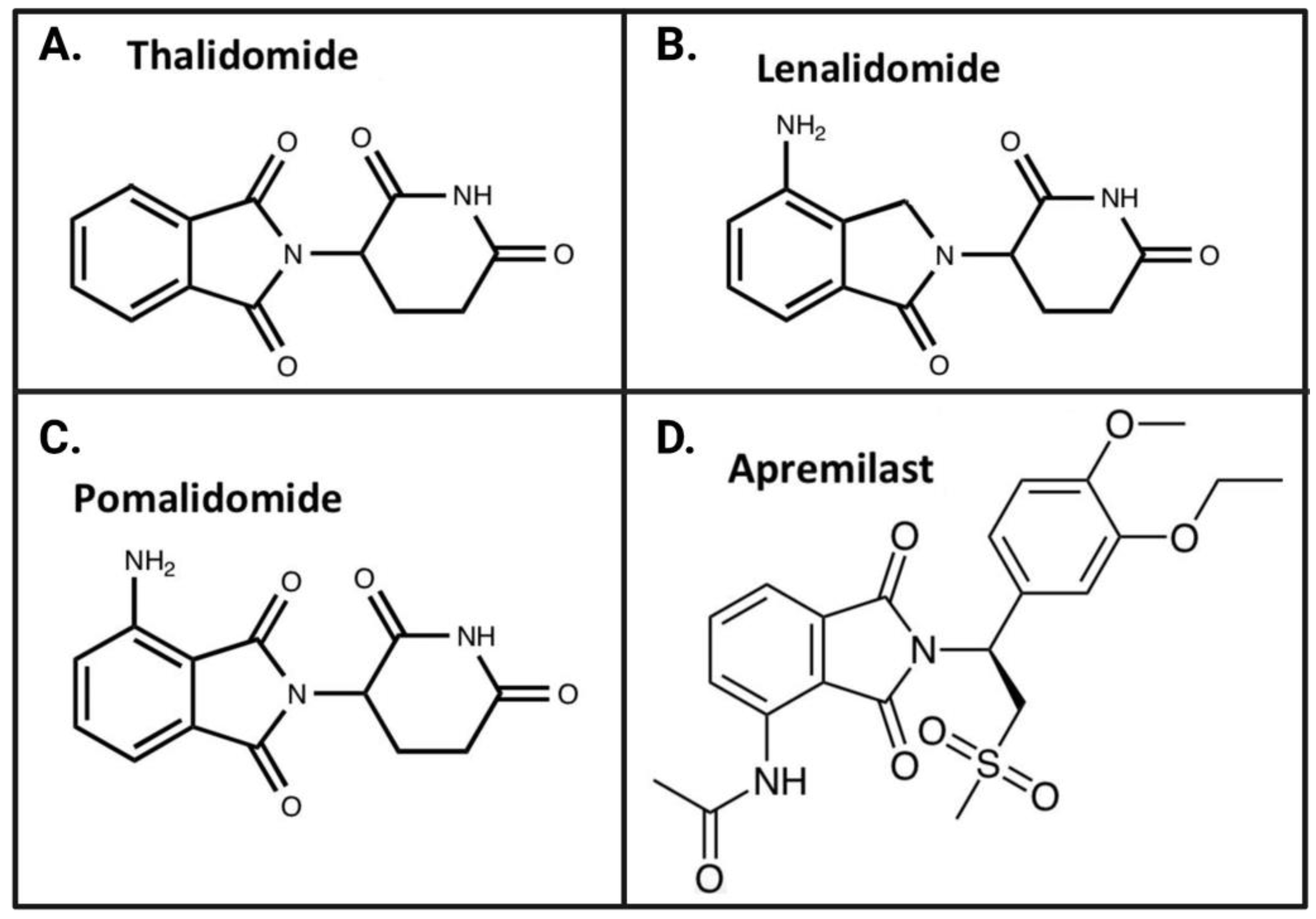
4.2. FDA-Approved IMiDs
4.2.1. Thalidomide
4.2.2. Lenalidomide
4.2.3. Pomalidomide
4.2.4. Apremilast
4.3. New Generation IMiDs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cereblon Binding? | Degradation of Neosubstrates? | Preclinical Outcomes | BBB Permeability Evaluation | Lipinski Rule of 5 |
|---|---|---|---|---|---|
3,6′-DP | Yes | No | Protects CNS cells in rat TBI models. See (a.) Lin et al., 2020, (b.) Huang et al., 2021, & (c.) Hsueh et al., 2022. | Brain/plasma ratio: 0.8 MPO score: 5.5 CLog P: 0.97 | Conforms |
| Anti-neuroinflammatory in rat ischemic stroke models. See (d.) Tsai et al., 2022. | |||||
| Anti-inflammatory and neuro- protective in 5xFAD mouse AD model. See (e.) Lecca et al., 2022. | |||||
1,6′-DP | Yes | Yes—Ikaros & Aiolos, at high concentrations only | Mitigates stroke and reduces pro- inflammatory factors in brain. See (d.) Tsai et al., 2022. | MPO score: 5.5 CLog P: 0.97 | Conforms |
NAP | No | No | Improves TBI-driven damage in rat model. See (f.) Hsueh et al., 2021. | MPO score: 3.7 CLog P: 3.86 | Conforms |
Pomalidomide | Yes | Yes—SALL4, Ikaros, Aiolos | Mitigates TBI-induced deficits andneuroinflammation at 5-fold higherdose vs. 3,6′-DP. See (a.) Lin et al., 2020 & (b.) Huang et al., 2021. | Brain/plasma ratio: 0.8 MPO score: 4.8 CLog P: −0.16 | Conforms |
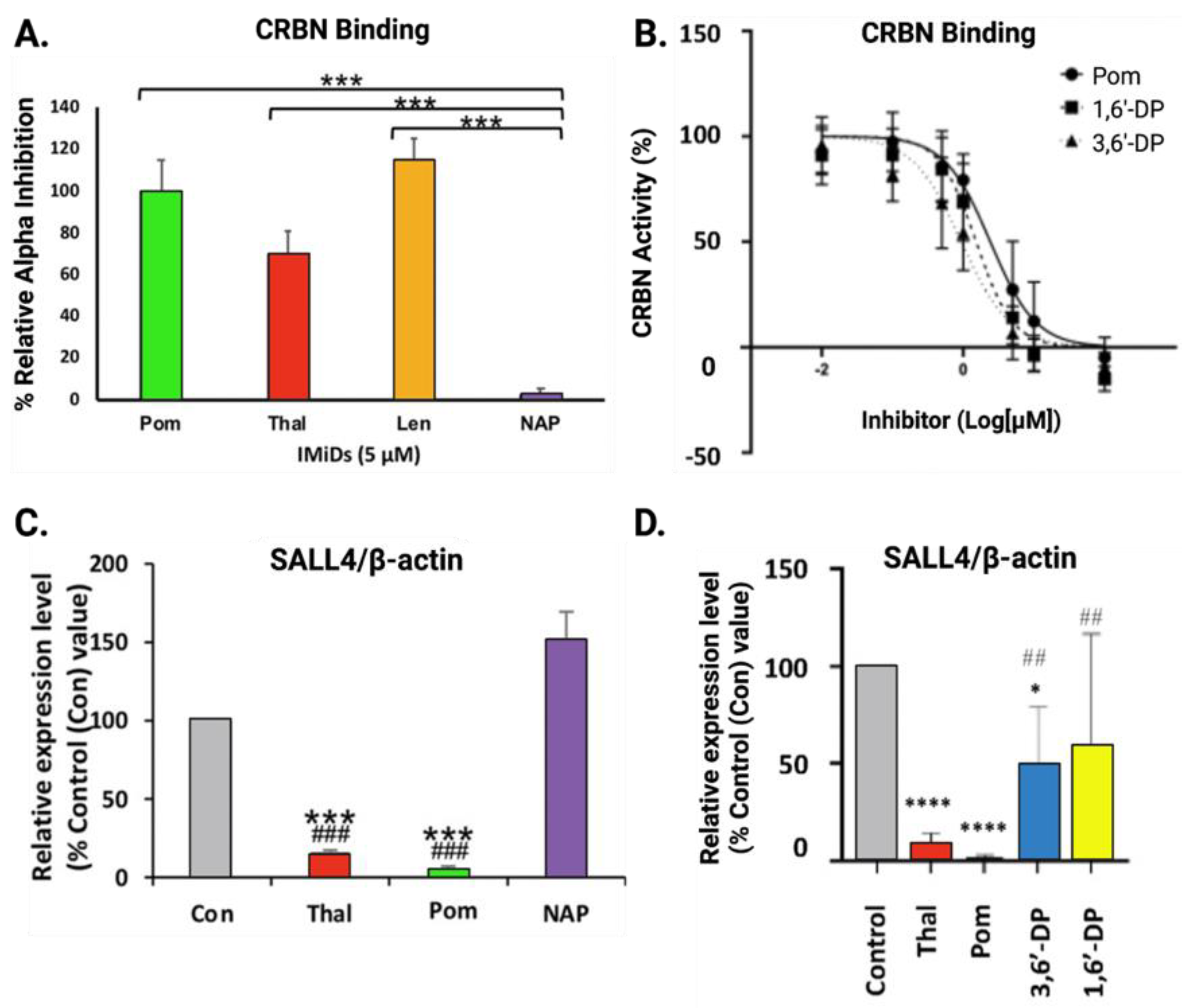
4.4. Cereblon Binding and Neosubstrate Degradation of New Generation IMiDs
5. 3,6′-DP and 1,6′-DP Experimental Data
5.1. TBI as a Model of Neurodegenerative Disease
5.2. Research Findings in TBI Models
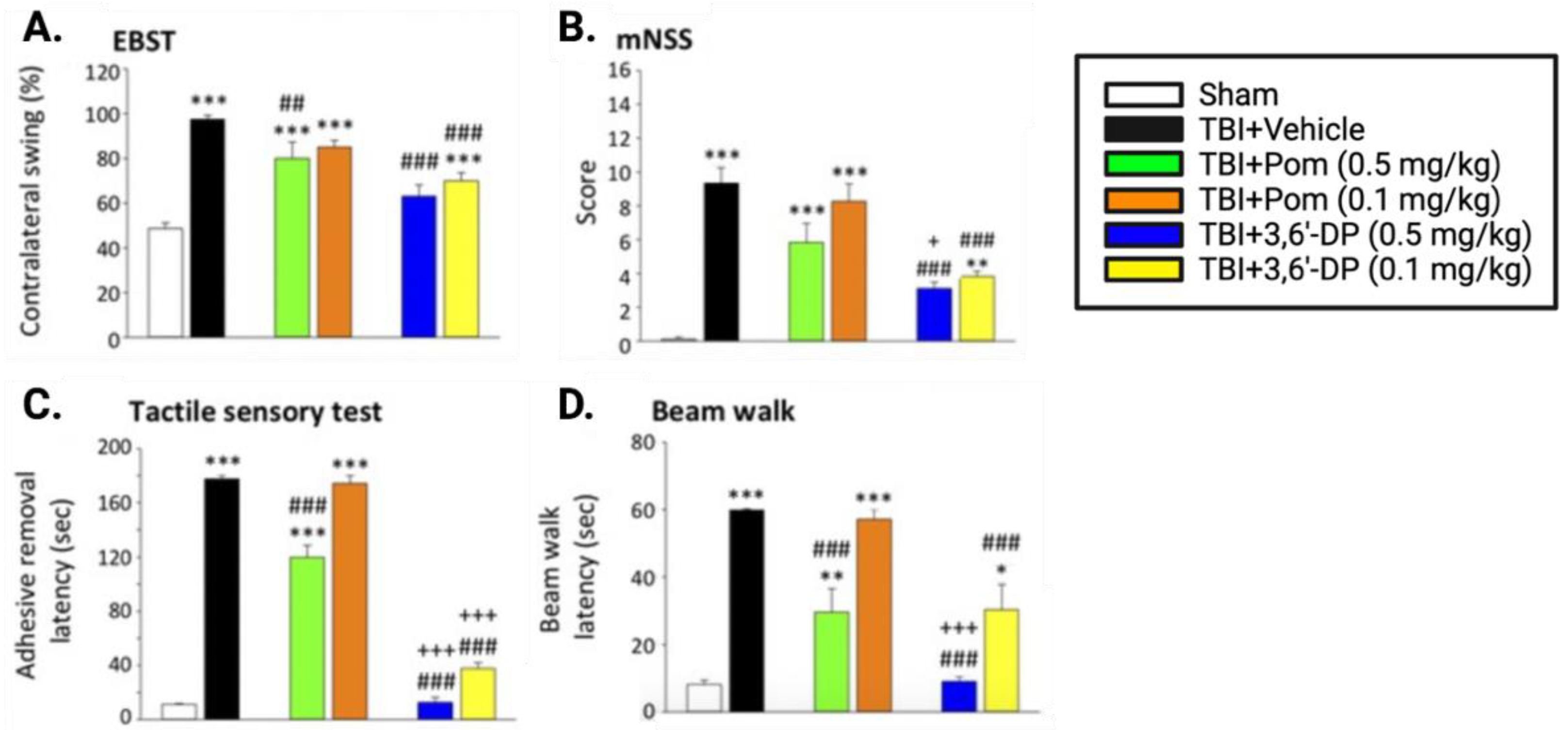
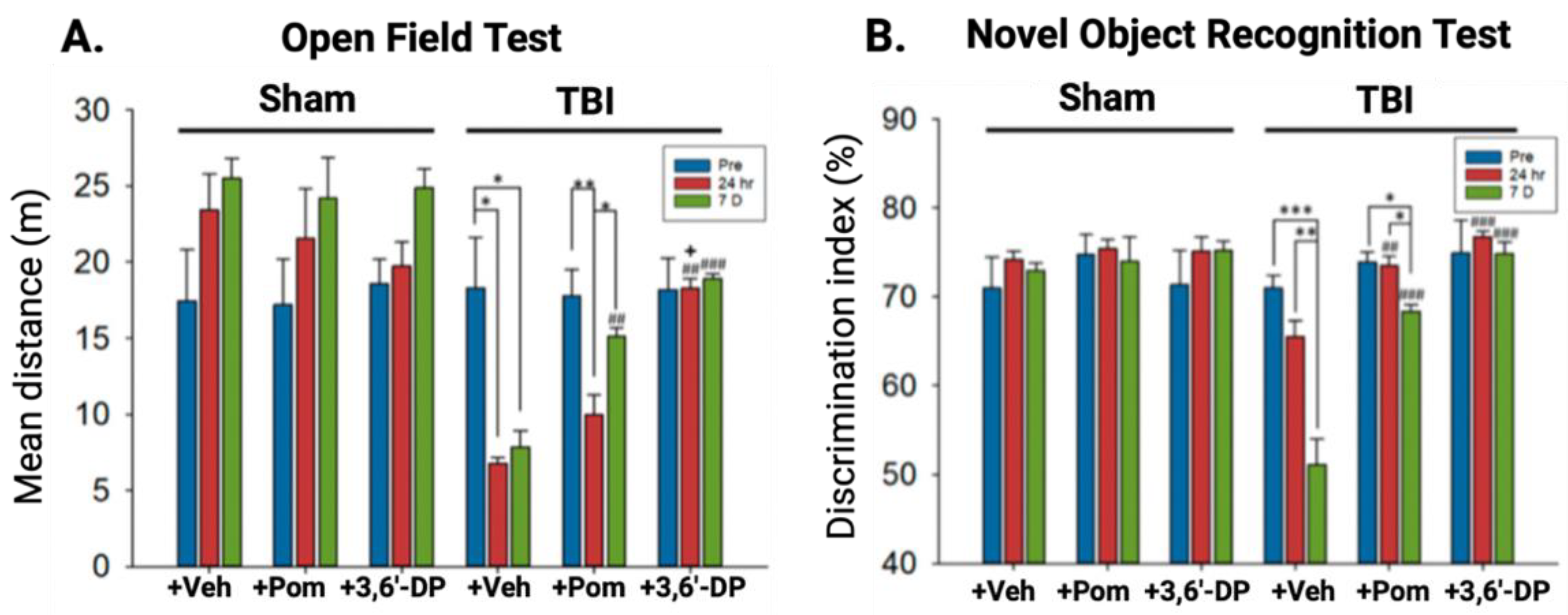
5.2.1. 3,6′-DP Mitigates Behavioral Impairments 24 h and Seven Days Post-TBI
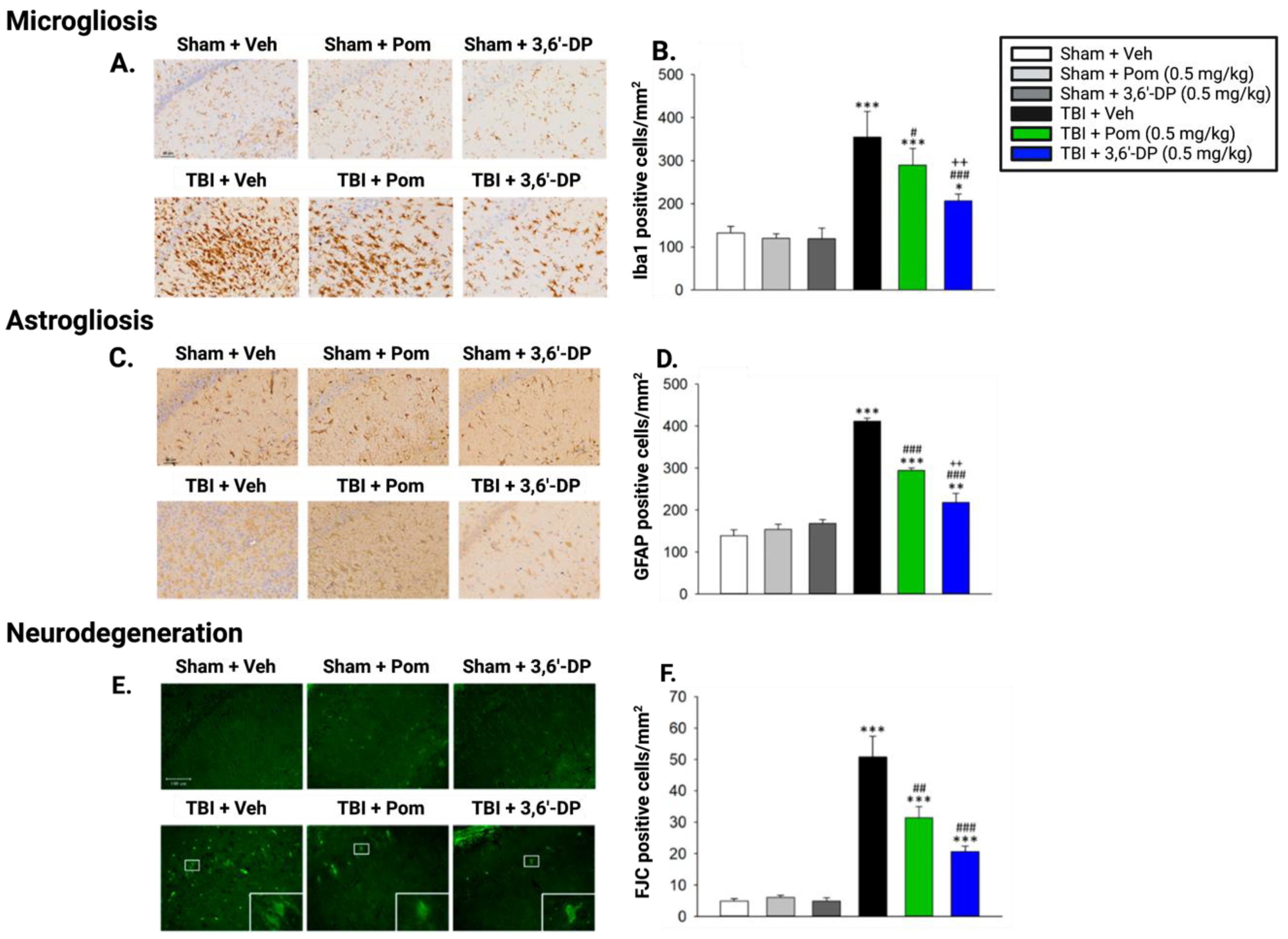
5.2.2. 3,6′-DP Mitigates Microgliosis and Neuroinflammation 24 h and 7 Days Post-TBI
5.2.3. 3,6′-DP Outperforms Pomalidomide in Minimizing TBI-Induced Astrogliosis 24 h and Seven Days Post TBI
5.2.4. 3,6′-DP Attenuates Neurodegeneration 24 h and 7 Days Post-TBI
5.2.5. F-3,6′-DP
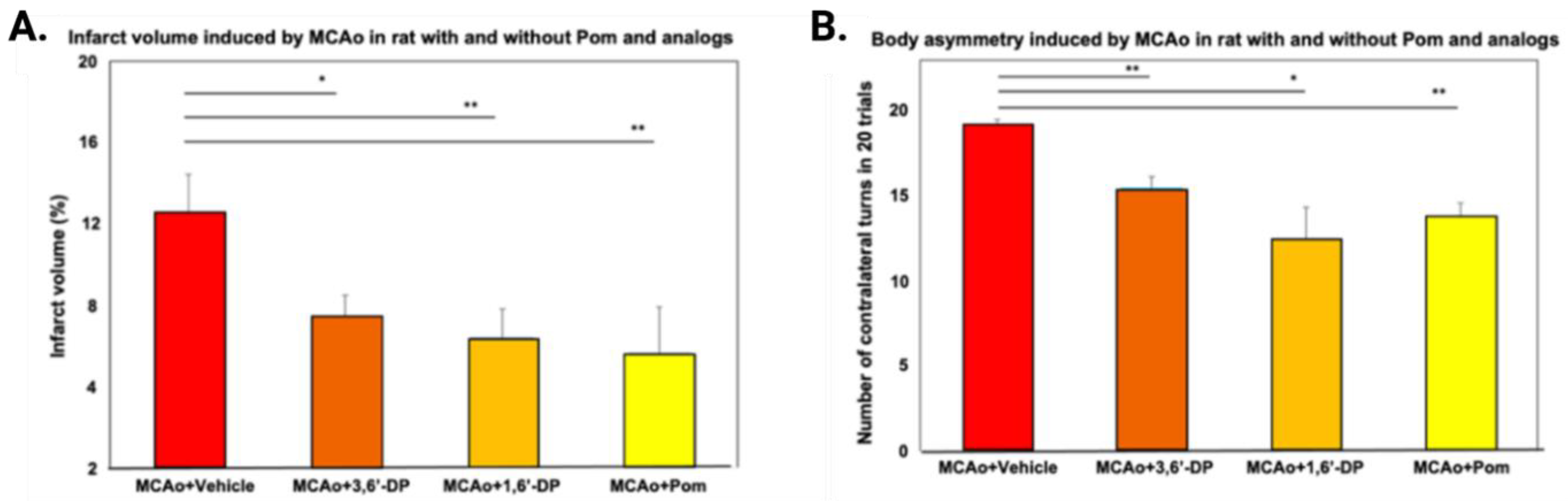
5.3. 3,6′-DP and 1,6′-DP as Ischemic Stroke Treatments
5.4. 3,6′-DP Treatment in an AD Mouse Model
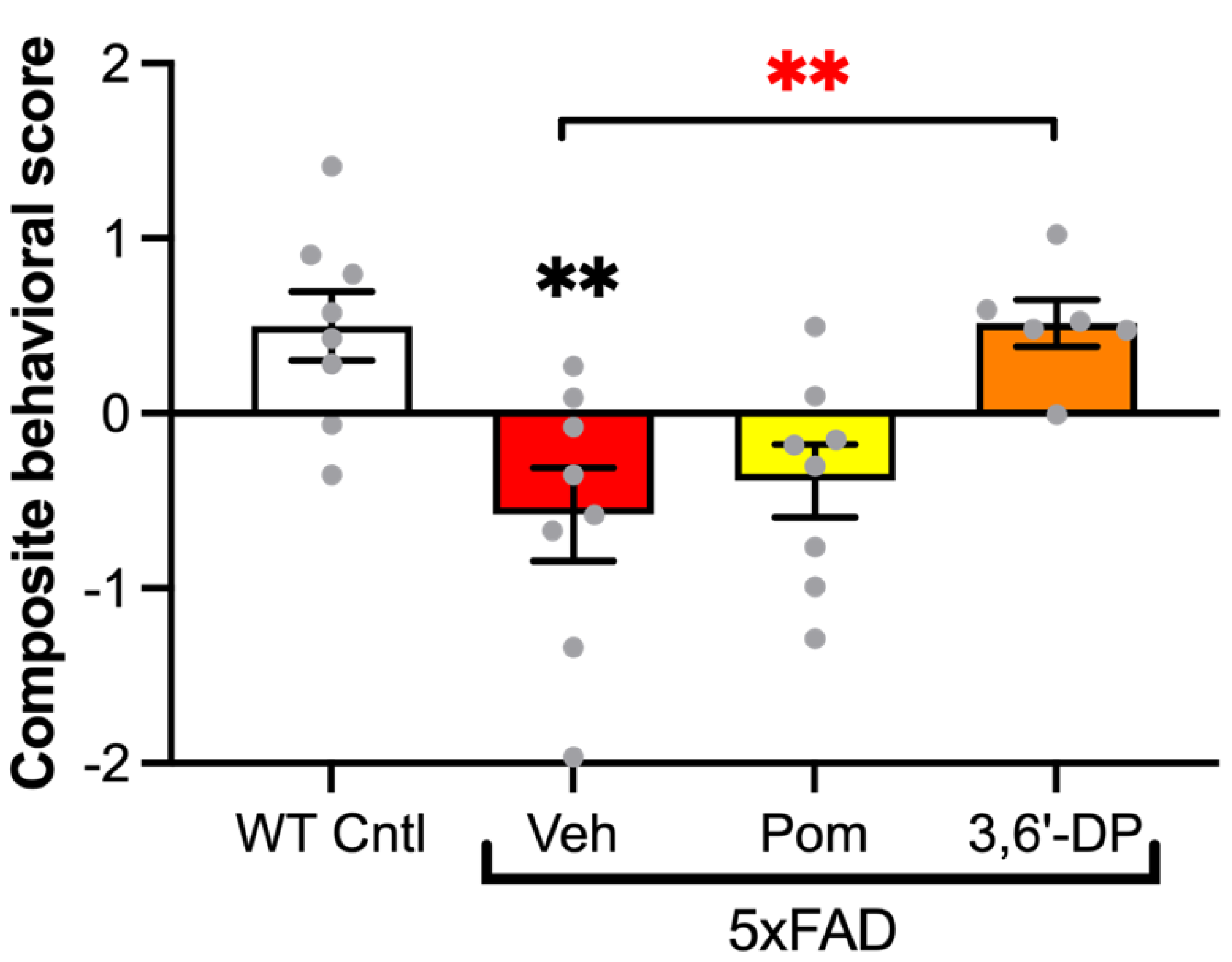
5.4.1. 3,6′-DP Treatment Mitigates Cognitive Decline in 5xFAD Mice
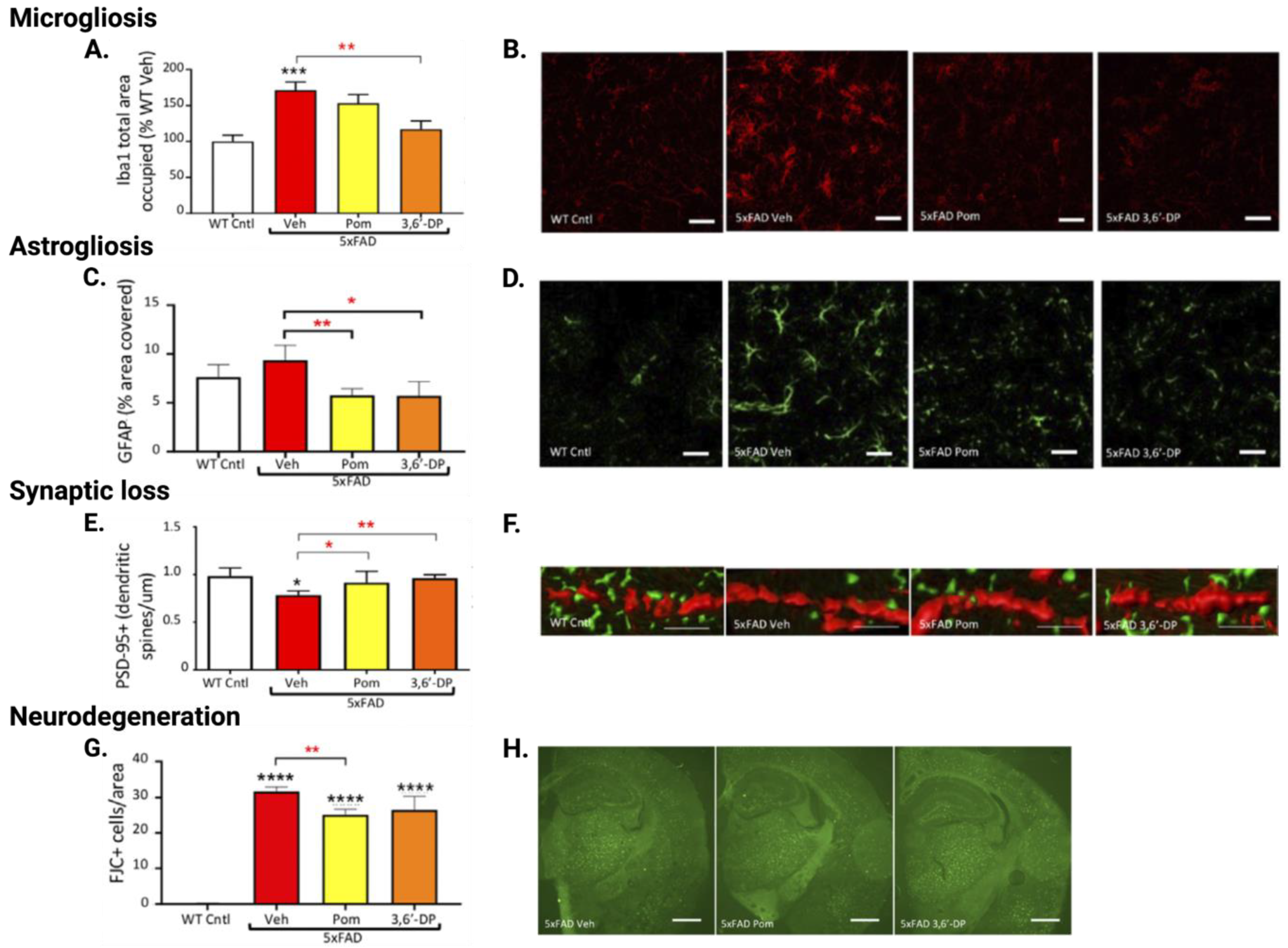
5.4.2. 3,6′-DP Mitigates Neuroinflammation in 5xFAD Mice
5.4.3. 3,6′-DP Mitigates Synaptic Loss and Neurodegeneration in 5xFAD Mice
5.4.4. Independence of Treatment Effects from Amyloid-β
5.5. 3,6′-DP Experimental Data Summary
6. NAP Experimental Data
6.1. NAP as a Treatment for Neuroinflammation in Preclinical Models
6.2. NAP as a Treatment for TBI-Induced Neuroinflammation and Neurodegeneration
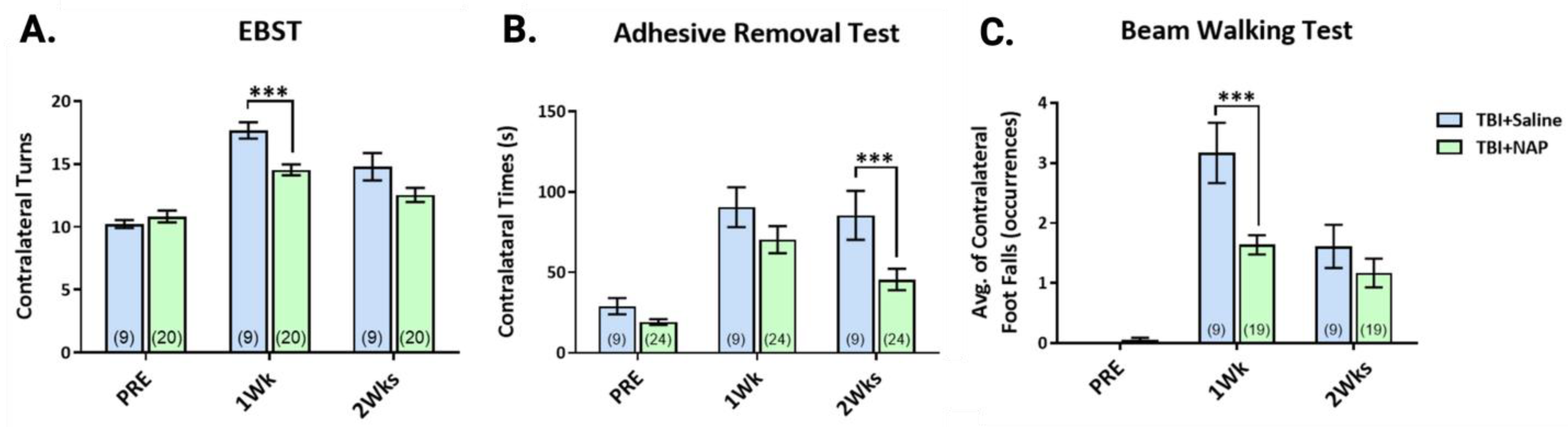
6.2.1. NAP Improves Behavioral Impairments Post-TBI
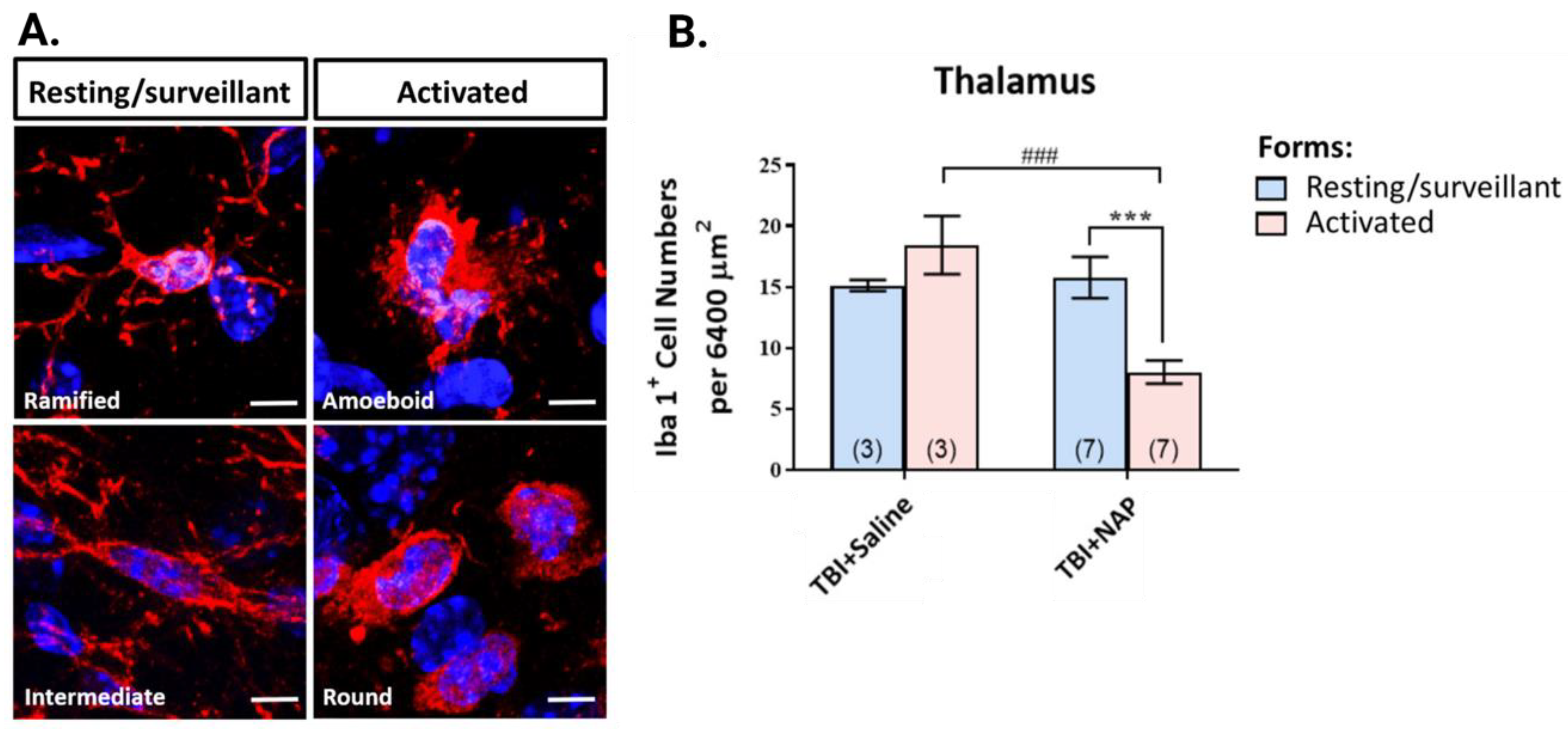
6.2.2. NAP Mitigates Neuroinflammation Post TBI
6.2.3. NAP Mitigates Synaptic Loss Post-TBI
6.3. A Newly Developed Non-Cereblon-Binding IMiD in Initial Phases of Study: Tetrafluorobornylphthalimide
6.4. NAP and TFBP Experimental Data Summary
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alam, Q.; Alam, M.Z.; Mushtaq, G.; Damanhouri, G.A.; Rasool, M.; Kamal, M.A.; Haque, A. Inflammatory Process in Alzheimer’s and Parkinson’s Diseases: Central Role of Cytokines. Curr. Pharm. Des. 2016, 22, 541–548. [Google Scholar] [CrossRef]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 74. [Google Scholar] [CrossRef] [PubMed]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson’s Disease. Front. Neurol. 2018, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Glotfelty, E.J.; Olson, L.; Karlsson, T.E.; Li, Y.Z.; Greig, N.H. Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson’s disease. Expert. Opin. Investig. Drugs 2020, 29, 595–602. [Google Scholar] [CrossRef]
