
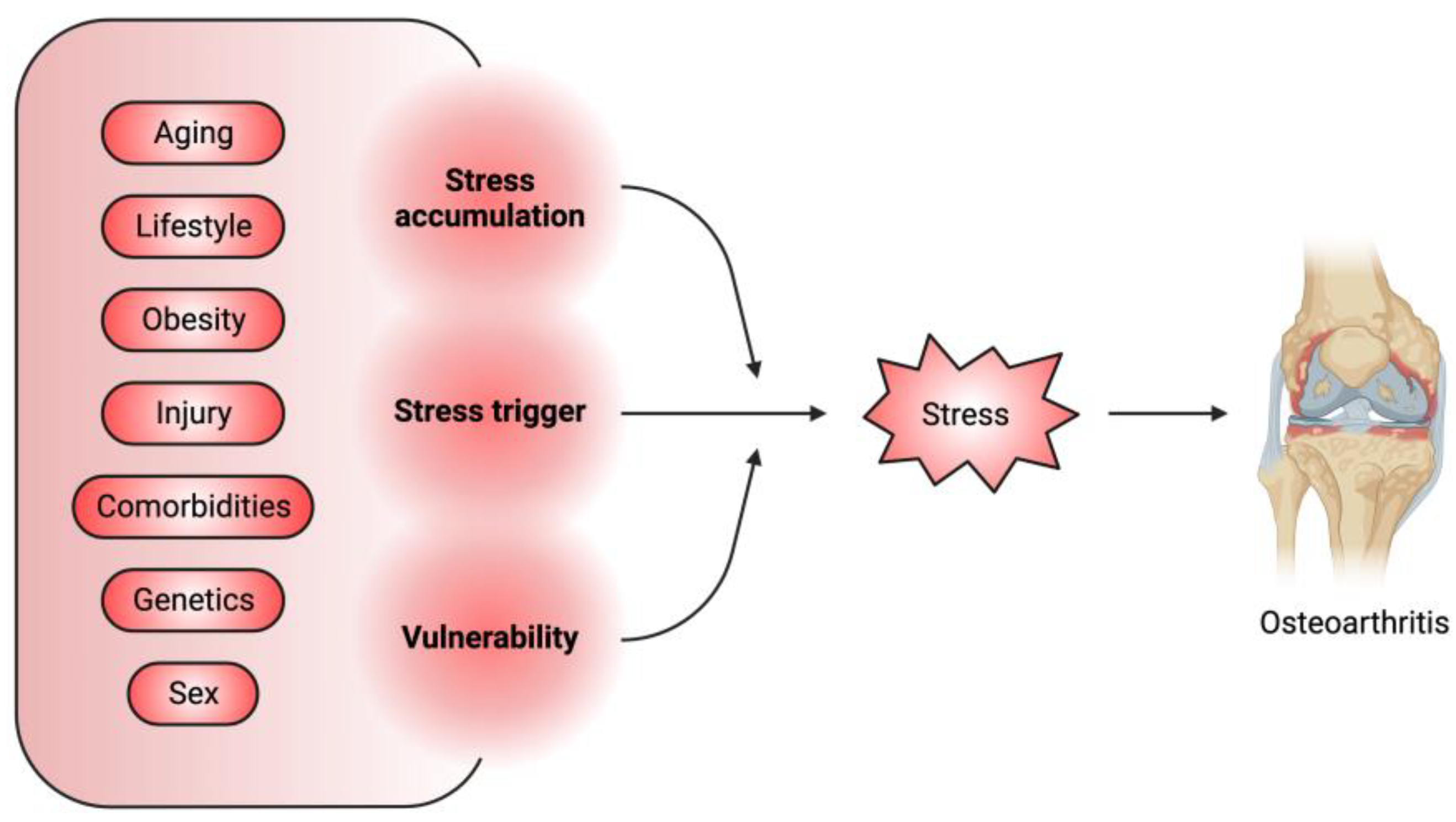
An Integrated View of Stressors as Causative Agents in OA Pathogenesis


{kind=link}
{kind=link}
Abstract
:1. Introduction
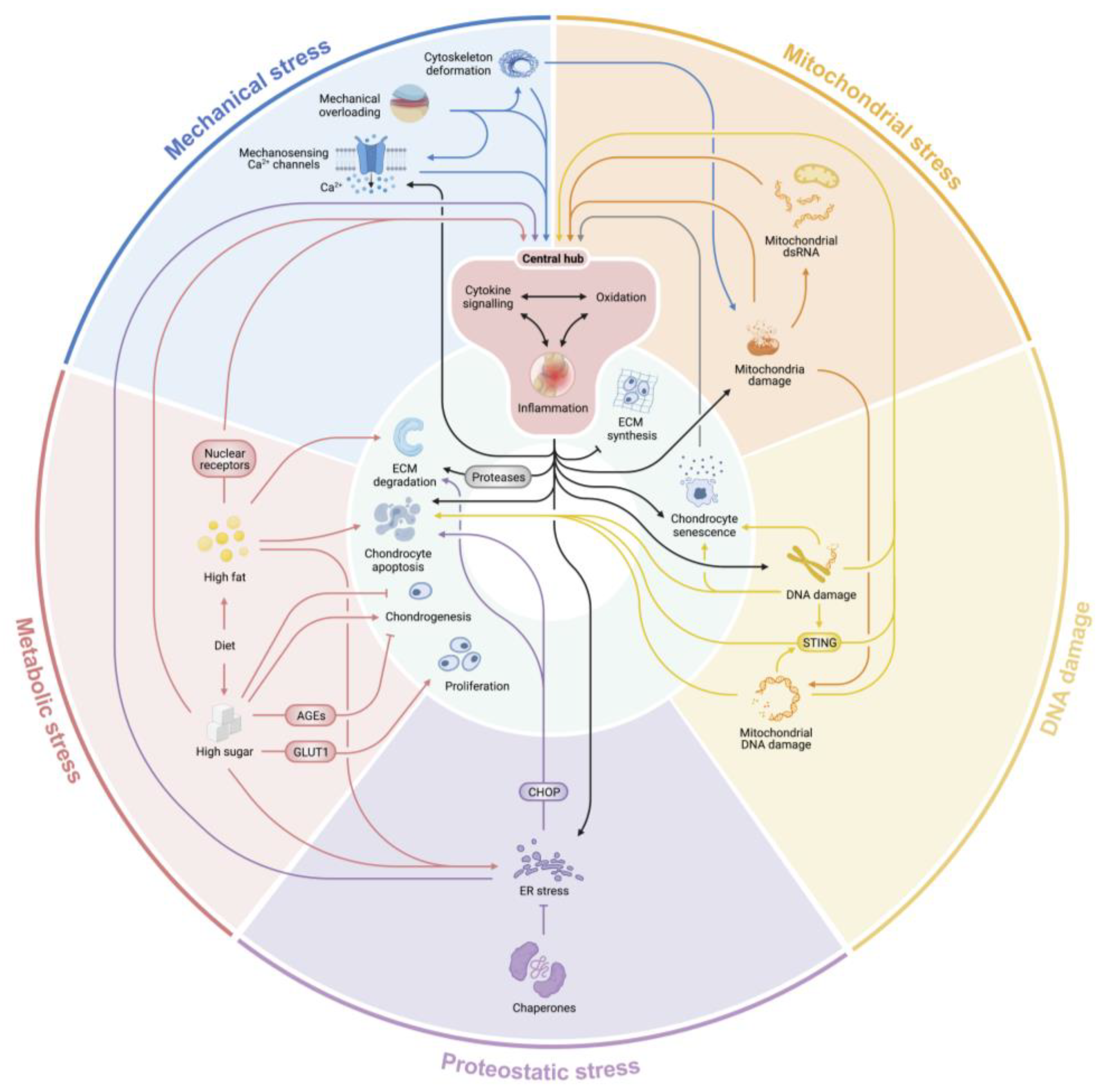
2. Primary Stressors in OA Pathogenesis
2.1. Mechanical Overloading
2.2. Oxidative Stress
2.3. DNA Damage
2.4. Proteostatic Stress
2.5. Metabolic Stress
3. An Integrated Schema of Stressors in OA and Joint Aging
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palazzo, C.; Nguyen, C.; Lefevre-Colau, M.M.; Rannou, F.; Poiraudeau, S. Risk factors and burden of osteoarthritis. Ann. Phys. Rehabil. Med. 2016, 59, 134–138. [Google Scholar] [CrossRef]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Long, H.; Liu, Q.; Yin, H.; Wang, K.; Diao, N.; Zhang, Y.; Lin, J.; Guo, A. Prevalence Trends of Site-Specific Osteoarthritis From 1990 to 2019: Findings from the Global Burden of Disease Study 2019. Arthritis Rheumatol. 2022, 74, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Grässel, S.; Muschter, D. Recent advances in the treatment of osteoarthritis. F1000Res 2020, 9, F1000 Faculty Rev-325. [Google Scholar] [CrossRef]
- Sophia Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of articular cartilage: Structure, composition, and function. Sport Health 2009, 1, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tuan, R.S. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat. Rev. Rheumatol. 2015, 11, 206–212. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, X.; Zhang, X.; Duan, X.; Yang, P.; Zhao, F.; Ao, Y. Effects of mechanical stress on chondrocyte phenotype and chondrocyte extracellular matrix expression. Sci. Rep. 2016, 6, 37268. [Google Scholar] [CrossRef]
- Brandl, A.; Hartmann, A.; Bechmann, V.; Graf, B.; Nerlich, M.; Angele, P. Oxidative stress induces senescence in chondrocytes. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2011, 29, 1114–1120. [Google Scholar] [CrossRef]
- Barbero, A.; Grogan, S.; Schafer, D.; Heberer, M.; Mainil-Varlet, P.; Martin, I. Age related changes in human articular chondrocyte yield, proliferation and post-expansion chondrogenic capacity. Osteoarthr. Cartil. 2004, 12, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Verbruggen, G.; Cornelissen, M.; Almqvist, K.F.; Wang, L.; Elewaut, D.; Broddelez, C.; de Ridder, L.; Veys, E.M. Influence of aging on the synthesis and morphology of the aggrecans synthesized by differentiated human articular chondrocytes. Osteoarthr. Cartil. 2000, 8, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Vinatier, C.; Dominguez, E.; Guicheux, J.; Carames, B. Role of the Inflammation-Autophagy-Senescence Integrative Network in Osteoarthritis. Front. Physiol. 2018, 9, 706. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Z.; Li, T.; Xu, H.; Zhang, H. Senescence in osteoarthritis: From mechanism to potential treatment. Arthritis Res. Ther. 2022, 24, 174. [Google Scholar] [CrossRef]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent cells and osteoarthritis: A painful connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Rellmann, Y.; Eidhof, E.; Dreier, R. Review: ER stress-induced cell death in osteoarthritic cartilage. Cell Signal. 2021, 78, 109880. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.Y.; Ahmad, N.; Haqqi, T.M. Oxidative stress and inflammation in osteoarthritis pathogenesis: Role of polyphenols. Biomed. Pharm. 2020, 129, 110452. [Google Scholar] [CrossRef]
- Fang, T.; Zhou, X.; Jin, M.; Nie, J.; Li, X. Molecular mechanisms of mechanical load-induced osteoarthritis. Int. Orthop. 2021, 45, 1125–1136. [Google Scholar] [CrossRef]
