
Myocardial Inflammation as a Manifestation of Genetic Cardiomyopathies: From Bedside to the Bench

 , ,
, ,  , , ,
, , ,
Abstract
:1. Myocarditis and Primary Cardiomyopathies
1.1. Introduction
1.2. Diagnosis of Myocarditis
1.3. Classification of Cardiomyopathies
2. Myocarditis as a Manifestation of Primary Cardiomyopathy: Clinical Scenarios
2.1. Familial Myocarditis
2.2. Autoptic Findings from Sudden Death Victims
2.3. Overlapping Phenotypes in Sporadic Disease
3. Modeling Myocardial Inflammation in Cardiomyopathies: Evidence from the Preclinical Setting
3.1. Molecular Mechanisms of Primary Cardiomyopathies
3.2. Role of Systemic and Local Inflammation
3.3. Inflammation Models in Genetic Etiologies Linked to DCM
3.4. Inflammation Models in Genetic Etiologies Linked to ACM
4. Clinical Implications and Conclusive Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Richardson, P.; McKenna, R.W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and Inflammatory Cardiomyopathy: Current Evidence and Future Directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Sala, S.; della Bella, P.; Basso, C.; Cooper, L.T. Reply: Genetic Basis for Acute Myocarditis Presenting with Ventricular Arrhythmias? J. Am. Coll. Cardiol. 2020, 76, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Sala, S.; Rizzo, S.; de Luca, G.; Campochiaro, C.; Sartorelli, S.; Benedetti, G.; Palmisano, A.; Esposito, A.; Tresoldi, M.; et al. Arrhythmias in Myocarditis: State of the Art. Heart Rhythm. 2019, 16, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Casella, M.; Merlo, M.; Benedetti, S.; Rizzo, S.; Cappelletto, C.; Di Resta, C.; Compagnucci, P.; De Gaspari, M.; Dello Russo, A.; et al. Inflammation on Endomyocardial Biopsy Predicts Risk of Subsequent Adverse Cardiovascular Events in Undefined Left Ventricular Arrhythmogenic Cardiomyopathy. JACC Clin. Electrophysiol. 2022, S2405-500X(22)00949-5. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current State of Knowledge on Aetiology, Diagnosis, Management, and Therapy of Myocarditis: A Position Statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy: An Expert Consensus Document. Circ. Heart Fail. 2020, 13, e007405. [Google Scholar] [CrossRef]
- Aretz, H.T.; Billingham, M.E.; Edwards, W.D.; Factor, S.M.; Fallon, J.T.; Fenoglio, J.J., Jr.; Olsen, E.G.; Schoen, F.J. Myocarditis. A Histopathologic Definition and Classification. Am. J. Cardiovasc. Pathol. 1987, 1, 3–14. [Google Scholar]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the Cardiomyopathies: A Position Statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary Definitions and Classification of the Cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [Green Version]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a Revised Definition of Dilated Cardiomyopathy, Hypokinetic Non-Dilated Cardiomyopathy, and Its Implications for Clinical Practice: A Position Statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS Expert Consensus Statement on Evaluation, Risk Stratification, and Management of Arrhythmogenic Cardiomyopathy. Heart Rhythm. 2019, 16, e301–e372. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; de Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of Arrhythmogenic Cardiomyopathy: The Padua Criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef]
- Peretto, G.; Cappelletti, A.M.; Spoladore, R.; Slavich, M.; Rizzo, S.; Palmisano, A.; Esposito, A.; de Cobelli, F.; Margonato, A.; Basso, C.; et al. Right Ventricular Endomyocardial Biopsy in Patients with Cardiac Magnetic Resonance Showing Left Ventricular Myocarditis. J. Cardiovasc. Med. 2021, 22, 560–566. [Google Scholar] [CrossRef]