- Joers, V.; Tansey, M.G.; Mulas, G.; Carta, A.R. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog. Neurobiol. 2017, 155, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Glotfelty, E.J.; Delgado, T.E.; Tovar-y-Romo, L.B.; Luo, Y.; Hoffer, B.J.; Olson, L.; Karlsson, T.E.; Mattson, M.P.; Harvey, B.K.; Tweedie, D.; et al. Incretin Mimetics as Rational Candidates for the Treatment of Traumatic Brain Injury. ACS Pharmacol. Transl. 2019, 2, 66–91. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Di Raimondo, D.; di Sciacca, R.; Pinto, A.; Licata, G. Inflammatory Cytokines in Acute Ischemic Stroke. Curr. Pharm. Des. 2008, 14, 3574–3589. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef]
- Xiao, T.S. Innate immunity and inflammation. Cell. Mol. Immunol. 2017, 14, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S.; Chen, S.N. Apoptotic cell: Linkage of inflammation and wound healing. Front. Pharmacol. 2014, 5, 1. [Google Scholar] [CrossRef]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Guzman-Martinez, L.; Cerda-Troncoso, C.; Farias, G.A.; Maccioni, R.B. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 2014, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Reich, N.; Holscher, C. The neuroprotective effects of glucagon-like peptide 1 in Alzheimer’s and Parkinson’s disease: An in-depth review. Front. Neurosci-Switz. 2022, 16, 970925. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Tweedie, D.; Scerba, M.T.; Greig, N.H. Neuroinflammation as a Factor of Neurodegenerative Disease: Thalidomide Analogs as Treatments. Front. Cell. Dev. Biol. 2019, 7, 313. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell. 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.E6. [Google Scholar] [CrossRef] [PubMed]
- Li, J.W.; Zong, Y.; Cao, X.P.; Tan, L.; Tan, L. Microglial priming in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 176. [Google Scholar] [CrossRef]
- Tweedie, D.; Sambamurti, K.; Greig, N.H. TNF-alpha inhibition as a treatment strategy for neurodegenerative disorders: New drug candidates and targets. Curr. Alzheimer Res. 2007, 4, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Tweedie, D.; Scerba, M.T.; Kim, D.S.; Palmas, M.F.; Pisanu, A.; Carta, A.R.; Greig, N.H. Repurposing Immunomodulatory Imide Drugs (IMiDs) in Neuropsychiatric and Neurodegenerative Disorders. Front. Neurosci. 2021, 15, 656921. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, S.C.; Luo, W.; Tweedie, D.; Kim, D.S.; Kim, Y.K.; Hwang, I.; Gil, J.E.; Han, B.S.; Chiang, Y.H.; Selman, W.; et al. N-Adamantyl Phthalimidine: A New Thalidomide-like Drug That Lacks Cereblon Binding and Mitigates Neuronal and Synaptic Loss, Neuroinflammation, and Behavioral Deficits in Traumatic Brain Injury and LPS Challenge. ACS Pharmacol. Transl. 2021, 4, 980–1000. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.N.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, S.; Lombardi, M.; Verderio, C.; Fumagalli, M. TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells 2020, 9, 2145. [Google Scholar] [CrossRef]
- Clark, I.A.; Vissel, B. Broader Insights into Understanding Tumor Necrosis Factor and Neurodegenerative Disease Pathogenesis Infer New Therapeutic Approaches. J. Alzheimers Dis. 2021, 79, 931–948. [Google Scholar] [CrossRef] [PubMed]
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Kopp, K.O.; Glotfelty, E.J.; Li, Y.; Greig, N.H. Glucagon-like peptide-1 (GLP-1) receptor agonists and neuroinflammation: Implications for neurodegenerative disease treatment. Pharmacol. Res. 2022, 186, 106550. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275, 316–327. [Google Scholar] [CrossRef]