- Eckstein, F.; Hudelmaier, M.; Putz, R. The effects of exercise on human articular cartilage. J. Anat. 2006, 208, 491–512. [Google Scholar] [CrossRef]
- Eskelinen, A.S.A.; Florea, C.; Tanska, P.; Hung, H.K.; Frank, E.H.; Mikkonen, S.; Nieminen, P.; Julkunen, P.; Grodzinsky, A.J.; Korhonen, R.K. Cyclic loading regime considered beneficial does not protect injured and interleukin-1-inflamed cartilage from post-traumatic osteoarthritis. J. Biomech. 2022, 141, 111181. [Google Scholar] [CrossRef]
- Fu, S.; Thompson, C.L.; Ali, A.; Wang, W.; Chapple, J.P.; Mitchison, H.M.; Beales, P.L.; Wann, A.K.T.; Knight, M.M. Mechanical loading inhibits cartilage inflammatory signalling via an HDAC6 and IFT-dependent mechanism regulating primary cilia elongation. Osteoarthr. Cartil. 2019, 27, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, X.; Li, J.; Wang, X.; Liu, D.; Zhai, L.; Ding, B.; Li, G.; Sun, Y.; Yokota, H.; et al. Mechanical loading mitigates osteoarthritis symptoms by regulating the inflammatory microenvironment in a mouse model. Ann. N. Y. Acad. Sci. 2022, 1512, 141–153. [Google Scholar] [CrossRef]
- Martin, J.A.; Anderson, D.D.; Goetz, J.E.; Fredericks, D.; Pedersen, D.R.; Ayati, B.P.; Marsh, J.L.; Buckwalter, J.A. Complementary models reveal cellular responses to contact stresses that contribute to post-traumatic osteoarthritis. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2017, 35, 515–523. [Google Scholar] [CrossRef] [PubMed]
- O’Conor, C.J.; Leddy, H.A.; Benefield, H.C.; Liedtke, W.B.; Guilak, F. TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc. Natl. Acad. Sci. USA 2014, 111, 1316–1321. [Google Scholar] [CrossRef]
- O’Conor, C.J.; Ramalingam, S.; Zelenski, N.A.; Benefield, H.C.; Rigo, I.; Little, D.; Wu, C.L.; Chen, D.; Liedtke, W.; McNulty, A.L.; et al. Cartilage-Specific Knockout of the Mechanosensory Ion Channel TRPV4 Decreases Age-Related Osteoarthritis. Sci. Rep. 2016, 6, 29053. [Google Scholar] [CrossRef]
- Lee, W.; Leddy, H.A.; Chen, Y.; Lee, S.H.; Zelenski, N.A.; McNulty, A.L.; Wu, J.; Beicker, K.N.; Coles, J.; Zauscher, S.; et al. Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. Proc. Natl. Acad. Sci. USA 2014, 111, E5114–E5122. [Google Scholar] [CrossRef]
- Bubolz, A.H.; Mendoza, S.A.; Zheng, X.; Zinkevich, N.S.; Li, R.; Gutterman, D.D.; Zhang, D.X. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: Role of Ca2+ entry and mitochondrial ROS signaling. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H634–H642. [Google Scholar] [CrossRef] [PubMed]
- Knight, M.M.; Bomzon, Z.; Kimmel, E.; Sharma, A.M.; Lee, D.A.; Bader, D.L. Chondrocyte Deformation Induces Mitochondrial Distortion and Heterogeneous Intracellular Strain Fields. Biomech. Model. Mechanobiol. 2006, 5, 180. [Google Scholar] [CrossRef]
- Koike, M.; Nojiri, H.; Ozawa, Y.; Watanabe, K.; Muramatsu, Y.; Kaneko, H.; Morikawa, D.; Kobayashi, K.; Saita, Y.; Sasho, T.; et al. Mechanical overloading causes mitochondrial superoxide and SOD2 imbalance in chondrocytes resulting in cartilage degeneration. Sci. Rep. 2015, 5, 11722. [Google Scholar] [CrossRef]
- Hirose, N.; Okamoto, Y.; Yanoshita, M.; Asakawa, Y.; Sumi, C.; Takano, M.; Nishiyama, S.; Su, S.C.; Mitsuyoshi, T.; Kunimatsu, R.; et al. Protective effects of cilengitide on inflammation in chondrocytes under excessive mechanical stress. Cell Biol. Int. 2020, 44, 966–974. [Google Scholar] [CrossRef]