- Ferreira, V.M.; Schulz-Menger, J.; Holmvang, G.; Kramer, C.M.; Carbone, I.; Sechtem, U.; Kindermann, I.; Gutberlet, M.; Cooper, L.T.; Liu, P.; et al. Cardiovascular Magnetic Resonance in Nonischemic Myocardial Inflammation: Expert Recommendations. J. Am. Coll. Cardiol. 2018, 72, 3158–3176. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Busnardo, E.; Ferro, P.; Palmisano, A.; Vignale, D.; Esposito, A.; de Luca, G.; Campochiaro, C.; Sartorelli, S.; de Gaspari, M.; et al. Clinical Applications of FDG-PET Scan in Arrhythmic Myocarditis. JACC Cardiovasc. Imaging 2022, 15, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ayala, J.M.; Pastor-Quirante, F.; Gonzalez-Carrillo, J.; Lopez-Cuenca, D.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Gimeno, J.R. Genetics of Myocarditis in Arrhythmogenic Right Ventricular Dysplasia. Heart Rhythm. 2015, 12, 766–773. [Google Scholar] [CrossRef]
- Piriou, N.; Marteau, L.; Kyndt, F.; Serfaty, J.M.; Toquet, C.; le Gloan, L.; Warin-Fresse, K.; Guijarro, D.; le Tourneau, T.; Conan, E.; et al. Familial Screening in Case of Acute Myocarditis Reveals Inherited Arrhythmogenic Left Ventricular Cardiomyopathies. ESC Heart Fail. 2020, 7, 1520–1533. [Google Scholar] [CrossRef]
- Bariani, R.; Cipriani, A.; Rizzo, S.; Celeghin, R.; Bueno Marinas, M.; Giorgi, B.; de Gaspari, M.; Rigato, I.; Leoni, L.; Zorzi, A.; et al. “Hot Phase” Clinical Presentation in Arrhythmogenic Cardiomyopathy. Europace 2021, 23, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Raimondi, F.; Piriou, N.; Sardo Infirri, L.; Mohiddin, S.A.; Mazzanti, A.; Shenoy, C.; Cavallari, U.A.; Imazio, M.; Aquaro, G.D.; et al. Acute Myocarditis Associated With Desmosomal Gene Variants. JACC Heart Fail. 2022, 10, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Hata, Y.; Hirono, K.; Yamaguchi, Y.; Ichida, F.; Oku, Y.; Nishida, N. Minimal Inflammatory Foci of Unknown Etiology May Be a Tentative Sign of Early Stage Inherited Cardiomyopathy. Mod. Pathol. 2019, 32, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Mahon, N.G.; Madden, B.P.; Caforio, A.L.P.; Elliott, P.M.; Haven, A.J.; Keogh, B.E.; Davies, M.J.; McKenna, W.J. Immunohistologic Evidence of Myocardial Disease in Apparently Healthy Relatives of Patients with Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2002, 39, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poller, W.; Escher, F.; Haas, J.; Heidecker, B.; Schultheiss, H.P.; Attanasio, P.; Skurk, C.; Haghikia, A.; Meder, B.; Klaassen, S. Missense Variant E1295K of Sodium Channel SCN5A Associated With Recurrent Ventricular Fibrillation and Myocardial Inflammation. JACC Case Rep. 2022, 4, 280–286. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated Cardiomyopathy: The Complexity of a Diverse Genetic Architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Casella, M.; Gasperetti, A.; Gaetano, F.; Busana, M.; Sommariva, E.; Catto, V.; Sicuso, R.; Rizzo, S.; Conte, E.; Mushtaq, S.; et al. Long-Term Follow-up Analysis of a Highly Characterized Arrhythmogenic Cardiomyopathy Cohort with Classical and Non-Classical Phenotypes-a Real-World Assessment of a Novel Prediction Model: Does the Subtype Really Matter. Europace 2020, 22, 797–805. [Google Scholar] [CrossRef]
- Peretto, G.; Mazzone, P. Arrhythmogenic Cardiomyopathy: One, None and a Hundred Thousand Diseases. J. Pers. Med. 2022, 12, 1256. [Google Scholar] [CrossRef]