- Russo, I.; Caracciolo, L.; Tweedie, D.; Choi, S.H.; Greig, N.H.; Barlati, S.; Bosetti, F. 3,6′-Dithiothalidomide, a new TNF-alpha synthesis inhibitor, attenuates the effect of A beta(1-42) intracerebroventricular injection on hippocampal neurogenesis and memory deficit. (vol 122, pg 1181, 2012). J. Neurochem. 2012, 123, 645. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Larke, L.E.C.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Adler, D.I.; Couthouis, J.; Liddelow, S.A.; Gitler, A.D.; Barres, B. Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Munch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Munch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.D.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Wang, J.; Tan, L.; Wang, H.F.; Tan, C.C.; Meng, X.F.; Wang, C.; Tang, S.W.; Yu, J.T. Anti-Inflammatory Drugs and Risk of Alzheimer’s Disease: An Updated Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2015, 44, 385–396. [Google Scholar] [CrossRef]
- Breitner, J.C.; Baker, L.D.; Montine, T.J.; Meinert, C.L.; Lyketsos, C.G.; Ashe, K.H.; Brandt, J.; Craft, S.; Evans, D.E.; Green, R.C.; et al. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement. 2011, 7, 402–411. [Google Scholar] [CrossRef]
- Stewart, W.F.; Kawas, C.; Corrada, M.; Metter, E.J. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997, 48, 626–632. [Google Scholar] [CrossRef]
- Jaturapatporn, D.; Isaac, M.G.; McCleery, J.; Tabet, N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2012, CD006378. [Google Scholar] [CrossRef]
- Klegeris, A.; McGeer, P.L. Non-steroidal anti-inflammatory drugs (NSAIDs) and other anti-inflammatory agents in the treatment of neurodegenerative disease. Curr. Alzheimer Res. 2005, 2, 355–365. [Google Scholar] [CrossRef]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Wang, Y.; Wang, D.Y.; Zhang, J.D.; Zhang, F.F. NSAID Exposure and Risk of Alzheimer’s Disease: An Updated Meta-Analysis From Cohort Studies. Front. Aging Neurosci. 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Gyengesi, E.; Munch, G. In search of an anti-inflammatory drug for Alzheimer disease. Nat. Rev. Neurol. 2020, 16, 131–132. [Google Scholar] [CrossRef] [PubMed]
- National Academies of Sciences, Engineering, and Medicine. Traumatic Brain Injury: A Roadmap for Accelerating Progress; The National Academies Press: Washington, DC, USA, 2022. [Google Scholar]
- Stein, D.G.; Sayeed, I. Repurposing and repositioning neurosteroids in the treatment of traumatic brain injury: A report from the trenches. Neuropharmacology 2019, 147, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug. Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C. Phenotypic vs. Target-Based Drug Discovery for First-in-Class Medicines. Clin. Pharmacol. Ther. 2013, 93, 299–301. [Google Scholar] [CrossRef]
- Tobinick, E. Perispinal etanercept a new therapeutic paradigm in neurology. Expert. Rev. Neurother. 2010, 10, 985–1002. [Google Scholar] [CrossRef]
- Tobinick, E. Perispinal etanercept advances as a neurotherapeutic. Expert. Rev. Neurother. 2018, 18, 453–455. [Google Scholar] [CrossRef]
- Clark, I.A.; Vissel, B. The Inflammatory Nature of Post-surgical Delirium Predicts Benefit of Agents With Anti-TNF Effects, Such as Dexmedetomidine. Front. Neurosci. 2018, 12, 257. [Google Scholar] [CrossRef]
- Shi, Q.L.; Chen, L.J. Cereblon: A Protein Crucial to the Multiple Functions of Immunomodulatory Drugs as well as Cell Metabolism and Disease Generation. J. Immunol. Res. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Peach, M.L.; Beedie, S.L.; Chau, C.H.; Collins, M.K.; Markolovic, S.; Luo, W.M.; Tweedie, D.; Steinebach, C.; Greig, N.H.; Gutschow, M.; et al. Antiangiogenic Activity and in Silico Cereblon Binding Analysis of Novel Thalidomide Analogs. Molecules 2020, 25, 5683. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug. Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Handa, H. Discovery of the target for immunomodulatory drugs (IMiDs). Rinsho Ketsueki 2016, 57, 556–562. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Lu, G.; Rychak, E.; Mendy, D.; Lu, C.C.; Rappley, I.; Fontanillo, C.; Cathers, B.E.; Daniel, T.O.; Hansen, J. CC-90009, a Novel Cereblon E3 Ligase Modulator, Targets GSPT1 for Degradation to Induce Potent Tumoricidal Activity Against Acute Myeloid Leukemia (AML). Blood 2019, 134, 2703. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.Y.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef]