- Bevill, S.; Boyer, K.; Andriacchi, T. The regional sensitivity of chondrocyte gene expression to coactive mechanical load and exogenous TNF-alpha stimuli. J. Biomech. Eng. 2014, 136, 091005. [Google Scholar] [CrossRef]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Vincent, T.L. Targeting mechanotransduction pathways in osteoarthritis: A focus on the pericellular matrix. Curr. Opin. Pharmacol. 2013, 13, 449–454. [Google Scholar] [CrossRef]
- Tang, X.; Muhammad, H.; McLean, C.; Miotla-Zarebska, J.; Fleming, J.; Didangelos, A.; Önnerfjord, P.; Leask, A.; Saklatvala, J.; Vincent, T.L. Connective tissue growth factor contributes to joint homeostasis and osteoarthritis severity by controlling the matrix sequestration and activation of latent TGFβ. Ann. Rheum. Dis. 2018, 77, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef]
- Lires-Deán, M.; Caramés, B.; Cillero-Pastor, B.; Galdo, F.; López-Armada, M.J.; Blanco, F.J. Anti-apoptotic effect of transforming growth factor-beta1 on human articular chondrocytes: Role of protein phosphatase 2A. Osteoarthr. Cartil. 2008, 16, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, S.; Xie, Z.; Shen, S.; Xu, W.; Chen, W.; Li, X.; Wu, Y.; Li, L.; Liu, B.; et al. TGFβ attenuates cartilage extracellular matrix degradation via enhancing FBXO6-mediated MMP14 ubiquitination. Ann. Rheum. Dis. 2020, 79, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Madej, W.; van Caam, A.; Blaney Davidson, E.N.; van der Kraan, P.M.; Buma, P. Physiological and excessive mechanical compression of articular cartilage activates Smad2/3P signaling. Osteoarthr. Cartil. 2014, 22, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef]
- Zhen, G.; Guo, Q.; Li, Y.; Wu, C.; Zhu, S.; Wang, R.; Guo, X.E.; Kim, B.C.; Huang, J.; Hu, Y.; et al. Mechanical stress determines the configuration of TGFbeta activation in articular cartilage. Nat. Commun. 2021, 12, 1706. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.L.; Sawaji, Y.; Burleigh, A.; McLean, C.; Inglis, J.; Saklatvala, J.; Vincent, T. Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS-5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheum. 2009, 60, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.S.; Clarke, A.R.; Duance, V.C.; Blain, E.J. Effects of Wnt3A and mechanical load on cartilage chondrocyte homeostasis. Arthritis Res. Ther. 2011, 13, R203. [Google Scholar] [CrossRef] [PubMed]
- De Palma, A.; Cheleschi, S.; Pascarelli, N.A.; Giannotti, S.; Galeazzi, M.; Fioravanti, A. Hydrostatic pressure as epigenetic modulator in chondrocyte cultures: A study on miRNA-155, miRNA-181a and miRNA-223 expression levels. J. Biomech. 2018, 66, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-κB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734. [Google Scholar] [CrossRef]
- Chang, S.H.; Mori, D.; Kobayashi, H.; Mori, Y.; Nakamoto, H.; Okada, K.; Taniguchi, Y.; Sugita, S.; Yano, F.; Chung, U.I.; et al. Excessive mechanical loading promotes osteoarthritis through the gremlin-1-NF-kappaB pathway. Nat. Commun. 2019, 10, 1442. [Google Scholar] [CrossRef]
- Devasagayam, T.P.; Tilak, J.C.; Boloor, K.K.; Sane, K.S.; Ghaskadbi, S.S.; Lele, R.D. Free radicals and antioxidants in human health: Current status and future prospects. J. Assoc. Physicians India 2004, 52, 794–804. [Google Scholar] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Blanco, F.J.; Rego, I.; Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 161–169. [Google Scholar] [CrossRef]