- Charron, P.; Arad, M.; Arbustini, E.; Basso, C.; Bilinska, Z.; Elliott, P.; Helio, T.; Keren, A.; McKenna, W.J.; Monserrat, L.; et al. Genetic Counselling and Testing in Cardiomyopathies: A Position Statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2010, 31, 2715–2728. [Google Scholar] [CrossRef] [Green Version]
- Kontorovich, A.R.; Patel, N.; Moscati, A.; Richter, F.; Peter, I.; Purevjav, E.; Selejan, S.R.; Kindermann, I.; Towbin, J.A.; Bohm, M.; et al. Myopathic Cardiac Genotypes Increase Risk for Myocarditis. JACC Basic Transl. Sci. 2021, 6, 584–592. [Google Scholar] [CrossRef]
- Artico, J.; Merlo, M.; Delcaro, G.; Cannatà, A.; Gentile, P.; de Angelis, G.; Paldino, A.; Bussani, R.; Ferro, M.D.; Sinagra, G. Lymphocytic Myocarditis: A Genetically Predisposed Disease? J. Am. Coll. Cardiol. 2020, 75, 3098–3100. [Google Scholar] [CrossRef]
- Cannata, A.; Artico, J.; Gentile, P.; Merlo, M.; Sinagra, G. Myocarditis Evolving in Cardiomyopathy: When Genetics and Offending Causes Work Together. Eur. Heart J. Suppl. 2019, 21, B90–B95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowlton, K.U. Myocarditis: An Intersection Between Genetic and Acquired Causes of Human Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 69, 1666–1668. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Fernández-Falgueras, A.; Sarquella-Brugada, G.; Sanchez, O.; Cesar, S.; Mademont, I.; Allegue, C.; Mates, J.; Pérez-Serra, A.; Coll, M.; et al. A Genetically Vulnerable Myocardium May Predispose to Myocarditis. J. Am. Coll. Cardiol. 2015, 66, 2913–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, D.; Lee, G.H.; Badorff, C.; Dorner, A.; Lee, S.; Wolf, P.; Knowlton, K.U. Dystrophin Deficiency Markedly Increases Enterovirus-Induced Cardiomyopathy: A Genetic Predisposition to Viral Heart Disease. Nat. Med. 2002, 8, 872–877. [Google Scholar] [CrossRef]
- Tiron, C.; Campuzano, O.; Fernández-Falgueras, A.; Alcalde, M.; Loma-Osorio, P.; Zamora, E.; Caballero, A.; Sarquella-Brugada, G.; Cesar, S.; Garcia-Cuenllas, L.; et al. Prevalence of Pathogenic Variants in Cardiomyopathy-Associated Genes in Myocarditis. Circ. Genom. Precis. Med. 2022, 15, E003408. [Google Scholar] [CrossRef]
- Seidel, F.; Laser, K.T.; Klingel, K.; Dartsch, J.; Theisen, S.; Pickardt, T.; Holtgrewe, M.; Gärtner, A.; Berger, F.; Beule, D.; et al. Pathogenic Variants in Cardiomyopathy Disorder Genes Underlie Pediatric Myocarditis-Further Impact of Heterozygous Immune Disorder Gene Variants? J. Cardiovasc. Dev. Dis. 2022, 9, 216. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.Y.; et al. Evidence From Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy: Associations of Circulating Anti-Heart and Anti-Intercalated Disk Autoantibodies with Disease Severity and Family History. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef]
- Peretto, G.; Sala, S.; de Luca, G.; Marcolongo, R.; Campochiaro, C.; Sartorelli, S.; Tresoldi, M.; Foppoli, L.; Palmisano, A.; Esposito, A.; et al. Immunosuppressive Therapy and Risk Stratification of Patients With Myocarditis Presenting With Ventricular Arrhythmias. JACC Clin. Electrophysiol. 2020, 6, 1221–1234. [Google Scholar] [CrossRef]
- Poller, W.; Haas, J.; Klingel, K.; Kühnisch, J.; Gast, M.; Kaya, Z.; Escher, F.; Kayvanpour, E.; Degener, F.; Opgen-Rhein, B.; et al. Familial Recurrent Myocarditis Triggered by Exercise in Patients With a Truncating Variant of the Desmoplakin Gene. J. Am. Heart Assoc. 2020, 9, e015289. [Google Scholar] [CrossRef]