- Eichner, R.; Heider, M.; Fernandez-Saiz, V.; van Bebber, F.; Garz, A.K.; Lemeer, S.; Rudelius, M.; Targosz, B.S.; Jacobs, L.; Knorn, A.M.; et al. Immunomodulatory drugs disrupt the cereblon-CD147-MCT1 axis to exert antitumor activity and teratogenicity. Nat. Med. 2016, 22, 735–743. [Google Scholar] [CrossRef]
- Moreira, A.L.; Sampaio, E.P.; Zmuidzinas, A.; Frindt, P.; Smith, K.A.; Kaplan, G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J. Exp. Med. 1993, 177, 1675–1680. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2013, 54, 683–687. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Beedie, S.L.; Huang, P.A.; Harris, E.M.; Strope, J.D.; Mahony, C.; Chau, C.H.; Vargesson, N.; Figg, W.D. Role of cereblon in angiogenesis and in mediating the antiangiogenic activity of immunomodulatory drugs. FASEB J. 2020, 34, 11395–11404. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Maiwald, S.; Steinebach, C.; Collins, M.K.; Strope, J.; Chau, C.H.; Figg, W.D.; Gutschow, M.; Hartmann, M.D. On the correlation of cereblon binding, fluorination and antiangiogenic properties of immunomodulatory drugs. Biochem. Biophys. Res. Commun. 2021, 534, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.; Wong, E.; Koipally, J.; Georgopoulos, K. Control of lymphocyte development by the Ikaros gene family. Curr. Opin. Immunol. 1999, 11, 167–171. [Google Scholar] [CrossRef]
- Sievers, Q.L.; Petzold, G.; Bunker, R.D.; Renneville, A.; Slabicki, M.; Liddicoat, B.J.; Abdulrahman, W.; Mikkelsen, T.; Ebert, B.L.; Thoma, N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362, eaat0572. [Google Scholar] [CrossRef]
- Quintana, F.J.; Jin, H.L.; Burns, E.J.; Nadeau, M.; Yeste, A.; Kumar, D.; Rangachari, M.; Zhu, C.; Xiao, S.; Kuchroo, V.K. Aiolos promotes TH17 cell differentiation by directly silencing Il2 expression. J. Neuroimmunol. 2012, 253, 75. [Google Scholar] [CrossRef]
- Davies, F.E.; Raje, N.; Hideshima, T.; Lentzsch, S.; Young, G.; Tai, Y.T.; Lin, B.; Podar, K.; Gupta, D.; Chauhan, D.; et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001, 98, 210–216. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Bach, C.; Bottcher, M.; Beier, F.; Rohner, L.; Stoll, A.; Rehli, M.; Gebhard, C.; Lischer, C.; Eberhardt, M.; et al. The IKZF1-IRF4/IRF5 Axis Controls Polarization of Myeloma-Associated Macrophages. Cancer Immunol. Res. 2021, 9, 265–278. [Google Scholar] [CrossRef]
- Fan, W.J.; Fan, Z.Q.; Yang, M.J.; Pan, Y.Z.; Bai, H. Molecular Mechanism of CRBN in the Activity of Lenalidomide against Myeloma—Review. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2018, 26, 1240–1243. [Google Scholar]
- Ito, T.; Handa, H. Molecular mechanisms of thalidomide and its derivatives. P. Jpn. Acad. B-Phys. 2020, 96, 189–203. [Google Scholar] [CrossRef]
- Vargesson, N. Thalidomide-induced teratogenesis: History and mechanisms. Birth Defects Res. C 2015, 105, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Vargesson, N. The teratogenic effects of thalidomide on limbs. J Hand Surg (Eur Vol) 2019, 44, 88–95. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Couto, S.; Zheng, X.D.; Lu, G.; Hui, J.L.; Stamp, K.; Drew, C.; Ren, Y.; Wang, M.; Carpenter, A.; et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol. 2018, 14, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Kohlhase, J.; Chitayat, D.; Kotzot, D.; Ceylaner, S.; Froster, U.G.; Fuchs, S.; Montgomery, T.; Rosler, B. SALL4 mutations in Okihiro syndrome (Duane-radial ray syndrome), acro-renal-ocular syndrome, and related disorders. Hum. Mutat. 2005, 26, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Donovan, K.A.; An, J.; Nowak, R.P.; Yuan, J.T.C.; Fink, E.C.; Berry, B.C.; Ebert, B.L.; Fischer, E.S. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. Elife 2018, 7, e38430. [Google Scholar] [CrossRef]
- Al-Baradie, R.; Yamada, K.; St Hilaire, C.; Chan, W.M.; Andrews, C.; McIntosh, N.; Nakano, M.; Martonyi, E.J.; Raymond, W.R.; Okumura, S.; et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 2002, 71, 1195–1199. [Google Scholar] [CrossRef]