- Dai, Y.; Liu, S.; Li, J.; Li, J.; Lan, Y.; Nie, H.; Zuo, Y. SIRT4 suppresses the inflammatory response and oxidative stress in osteoarthritis. Am. J. Transl. Res. 2020, 12, 1965–1975. [Google Scholar]
- Altindag, O.; Erel, O.; Aksoy, N.; Selek, S.; Celik, H.; Karaoglanoglu, M. Increased oxidative stress and its relation with collagen metabolism in knee osteoarthritis. Rheumatol. Int. 2007, 27, 339–344. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Wegner, A.M.; Campos, N.R.; Robbins, M.A.; Haddad, A.F.; Cunningham, H.C.; Yik, J.H.N.; Christiansen, B.A.; Haudenschild, D.R. Acute Changes in NADPH Oxidase 4 in Early Post-Traumatic Osteoarthritis. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2019, 37, 2429–2436. [Google Scholar] [CrossRef] [PubMed]
- Hiran, T.S.; Moulton, P.J.; Hancock, J.T. Detection of superoxide and NADPH oxidase in porcine articular chondrocytes. Free Radic. Biol. Med. 1997, 23, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Mendes, A.F.; Carvalho, A.P.; Caramona, M.M.; Lopes, M.C. Role of nitric oxide in the activation of NF-kappaB, AP-1 and NOS II expression in articular chondrocytes. Inflamm. Res. 2002, 51, 369–375. [Google Scholar] [CrossRef]
- van den Berg, W.B.; van de Loo, F.; Joosten, L.A.; Arntz, O.J. Animal models of arthritis in NOS2-deficient mice. Osteoarthr. Cartil. 1999, 7, 413–415. [Google Scholar] [CrossRef]
- Ahmad, N.; Ansari, M.Y.; Bano, S.; Haqqi, T.M. Imperatorin suppresses IL-1beta-induced iNOS expression via inhibiting ERK-MAPK/AP1 signaling in primary human OA chondrocytes. Int. Immunopharmacol. 2020, 85, 106612. [Google Scholar] [CrossRef]
- Fermor, B.; Weinberg, J.B.; Pisetsky, D.S.; Misukonis, M.A.; Banes, A.J.; Guilak, F. The effects of static and intermittent compression on nitric oxide production in articular cartilage explants. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2001, 19, 729–737. [Google Scholar] [CrossRef]
- Blanco, F.J.; Ochs, R.L.; Schwarz, H.; Lotz, M. Chondrocyte apoptosis induced by nitric oxide. Am. J. Pathol. 1995, 146, 75–85. [Google Scholar]
- Charlier, E.; Relic, B.; Deroyer, C.; Malaise, O.; Neuville, S.; Collée, J.; Malaise, M.G.; De Seny, D. Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 2146. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Grishko, V.I.; Ho, R.; Wilson, G.L.; Pearsall, A.W.T. Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthr. Cartil. 2009, 17, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Luo, P.; Yang, M.; Wang, J.; Hou, W.; Xu, P. The role of oxidative stress in the development of knee osteoarthritis: A comprehensive research review. Front. Mol. Biosci. 2022, 9, 1001212. [Google Scholar] [CrossRef]
- Johnson, K.; Jung, A.; Murphy, A.; Andreyev, A.; Dykens, J.; Terkeltaub, R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000, 43, 1560–1570. [Google Scholar] [CrossRef]
- Collins, J.A.; Wood, S.T.; Nelson, K.J.; Rowe, M.A.; Carlson, C.S.; Chubinskaya, S.; Poole, L.B.; Furdui, C.M.; Loeser, R.F. Oxidative Stress Promotes Peroxiredoxin Hyperoxidation and Attenuates Pro-survival Signaling in Aging Chondrocytes. J. Biol. Chem. 2016, 291, 6641–6654. [Google Scholar] [CrossRef]
- Kim, S.; Lee, K.; Choi, Y.S.; Ku, J.; Kim, H.; Kharbash, R.; Yoon, J.; Lee, Y.S.; Kim, J.H.; Lee, Y.J.; et al. Mitochondrial double-stranded RNAs govern the stress response in chondrocytes to promote osteoarthritis development. Cell Rep. 2022, 40, 111178. [Google Scholar] [CrossRef]
- Yin, W.; Park, J.I.; Loeser, R.F. Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-Kinase-Akt and MEK-ERK MAPK signaling pathways. J. Biol. Chem. 2009, 284, 31972–31981. [Google Scholar] [CrossRef]