- Chatterjee, D.; Fatah, M.; Akdis, D.; Spears, D.A.; Koopmann, T.T.; Mittal, K.; Rafiq, M.A.; Cattanach, B.M.; Zhao, Q.; Healey, J.S.; et al. An Autoantibody Identifies Arrhythmogenic Right Ventricular Cardiomyopathy and Participates in Its Pathogenesis. Eur. Heart J. 2018, 39, 3932–3944. [Google Scholar] [CrossRef] [Green Version]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 2018, 72, e91–e220. [Google Scholar] [CrossRef]
- Rizzo, S.; Carturan, E.; de Gaspari, M.; Pilichou, K.; Thiene, G.; Basso, C. Update on Cardiomyopathies and Sudden Cardiac Death. Forensic Sci. Res. 2019, 4, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Leone, O.; Veinot, J.P.; Angelini, A.; Baandrup, U.T.; Basso, C.; Berry, G.; Bruneval, P.; Burke, M.; Butany, J.; Calabrese, F.; et al. 2011 Consensus Statement on Endomyocardial Biopsy from the Association for European Cardiovascular Pathology and the Society for Cardiovascular Pathology. Cardiovasc. Pathol. 2012, 21, 245–274. [Google Scholar] [CrossRef] [PubMed]
- de Luca, G.; Campochiaro, C.; de Santis, M.; Sartorelli, S.; Peretto, G.; Sala, S.; Canestrari, G.; de Lorenzis, E.; Basso, C.; Rizzo, S.; et al. Systemic Sclerosis Myocarditis Has Unique Clinical, Histological and Prognostic Features: A Comparative Histological Analysis. Rheumatology 2020, 59, 2523–2533. [Google Scholar] [CrossRef]
- Lobo, V.F.; Silver, M.D.; Butany, J.; Heggtveit, H.A. Left Ventricular Involvement in Right Ventricular Dysplasia/Cardiomyopathy. Can. J. Cardiol. 1999, 15, 1239–1247. [Google Scholar] [PubMed]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic Right Ventricular Cardiomyopathy. Dysplasia, Dystrophy, or Myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef]
- di Resta, C.; Berg, J.; Villatore, A.; Maia, M.; Pili, G.; Fioravanti, F.; Tomaiuolo, R.; Sala, S.; Benedetti, S.; Peretto, G. Concealed Substrates in Brugada Syndrome: Isolated Channelopathy or Associated Cardiomyopathy? Genes 2022, 13, 1755. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, F.; Zanatta, A.; Basso, C.; Thiene, G. Cardiovascular Medicine in Morgagni’s De Sedibus: Dawn of Cardiovascular Pathology. Cardiovasc. Pathol. 2016, 25, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef]
- Cipriani, A.; Bauce, B.; de Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis With Dilated Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e014628. [Google Scholar] [CrossRef] [PubMed]
- Augusto, J.B.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; Treibel, T.A.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated Cardiomyopathy and Arrhythmogenic Left Ventricular Cardiomyopathy: A Comprehensive Genotype-Imaging Phenotype Study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef]
- Peretto, G.; di Resta, C.; Perversi, J.; Forleo, C.; Maggi, L.; Politano, L.; Barison, A.; Previtali, S.C.; Carboni, N.; Brun, F.; et al. Cardiac and Neuromuscular Features of Patients With LMNA-Related Cardiomyopathy. Ann. Intern. Med. 2019, 171, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Sala, S.; Lazzeroni, D.; Palmisano, A.; Gigli, L.; Esposito, A.; de Cobelli, F.; Camici, P.G.; Mazzone, P.; Basso, C.; et al. Septal Late Gadolinium Enhancement and Arrhythmic Risk in Genetic and Acquired Non-Ischaemic Cardiomyopathies. Heart Lung Circ. 2020, 29, 1356–1365. [Google Scholar] [CrossRef]