- Zhu, X.X.; Giordano, T.; Yu, Q.S.; Holloway, H.W.; Perry, T.A.; Lahiri, D.K.; Brossi, A.; Greig, N.H. Thiothalidomides: Novel isosteric analogues of thalidomide with enhanced TNF-alpha inhibitory activity. J. Med. Chem. 2003, 46, 5222–5229. [Google Scholar] [CrossRef]
- Sheskin, J. Thalidomide in the Treatment of Lepra Reactions. Clin. Pharmacol. Ther. 1965, 6, 303–306. [Google Scholar] [CrossRef]
- Sheskin, J. The Treatment of Lepra Reaction in Lepromatous Leprosy—15 Years Experience with Thalidomide. Int. J. Dermatol. 1980, 19, 318–322. [Google Scholar] [CrossRef]
- Teo, S.K.; Stirling, D.I.; Zeldis, J.B. Thalidomide as a novel therapeutic agent: New uses for an old product. Drug. Discov. Today 2005, 10, 107–114. [Google Scholar] [CrossRef]
- Gutierrez-Rodriguez, O. Thalidomide—A Promising New Treatment for Rheumatoid-Arthritis. Arthritis Rheum. 1984, 27, 1118–1121. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, E.P.; Sarno, E.N.; Galilly, R.; Cohn, Z.A.; Kaplan, G. Thalidomide Selectively Inhibits Tumor-Necrosis-Factor-Alpha Production by Stimulated Human Monocytes. J. Exp. Med. 1991, 173, 699–703. [Google Scholar] [CrossRef]
- Zeldis, J.B.; Knight, R.; Hussein, M.; Chopra, R.; Muller, G. A review of the history, properties, and use of the immunomodulatory compound lenalidomide. Ann. N. Y. Acad. Sci. 2011, 1222, 76–82. [Google Scholar] [CrossRef]
- Damato, R.J.; Loughnan, M.S.; Flynn, E.; Folkman, J. Thalidomide Is an Inhibitor of Angiogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef]
- Gao, S.B.; Wang, S.C.; Fan, R.H.; Hu, J.Y. Recent advances in the molecular mechanism of thalidomide teratogenicity. Biomed. Pharmacother. 2020, 127, 110114. [Google Scholar] [CrossRef] [PubMed]
- Corral, L.G.; Muller, G.W.; Moreira, A.L.; Chen, Y.X.; Wu, M.D.; Stirling, D.; Kaplan, G. Selection of novel analogs of thalidomide with enhanced tumor necrosis factor alpha inhibitory activity. Mol. Med. 1996, 2, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Palumbo, A.; Raje, N.; Boccadoro, M.; Anderson, K.C. Thalidomide and lenalidomide: Mechanism-based potential drug combinations. Leuk. Lymphoma 2008, 49, 1238–1245. [Google Scholar] [CrossRef]
- Hofmeister, C.C.; Yang, X.X.; Pichiorri, F.; Chen, P.; Rozewski, D.M.; Johnson, A.J.; Lee, S.; Liu, Z.F.; Garr, C.L.; Hade, E.M.; et al. Phase I Trial of Lenalidomide and CCI-779 in Patients With Relapsed Multiple Myeloma: Evidence for Lenalidomide-CCI-779 Interaction via P-Glycoprotein. J. Clin. Oncol. 2011, 29, 3427–3434. [Google Scholar] [CrossRef]
- Schinkel, A.H. P-glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug. Deliver Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Corral, L.G.; Haslett, P.A.J.; Muller, G.W.; Chen, R.; Wong, L.M.; Ocampo, C.J.; Patterson, R.T.; Stirling, D.I.; Kaplan, G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J. Immunol. 1999, 163, 380–386. [Google Scholar] [CrossRef]
- Tsai, Y.R.; Tweedie, D.; Navas-Enamorado, I.; Scerba, M.T.; Chang, C.F.; Lai, J.H.; Wu, J.C.C.; Chen, Y.H.; Kang, S.J.; Hoffer, B.J.; et al. Pomalidomide Reduces Ischemic Brain Injury in Rodents. Cell. Transplant. 2019, 28, 439–450. [Google Scholar] [CrossRef]
- Schafer, P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem. Pharmacol. 2012, 83, 1583–1590. [Google Scholar] [CrossRef]
- Scerba, M.T.; Siegler, M.A.; Greig, N.H. Thionation of Aminophthalimide Hindered Carbonyl Groups and Application to the Synthesis of 3,6′-Dithionated Pomalidomides. Synlett 2021, 32, 917–922. [Google Scholar] [CrossRef]
- Lin, C.T.; Lecca, D.; Yang, L.Y.; Luo, W.M.; Scerba, M.T.; Tweedie, D.; Huang, P.S.; Jung, Y.J.; Kim, D.S.; Yang, C.H.; et al. 3,6′-dithiopomalidomide reduces neural loss, inflammation, behavioral deficits in brain injury and microglial activation. Elife 2020, 9, e54726. [Google Scholar] [CrossRef]