- Yasuhara, R.; Miyamoto, Y.; Akaike, T.; Akuta, T.; Nakamura, M.; Takami, M.; Morimura, N.; Yasu, K.; Kamijo, R. Interleukin-1beta induces death in chondrocyte-like ATDC5 cells through mitochondrial dysfunction and energy depletion in a reactive nitrogen and oxygen species-dependent manner. Biochem. J. 2005, 389, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Joos, H.; Wildner, A.; Hogrefe, C.; Reichel, H.; Brenner, R.E. Interleukin-1 beta and tumor necrosis factor alpha inhibit migration activity of chondrogenic progenitor cells from non-fibrillated osteoarthritic cartilage. Arthritis Res. Ther. 2013, 15, R119. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free. Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef]
- Aivaliotis, I.L.; Pateras, I.S.; Papaioannou, M.; Glytsou, C.; Kontzoglou, K.; Johnson, E.O.; Zoumpourlis, V. How Do Cytokines Trigger Genomic Instability? J. Biomed. Biotechnol. 2012, 2012, 536761. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Copp, M.E.; Chubinskaya, S.; Bracey, D.N.; Shine, J.; Sessions, G.; Loeser, R.F.; Diekman, B.O. Comet assay for quantification of the increased DNA damage burden in primary human chondrocytes with aging and osteoarthritis. Aging Cell 2022, 21, e13698. [Google Scholar] [CrossRef]
- Chen, A.F.; Davies, C.M.; De Lin, M.; Fermor, B. Oxidative DNA damage in osteoarthritic porcine articular cartilage. J. Cell. Physiol. 2008, 217, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Copp, M.E.; Flanders, M.C.; Gagliardi, R.; Gilbertie, J.M.; Sessions, G.A.; Chubinskaya, S.; Loeser, R.F.; Schnabel, L.V.; Diekman, B.O. The combination of mitogenic stimulation and DNA damage induces chondrocyte senescence. Osteoarthr. Cartil. 2021, 29, 402–412. [Google Scholar] [CrossRef]
- Kim, J.; Xu, M.; Xo, R.; Mates, A.; Wilson, G.L.; Pearsall, A.W.t.; Grishko, V. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthr. Cartil. 2010, 18, 424–432. [Google Scholar] [CrossRef]
- Davies, C.M.; Guilak, F.; Weinberg, J.B.; Fermor, B. Reactive nitrogen and oxygen species in interleukin-1-mediated DNA damage associated with osteoarthritis. Osteoarthr. Cartil. 2008, 16, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.; Kang, D.; Jung, J.; Yoo, T.J.; Shim, M.S.; Gladyshev, V.N.; Tsuji, P.A.; Hatfield, D.L.; Kim, J.H.; Lee, B.J. SEPHS1: Its evolution, function and roles in development and diseases. Arch. Biochem. Biophys. 2022, 730, 109426. [Google Scholar] [CrossRef]
- Kang, D.; Lee, J.; Jung, J.; Carlson, B.A.; Chang, M.J.; Chang, C.B.; Kang, S.B.; Lee, B.C.; Gladyshev, V.N.; Hatfield, D.L.; et al. Selenophosphate synthetase 1 deficiency exacerbates osteoarthritis by dysregulating redox homeostasis. Nat. Commun. 2022, 13, 779. [Google Scholar] [CrossRef]
- Takayama, K.; Kawakami, Y.; Lee, S.; Greco, N.; Lavasani, M.; Mifune, Y.; Cummins, J.H.; Yurube, T.; Kuroda, R.; Kurosaka, M.; et al. Involvement of ERCC1 in the pathogenesis of osteoarthritis through the modulation of apoptosis and cellular senescence. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2014, 32, 1326–1332. [Google Scholar] [CrossRef]
- Guo, Q.; Chen, X.; Chen, J.; Zheng, G.; Xie, C.; Wu, H.; Miao, Z.; Lin, Y.; Wang, X.; Gao, W.; et al. STING promotes senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the NF-κB signaling pathway. Cell Death Dis. 2021, 12, 13. [Google Scholar] [CrossRef]