- Protonotarios, A.; Wicks, E.; Ashworth, M.; Stephenson, E.; Guttmann, O.; Savvatis, K.; Sekhri, N.; Mohiddin, S.A.; Syrris, P.; Menezes, L.; et al. Prevalence of 18F-Fluorodeoxyglucose Positron Emission Tomography Abnormalities in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy. Int. J. Cardiol. 2019, 284, 99–104. [Google Scholar] [CrossRef]
- Peretto, G.; Sala, S.; Rizzo, S.; Palmisano, A.; Esposito, A.; de Cobelli, F.; Campochiaro, C.; de Luca, G.; Foppoli, L.; Dagna, L.; et al. Ventricular Arrhythmias in Myocarditis: Characterization and Relationships With Myocardial Inflammation. J. Am. Coll. Cardiol. 2020, 75, 1046–1057. [Google Scholar] [CrossRef]
- Cronin, E.M.; Bogun, F.M.; Maury, P.; Peichl, P.; Chen, M.; Namboodiri, N.; Aguinaga, L.; Leite, L.R.; Al-Khatib, S.M.; Anter, E.; et al. 2019 HRS/EHRA/APHRS/LAHRS Expert Consensus Statement on Catheter Ablation of Ventricular Arrhythmias. Europace 2019, 21, 1143–1144. [Google Scholar] [CrossRef]
- Peretto, G.; Sala, S.; Basso, C.; Rizzo, S.; Radinovic, A.; Frontera, A.; Limite, L.R.; Paglino, G.; Bisceglia, C.; de Luca, G.; et al. Inflammation as a Predictor of Recurrent Ventricular Tachycardia After Ablation in Patients With Myocarditis. J. Am. Coll. Cardiol. 2020, 76, 1644–1656. [Google Scholar] [CrossRef] [PubMed]
- Bowles, N.E.; Bowles, K.R.; Towbin, J.A. The “Final Common Pathway” Hypothesis and Inherited Cardiovascular Disease. The Role of Cytoskeletal Proteins in Dilated Cardiomyopathy. Herz 2000, 25, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Lorts, A. Arrhythmias and Dilated Cardiomyopathy Common Pathogenetic Pathways? J. Am. Coll. Cardiol. 2011, 57, 2169–2171. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, V.; Nijenkamp, L.L.A.M.; Regan, J.A.; van der Velden, J. The Physiological Role of Cardiac Cytoskeleton and Its Alterations in Heart Failure. Biochim. Biophys. Acta 2014, 1838, 700–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.Á.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef]
- Towbin, J.A.; Hejtmancik, J.F.; Brink, P.; Gelb, B.; Zhu, X.M.; Chamberlain, J.S.; McCabe, E.R.B.; Swift, M. X-Linked Dilated Cardiomyopathy. Molecular Genetic Evidence of Linkage to the Duchenne Muscular Dystrophy (Dystrophin) Gene at the Xp21 Locus. Circulation 1993, 87, 1854–1865. [Google Scholar] [CrossRef] [Green Version]
- Vatta, M.; Mohapatra, B.; Jimenez, S.; Sanchez, X.; Faulkner, G.; Perles, Z.; Sinagra, G.; Lin, J.H.; Vu, T.M.; Zhou, Q.; et al. Mutations in Cypher/ZASP in Patients with Dilated Cardiomyopathy and Left Ventricular Non-Compaction. J. Am. Coll. Cardiol. 2003, 42, 2014–2027. [Google Scholar] [CrossRef] [Green Version]
- Arimura, T.; Ishikawa, T.; Nunoda, S.; Kawai, S.; Kimura, A. Dilated Cardiomyopathy-Associated BAG3 Mutations Impair Z-Disc Assembly and Enhance Sensitivity to Apoptosis in Cardiomyocytes. Hum. Mutat. 2011, 32, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Vite, A.; Radice, G.L. N-Cadherin/Catenin Complex as a Master Regulator of Intercalated Disc Function. Cell Commun. Adhes. 2014, 21, 169–179. [Google Scholar] [CrossRef]
- Kamisago, M.; Sharma, S.D.; DePalma, S.R.; Solomon, S.; Sharma, P.; McDonough, B.; Smoot, L.; Mullen, M.P.; Woolf, P.K.; Wigle, E.D.; et al. Mutations in Sarcomere Protein Genes as a Cause of Dilated Cardiomyopathy. N. Engl. J. Med. 2000, 343, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.P.; Kamisago, M.; Asahi, M.; Hua Li, G.; Ahmad, F.; Mende, U.; Kranias, E.G.; MacLennan, D.H.; Seidman, J.G.; Seidman, C.E. Dilated Cardiomyopathy and Heart Failure Caused by a Mutation in Phospholamban. Science 2003, 299, 1410–1413. [Google Scholar] [CrossRef]