- Huang, P.S.; Tsai, P.Y.; Yang, L.Y.; Lecca, D.; Luo, W.M.; Kim, D.S.; Hoffer, B.J.; Chiang, Y.H.; Greig, N.H.; Wang, J.Y. 3,6′-Dithiopomalidomide Ameliorates Hippocampal Neurodegeneration, Microgliosis and Astrogliosis and Improves Cognitive Behaviors in Rats with a Moderate Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 8276. [Google Scholar] [CrossRef]
- Hsueh, S.C.; Scerba, M.T.; Tweedie, D.; Lecca, D.; Kim, D.S.; Baig, A.M.; Kim, Y.K.; Hwang, I.; Kim, S.; Selman, W.R.; et al. Activity of a Novel Anti-Inflammatory Agent F-3,6′-dithiopomalidomide as a Treatment for Traumatic Brain Injury. Biomedicines 2022, 10, 2449. [Google Scholar] [CrossRef]
- Tsai, Y.R.; Kim, D.S.; Hsueh, S.C.; Chen, K.Y.; Wu, J.C.C.; Wang, J.Y.; Tsou, Y.S.; Hwang, I.; Kim, Y.; Gil, D.; et al. 3,6′- and 1,6′-Dithiopomalidomide Mitigate Ischemic Stroke in Rats and Blunt Inflammation. Pharmaceutics 2022, 14, 950. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Jung, Y.J.; Scerba, M.T.; Hwang, I.; Kim, Y.K.; Kim, S.; Modrow, S.; Tweedie, D.; Hsueh, S.C.; Liu, D.; et al. Role of chronic neuroinflammation in neuroplasticity and cognitive function: A hypothesis. Alzheimers Dement. 2022, 18, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central nervous system multiparameter optimization desirability: Application in drug discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug. Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef]
- Chamberlain, P.P.; Cathers, B.E. Cereblon modulators: Low molecular weight inducers of protein degradation. Drug. Discov. Today Technol. 2019, 31, 29–34. [Google Scholar] [CrossRef]
- Crane, P.K.; Gibbons, L.E.; Dams-O’Connor, K.; Trittschuh, E.; Leverenz, J.B.; Keene, C.D.; Sonnen, J.; Montine, T.J.; Bennett, D.A.; Leurgans, S.; et al. Association of Traumatic Brain Injury With Late-Life Neurodegenerative Conditions and Neuropathologic Findings. JAMA Neurol. 2016, 73, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Fann, J.R.; Ribe, A.R.; Pedersen, H.S.; Fenger-Gron, M.; Christensen, J.; Benros, M.E.; Vestergaard, M. Long-term risk of dementia among people with traumatic brain injury in Denmark: A population-based observational cohort study. Lancet Psychiat 2018, 5, 424–431. [Google Scholar] [CrossRef]
- Schaffert, J.; Lobue, C.; White, C.L.; Chiang, H.S.; Didehbani, N.; Lacritz, L.; Rossetti, H.; Dieppa, M.; Hart, J.; Cullum, C.M. Traumatic Brain Injury History Is Associated With an Earlier Age of Dementia Onset in Autopsy-Confirmed Alzheimer’s Disease. Neuropsychology 2018, 32, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.C.; Byers, A.L.; Barnes, D.E.; Li, Y.X.; Boscardin, J.; Yaffe, K. Mild TBI and risk of Parkinson disease A Chronic Effects of Neurotrauma Consortium Study. Neurology 2018, 90, E1771–E1779. [Google Scholar] [CrossRef]
- Goldman, S.M.; Tanner, C.M.; Oakes, D.; Bhudhikanok, G.S.; Gupta, A.R.; Langston, J.W. Head injury and Parkinson’s disease risk in twins. Ann. Neurol. 2006, 60, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotraum 2015, 32, 1834–1848. [Google Scholar] [CrossRef] [PubMed]
- Andriessen, T.M.J.C.; Jacobs, B.; Vos, P.E. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J. Cell. Mol. Med. 2010, 14, 2381–2392. [Google Scholar] [CrossRef]
- Onyango, I.G.; Jauregui, G.V.; Carna, M.; Bennett, J.P.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef]
- Greig, N.H.; Lecca, D.; Hsueh, S.C.; Nogueras-Ortiz, C.; Kapogiannis, D.; Tweedie, D.; Glotfelty, E.J.; Becker, R.E.; Chiang, Y.H.; Hoffer, B.J. (-)-Phenserine tartrate (PhenT) as a treatment for traumatic brain injury. CNS Neurosci. Ther. 2020, 26, 636–649. [Google Scholar] [CrossRef]