- Zhang, X.; Xiang, S.; Zhang, Y.; Liu, S.; Lei, G.; Hines, S.; Wang, N.; Lin, H. In vitro study to identify ligand-independent function of estrogen receptor-alpha in suppressing DNA damage-induced chondrocyte senescence. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2023, 37, e22746. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, X.; Rothrauff, B.B.; Fritch, M.R.; Chang, A.; He, Y.; Yeung, M.; Liu, S.; Lipa, K.E.; Lei, G.; et al. Novel role of estrogen receptor-alpha on regulating chondrocyte phenotype and response to mechanical loading. Osteoarthr. Cartil. 2022, 30, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Kung, L.H.; Rajpar, M.H.; Briggs, M.D.; Boot-Handford, R.P. Hypertrophic chondrocytes have a limited capacity to cope with increases in endoplasmic reticulum stress without triggering the unfolded protein response. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2012, 60, 734–748. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Piróg, K.A.; Jaka, O.; Katakura, Y.; Meadows, R.S.; Kadler, K.E.; Boot-Handford, R.P.; Briggs, M.D. A mouse model offers novel insights into the myopathy and tendinopathy often associated with pseudoachondroplasia and multiple epiphyseal dysplasia. Hum. Mol. Genet. 2010, 19, 52–64. [Google Scholar] [CrossRef]
- Nugent, A.E.; Speicher, D.M.; Gradisar, I.; McBurney, D.L.; Baraga, A.; Doane, K.J.; Horton, W.E., Jr. Advanced osteoarthritis in humans is associated with altered collagen VI expression and upregulation of ER-stress markers Grp78 and bag-1. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2009, 57, 923–931. [Google Scholar] [CrossRef]
- Yang, L.; McBurney, D.; Tang, S.C.; Carlson, S.G.; Horton, W.E., Jr. A novel role for Bcl-2 associated-athanogene-1 (Bag-1) in regulation of the endoplasmic reticulum stress response in mammalian chondrocytes. J. Cell. Biochem. 2007, 102, 786–800. [Google Scholar] [CrossRef]
- Hamamura, K.; Goldring, M.B.; Yokota, H. Involvement of p38 MAPK in regulation of MMP13 mRNA in chondrocytes in response to surviving stress to endoplasmic reticulum. Arch. Oral Biol. 2009, 54, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Hirose, J.; Senba, K.; Yamabe, S.; Oike, Y.; Gotoh, T.; Mizuta, H. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int. J. Exp. Pathol. 2011, 92, 232–242. [Google Scholar] [CrossRef]
- Tan, L.; Register, T.C.; Yammani, R.R. Age-Related Decline in Expression of Molecular Chaperones Induces Endoplasmic Reticulum Stress and Chondrocyte Apoptosis in Articular Cartilage. Aging Dis. 2020, 11, 1091–1102. [Google Scholar] [CrossRef]
- Feng, Y.; Li, B.; Li, S.J.; Yang, X.C.; Lv, T.T.; Shang, H.; Wu, Z.B.; Zhang, Y. Skp2/p27 axis regulates chondrocyte proliferation under high glucose induced endoplasmic reticulum stress. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9129–9138. [Google Scholar] [CrossRef]
- Tan, L.; Harper, L.; McNulty, M.A.; Carlson, C.S.; Yammani, R.R. High-fat diet induces endoplasmic reticulum stress to promote chondrocyte apoptosis in mouse knee joints. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 5818–5826. [Google Scholar] [CrossRef] [PubMed]
- Eymard, F.; Parsons, C.; Edwards, M.H.; Petit-Dop, F.; Reginster, J.Y.; Bruyère, O.; Richette, P.; Cooper, C.; Chevalier, X. Diabetes is a risk factor for knee osteoarthritis progression. Osteoarthr. Cartil. 2015, 23, 851–859. [Google Scholar] [CrossRef]
- Xie, Y.; Zhou, W.; Zhong, Z.; Zhao, Z.; Yu, H.; Huang, Y.; Zhang, P. Metabolic syndrome, hypertension, and hyperglycemia were positively associated with knee osteoarthritis, while dyslipidemia showed no association with knee osteoarthritis. Clin. Rheumatol. 2021, 40, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Courties, A.; Gualillo, O.; Berenbaum, F.; Sellam, J. Metabolic stress-induced joint inflammation and osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1955–1965. [Google Scholar] [CrossRef] [PubMed]
- Gierman, L.M.; van der Ham, F.; Koudijs, A.; Wielinga, P.Y.; Kleemann, R.; Kooistra, T.; Stoop, R.; Kloppenburg, M.; van Osch, G.J.; Stojanovic-Susulic, V.; et al. Metabolic stress-induced inflammation plays a major role in the development of osteoarthritis in mice. Arthritis Rheum. 2012, 64, 1172–1181. [Google Scholar] [CrossRef]
- Ratneswaran, A.; Sun, M.M.; Dupuis, H.; Sawyez, C.; Borradaile, N.; Beier, F. Nuclear receptors regulate lipid metabolism and oxidative stress markers in chondrocytes. J. Mol. Med. 2017, 95, 431–444. [Google Scholar] [CrossRef]
- Alvarez-Garcia, O.; Rogers, N.H.; Smith, R.G.; Lotz, M.K. Palmitate has proapoptotic and proinflammatory effects on articular cartilage and synergizes with interleukin-1. Arthritis Rheumatol. 2014, 66, 1779–1788. [Google Scholar] [CrossRef]
- Lee, S.W.; Rho, J.H.; Lee, S.Y.; Chung, W.T.; Oh, Y.J.; Kim, J.H.; Yoo, S.H.; Kwon, W.Y.; Bae, J.Y.; Seo, S.Y.; et al. Dietary fat-associated osteoarthritic chondrocytes gain resistance to lipotoxicity through PKCK2/STAMP2/FSP27. Bone Res. 2018, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- van Gastel, N.; Stegen, S.; Eelen, G.; Schoors, S.; Carlier, A.; Daniëls, V.W.; Baryawno, N.; Przybylski, D.; Depypere, M.; Stiers, P.J.; et al. Lipid availability determines fate of skeletal progenitor cells via SOX9. Nature 2020, 579, 111–117. [Google Scholar] [CrossRef]
- Donovan, E.L.; Lopes, E.B.P.; Batushansky, A.; Kinter, M.; Griffin, T.M. Independent effects of dietary fat and sucrose content on chondrocyte metabolism and osteoarthritis pathology in mice. Dis. Model. Mech. 2018, 11, dmm034827. [Google Scholar] [CrossRef]
- Lee, S.Y.; Abel, E.D.; Long, F. Glucose metabolism induced by Bmp signaling is essential for murine skeletal development. Nat. Commun. 2018, 9, 4831. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, L.; Chen, D. Growth factor signalling in osteoarthritis. Growth Factors 2018, 36, 187–195. [Google Scholar] [CrossRef]
- Suzuki, A.; Yabu, A.; Nakamura, H. Advanced glycation end products in musculoskeletal system and disorders. Methods 2020, 203, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chan, D.C.; Chiang, C.K.; Wang, C.C.; Yang, T.H.; Lan, K.C.; Chao, S.C.; Tsai, K.S.; Yang, R.S.; Liu, S.H. Advanced glycation end-products induced VEGF production and inflammatory responses in human synoviocytes via RAGE-NF-κB pathway activation. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2016, 34, 791–800. [Google Scholar] [CrossRef]
- Yamabe, S.; Hirose, J.; Uehara, Y.; Okada, T.; Okamoto, N.; Oka, K.; Taniwaki, T.; Mizuta, H. Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. FEBS J. 2013, 280, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960. [Google Scholar] [CrossRef]
- Lee, W.; Nims, R.J.; Savadipour, A.; Zhang, Q.; Leddy, H.A.; Liu, F.; McNulty, A.L.; Chen, Y.; Guilak, F.; Liedtke, W.B. Inflammatory signaling sensitizes Piezo1 mechanotransduction in articular chondrocytes as a pathogenic feed-forward mechanism in osteoarthritis. Proc. Natl. Acad. Sci. USA 2021, 118, e2001611118. [Google Scholar] [CrossRef]