- Tweedie, D.; Karnati, H.K.; Mullins, R.; Pick, C.G.; Hoffer, B.J.; Goetzl, E.J.; Kapogiannis, D.; Greig, N.H. Time-dependent cytokine and chemokine changes in mouse cerebral cortex following a mild traumatic brain injury. Elife 2020, 9, e55827. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Acosta, S.; Tuazon, J.P.; Xu, K.; Nguyen, H.; Lippert, T.; Liska, M.G.; Semechkin, A.; Garitaonandia, I.; Gonzalez, R.; et al. Human parthenogenetic neural stem cell grafts promote multiple regenerative processes in a traumatic brain injury model. Theranostics 2019, 9, 1029–1046. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Kaneko, Y.; Bae, E.; Stahl, C.E.; Wang, Y.; van Loveren, H.; Sanberg, P.R.; Borlongan, C.V. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. 2009, 1287, 157–163. [Google Scholar] [CrossRef]
- Scherbel, U.; Raghupathi, R.; Nakamura, M.; Saatman, K.E.; Trojanowski, J.Q.; Neugebauer, E.; Marino, M.W.; McIntosh, T.K. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc. Natl. Acad. Sci. USA 1999, 96, 8721–8726. [Google Scholar] [CrossRef] [PubMed]
- Hopperton, K.; Mohammad, D.; Trepanier, M.; Giuliano, V.; Bazinet, R.P. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: A systematic review. Mol. Psychiatr. 2018, 23, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of fluorine-containing drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Lai, J.H.; Wu, J.C.; Greig, N.H.; Becker, R.E.; Luo, Y.; Chen, Y.H.; Kang, S.J.; Chiang, Y.H.; Chen, K.Y. (-)-Phenserine inhibits neuronal apoptosis following ischemia/reperfusion injury. Brain Res. 2017, 1677, 118–128. [Google Scholar] [CrossRef]
- Veenith, T.V.; Carter, E.L.; Geeraerts, T.; Grossac, J.; Newcombe, V.F.; Outtrim, J.; Gee, G.S.; Lupson, V.; Smith, R.; Aigbirhio, F.I.; et al. Pathophysiologic Mechanisms of Cerebral Ischemia and Diffusion Hypoxia in Traumatic Brain Injury. JAMA Neurol. 2016, 73, 542–550. [Google Scholar] [CrossRef]
- Graham, D.I.; Ford, I.; Adams, J.H.; Doyle, D.; Teasdale, G.M.; Lawrence, A.E.; Mclellan, D.R. Ischemic Brain-Damage Is Still Common in Fatal Non-Missile Head-Injury. J. Neurol. Neurosur. Ps. 1989, 52, 346–350. [Google Scholar] [CrossRef]
- Vespa, P.M. Brain Hypoxia and Ischemia After Traumatic Brain Injury: Is Oxygen the Right Metabolic Target? JAMA Neurol. 2016, 73, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Jiang, J.; He, B.; Lin, W.J.; Li, Y.; Duan, J.; Li, H.; Huang, X.; Cai, J.; Xie, J.; et al. A phase 2 study of thalidomide for the treatment of radiation-induced blood-brain barrier injury. Sci. Transl. Med. 2023, 15, eabm6543. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Lee, J.H.; Tweedie, D.; Mughal, M.R.; Chigurupati, S.; Greig, N.H.; Mattson, M.P. 3,6′-dithiothalidomide improves experimental stroke outcome by suppressing neuroinflammation. J. Neurosci. Res. 2013, 91, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.Y.; Schneider, L.S.; Howard, R. The need to show minimum clinically important differences in Alzheimer’s disease trials. Lancet Psychiat 2021, 8, 1013–1016. [Google Scholar] [CrossRef]
- Tampi, R.R.; Forester, B.P.; Agronin, M. Aducanumab: Evidence from clinical trial data and controversies. Drugs Context. 2021, 10, 1–9. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef]
- Thambisetty, M.; Howard, R. Lecanemab trial in AD brings hope but requires greater clarity. Nat. Rev. Neurol. 2023, 19, 132–133. [Google Scholar] [CrossRef]
- Poon, V.Y.; Choi, S.; Park, M. Growth factors in synaptic function. Front. Synaptic Neurosci. 2013, 5, 6. [Google Scholar] [CrossRef]
- Rizzo, F.R.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Vanni, V.; Guadalupi, L.; Stampanoni Bassi, M.; Buttari, F.; Mandolesi, G.; et al. Tumor Necrosis Factor and Interleukin-1beta Modulate Synaptic Plasticity during Neuroinflammation. Neural Plast. 2018, 2018, 8430123. [Google Scholar] [CrossRef]
- Bourgognon, J.M.; Cavanagh, J. The role of cytokines in modulating learning and memory and brain plasticity. Brain Neurosci. Adv. 2020, 4, 2398212820979802. [Google Scholar] [CrossRef]