- Feng, K.; Chen, Z.; Pengcheng, L.; Zhang, S.; Wang, X. Quercetin attenuates oxidative stress-induced apoptosis via SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and prevents the progression of osteoarthritis in a rat model. J. Cell. Physiol. 2019, 234, 18192–18205. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Ge, Y.; Chen, Z.; Li, X.; Liu, Z.; Li, X.; Li, H.; Tang, T.; Yang, F.; Wang, X. Curcumin Inhibits the PERK-eIF2α-CHOP Pathway through Promoting SIRT1 Expression in Oxidative Stress-induced Rat Chondrocytes and Ameliorates Osteoarthritis Progression in a Rat Model. Oxidative Med. Cell. Longev. 2019, 2019, 8574386. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.R.; Deshpande, B.R.; Collins, J.E.; Katz, J.N.; Losina, E. Comparative pain reduction of oral non-steroidal anti-inflammatory drugs and opioids for knee osteoarthritis: Systematic analytic review. Osteoarthr. Cartil. 2016, 24, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Nixon, A.J.; Grol, M.W.; Lang, H.M.; Ruan, M.Z.C.; Stone, A.; Begum, L.; Chen, Y.; Dawson, B.; Gannon, F.; Plutizki, S.; et al. Disease-Modifying Osteoarthritis Treatment with Interleukin-1 Receptor Antagonist Gene Therapy in Small and Large Animal Models. Arthritis Rheumatol. 2018, 70, 1757–1768. [Google Scholar] [CrossRef]
- Stone, A.; Grol, M.W.; Ruan, M.Z.C.; Dawson, B.; Chen, Y.; Jiang, M.M.; Song, I.W.; Jayaram, P.; Cela, R.; Gannon, F.; et al. Combinatorial Prg4 and Il-1ra Gene Therapy Protects Against Hyperalgesia and Cartilage Degeneration in Post-Traumatic Osteoarthritis. Hum. Gene 2019, 30, 225–235. [Google Scholar] [CrossRef]
- Watson Levings, R.S.; Smith, A.D.; Broome, T.A.; Rice, B.L.; Gibbs, E.P.; Myara, D.A.; Hyddmark, E.V.; Nasri, E.; Zarezadeh, A.; Levings, P.P.; et al. Self-Complementary Adeno-Associated Virus-Mediated Interleukin-1 Receptor Antagonist Gene Delivery for the Treatment of Osteoarthritis: Test of Efficacy in an Equine Model. Hum. Gene Clin. Dev. 2018, 29, 101–112. [Google Scholar] [CrossRef]
- Colletti, A.; Cicero, A.F.G. Nutraceutical Approach to Chronic Osteoarthritis: From Molecular Research to Clinical Evidence. Int. J. Mol. Sci. 2021, 22, 2920. [Google Scholar] [CrossRef]
- Mariano, A.; Bigioni, I.; Misiti, F.; Fattorini, L.; Scotto D’Abusco, A.; Rodio, A. The Nutraceuticals as Modern Key to Achieve Erythrocyte Oxidative Stress Fighting in Osteoarthritis. Curr. Issues Mol. Biol. 2022, 44, 3481–3495. [Google Scholar] [CrossRef] [PubMed]
- Yamada, E.F.; Olin, L.C.; Pontel, C.L.; da Rosa, H.S.; Folmer, V.; da Silva, M.D. Sida tuberculata reduces oxidative stress and pain caused by the knee osteoarthritis. J. Ethnopharmacol. 2020, 248, 112277. [Google Scholar] [CrossRef]
- Kumar, S.; Adjei, I.M.; Brown, S.B.; Liseth, O.; Sharma, B. Manganese dioxide nanoparticles protect cartilage from inflammation-induced oxidative stress. Biomaterials 2019, 224, 119467. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Floramo, J.S.; Molchanov, V.; Liu, H.; Liu, Y.; Craig, S.E.L.; Yang, T. An Integrated View of Stressors as Causative Agents in OA Pathogenesis. Biomolecules 2023, 13, 721. https://doi.org/10.3390/biom13050721
Floramo JS, Molchanov V, Liu H, Liu Y, Craig SEL, Yang T. An Integrated View of Stressors as Causative Agents in OA Pathogenesis. Biomolecules. 2023; 13(5):721. https://doi.org/10.3390/biom13050721
Chicago/Turabian StyleFloramo, Joseph S., Vladimir Molchanov, Huadie Liu, Ye Liu, Sonya E. L. Craig, and Tao Yang. 2023. "An Integrated View of Stressors as Causative Agents in OA Pathogenesis" Biomolecules 13, no. 5: 721. https://doi.org/10.3390/biom13050721