- Clark, I.A.; Vissel, B. A Neurologist’s Guide to TNF Biology and to the Principles behind the Therapeutic Removal of Excess TNF in Disease. Neural Plast. 2015, 2015, 358263. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Spires-Jones, T.; Zetterberg, H.; Blennow, K.; Caggiano, A.; DeKosky, S.T.; Fillit, H.; Harrison, J.E.; Schneider, L.S.; Scheltens, P.; et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Hsueh, S.C.; Luo, W.; Tweedie, D.; Kim, D.S.; Baig, A.M.; Vargesson, N.; Kim, Y.K.; Hwang, I.; Kim, S.; et al. Novel, thalidomide-like, non-cereblon binding drug tetrafluorobornylphthalimide mitigates inflammation and brain injury. J. Biomed. Sci. 2023, 30, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Wi, S.M.; Kang, J.A.; Yang, T.; Park, C.S.; Park, S.G.; Chung, S.; Shim, J.H.; Chun, E.; Lee, K.Y. Cereblon negatively regulates TLR4 signaling through the attenuation of ubiquitination of TRAF6. Cell. Death Dis. 2016, 7, e2313. [Google Scholar] [CrossRef] [PubMed]
- Leprosy. Available online: https://www.who.int/news-room/fact-sheets/detail/leprosy (accessed on 16 February 2023).
- Crawford, A. Brazil’s New Generation of Thalidomide Babies. Available online: https://www.bbc.com/news/magazine-23418102 (accessed on 16 February 2023).
- Jiang, T.; Su, H.; Li, Y.; Wu, Y.; Ming, Y.; Li, C.; Fu, R.; Feng, L.; Li, Z.; Li, L.; et al. Post-marketing safety of immunomodulatory drugs in multiple myeloma: A pharmacovigilance investigation based on the FDA adverse event reporting system. Front. Pharmacol. 2022, 13, 989032. [Google Scholar] [CrossRef]
- Hirose, Y.; Kitazono, T.; Sezaki, M.; Abe, M.; Sakimura, K.; Funato, H.; Handa, H.; Vogt, K.E.; Yanagisawa, M. Hypnotic effect of thalidomide is independent of teratogenic ubiquitin/proteasome pathway. Proc. Natl. Acad. Sci. USA 2020, 117, 23106–23112. [Google Scholar] [CrossRef]
- Asatsuma-Okumura, T.; Ando, H.; De Simone, M.; Yamamoto, J.; Sato, T.; Shimizu, N.; Asakawa, K.; Yamaguchi, Y.; Ito, T.; Guerrini, L.; et al. p63 is a cereblon substrate involved in thalidomide teratogenicity. Nat. Chem. Biol. 2019, 15, 1077–1084. [Google Scholar] [CrossRef]
- Yamanaka, S.; Murai, H.; Saito, D.; Abe, G.; Tokunaga, E.; Iwasaki, T.; Takahashi, H.; Takeda, H.; Suzuki, T.; Shibata, N.; et al. Thalidomide and its metabolite 5-hydroxythalidomide induce teratogenicity via the cereblon neosubstrate PLZF. EMBO J. 2021, 40, e105375. [Google Scholar] [CrossRef]
- Yamamoto, J.; Ito, T.; Yamaguchi, Y.; Handa, H. Discovery of CRBN as a target of thalidomide: A breakthrough for progress in the development of protein degraders. Chem. Soc. Rev. 2022, 51, 6234–6250. [Google Scholar] [CrossRef]
- Mahony, C.; Erskine, L.; Niven, J.; Greig, N.H.; Figg, W.D.; Vargesson, N. Pomalidomide is nonteratogenic in chicken and zebrafish embryos and nonneurotoxic in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 12703–12708. [Google Scholar] [CrossRef] [PubMed]














| (N = 4/group) | Sham Control | MCAo + Vehicle | MCAo + 3,6′-DP | MCAo + 1,6′-DP | MCAo + POM |
|---|---|---|---|---|---|
| IL-1β (pg/mL) | 6.58 ± 2.08 | 10.20 ± 0.76 | 4.06 ± 0.95 ** | 2.93 ± 0.21 ** | 4.76 ± 1.01 * |
| TNF-α (pg/mL) | 4.08 ± 0.88 * | 7.62 ± 1.04 | 3.39 ± 0.15 ** | <2.22 | 2.59 ± 0.14 ** |
| IL-10 (pg/mL) | 14.28 ± 3.16 | 12.51 ± 0.76 | 26.49 ± 0 86 ** | 11.59 ± 3.78 | 16.62 ± 3.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopp, K.O.; Greer, M.E.; Glotfelty, E.J.; Hsueh, S.-C.; Tweedie, D.; Kim, D.S.; Reale, M.; Vargesson, N.; Greig, N.H. A New Generation of IMiDs as Treatments for Neuroinflammatory and Neurodegenerative Disorders. Biomolecules 2023, 13, 747. https://doi.org/10.3390/biom13050747
Kopp KO, Greer ME, Glotfelty EJ, Hsueh S-C, Tweedie D, Kim DS, Reale M, Vargesson N, Greig NH. A New Generation of IMiDs as Treatments for Neuroinflammatory and Neurodegenerative Disorders. Biomolecules. 2023; 13(5):747. https://doi.org/10.3390/biom13050747
Chicago/Turabian StyleKopp, Katherine O., Margaret E. Greer, Elliot J. Glotfelty, Shih-Chang Hsueh, David Tweedie, Dong Seok Kim, Marcella Reale, Neil Vargesson, and Nigel H. Greig. 2023. "A New Generation of IMiDs as Treatments for Neuroinflammatory and Neurodegenerative Disorders" Biomolecules 13, no. 5: 747. https://doi.org/10.3390/biom13050747