- Vermij, S.H.; Abriel, H.; van Veen, T.A.B. Refining the Molecular Organization of the Cardiac Intercalated Disc. Cardiovasc. Res. 2017, 113, 259–275. [Google Scholar] [CrossRef]
- Rizzo, S.; Lodder, E.M.; Verkerk, A.O.; Wolswinkel, R.; Beekman, L.; Pilichou, K.; Basso, C.; Remme, C.A.; Thiene, G.; Bezzina, C.R. Intercalated Disc Abnormalities, Reduced Na(+) Current Density, and Conduction Slowing in Desmoglein-2 Mutant Mice Prior to Cardiomyopathic Changes. Cardiovasc. Res. 2012, 95, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the Desmosomal Protein Plakophilin-2 Are Common in Arrhythmogenic Right Ventricular Cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- te Riele, A.S.J.M.; James, C.A.; Sawant, A.C.; Bhonsale, A.; Groeneweg, J.A.; Mast, T.P.; Murray, B.; Tichnell, C.; Dooijes, D.; van Tintelen, J.P.; et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy in the Pediatric Population: Clinical Characterization and Comparison With Adult-Onset Disease. JACC Clin. Electrophysiol. 2015, 1, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhu, W.; Wang, C.; Huang, L.; Zhou, Q.; Hu, J.; Cheng, X.; Hong, K. Genotype-Phenotype Relationship in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy Caused by Desmosomal Gene Mutations: A Systematic Review and Meta-Analysis. Sci. Rep. 2017, 7, srep41387. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Asatryan, B.; Wehrens, X.H.T. Genetic Basis and Molecular Biology of Cardiac Arrhythmias in Cardiomyopathies. Cardiovasc. Res. 2020, 116, 1600–1619. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Tandri, H.; Huang, H.; Halushka, M.K.; Gautam, S.; Basso, C.; Thiene, G.; Tsatsopoulou, A.; Protonotarios, N.; McKenna, W.J.; et al. A New Diagnostic Test for Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2009, 360, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Tedgui, A.; Fontaliran, F.; Frank, R.; Durigon, M.; Fontaine, G. Evidence of Apoptosis in Arrhythmogenic Right Ventricular Dysplasia. N. Engl. J. Med. 1996, 335, 1190–1197. [Google Scholar] [CrossRef]
- Rook, M.B.; Evers, M.M.; Vos, M.A.; Bierhuizen, M.F.A. Biology of Cardiac Sodium Channel Nav1.5 Expression. Cardiovasc. Res. 2012, 93, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Burke, B.; Stewart, C.L. The Nuclear Lamins: Flexibility in Function. Nat. Rev. Mol. Cell Biol. 2013, 14, 13–24. [Google Scholar] [CrossRef]
- Peretto, G.; Sala, S.; Benedetti, S.; di Resta, C.; Gigli, L.; Ferrari, M.; Bella, P.D. Updated Clinical Overview on Cardiac Laminopathies: An Electrical and Mechanical Disease. Nucleus 2018, 9, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.R.G.; Slavov, D.; Gajewski, A.; Vlcek, S.; Ku, L.; Fain, P.R.; Carniel, E.; di Lenarda, A.; Sinagra, G.; Boucek, M.M.; et al. Thymopoietin (Lamina-Associated Polypeptide 2) Gene Mutation Associated with Dilated Cardiomyopathy. Hum. Mutat. 2005, 26, 566–574. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; McKenna, W.J. Genetics of Right Ventricular Cardiomyopathy. J. Cardiovasc. Electrophysiol. 2005, 16, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Savvatis, K.; Müller, I.; Fröhlich, M.; Pappritz, K.; Zietsch, C.; Hamdani, N.; Grote, K.; Schieffer, B.; Klingel, K.; van Linthout, S.; et al. Interleukin-6 Receptor Inhibition Modulates the Immune Reaction and Restores Titin Phosphorylation in Experimental Myocarditis. Basic Res. Cardiol. 2014, 109, 449. [Google Scholar] [CrossRef] [PubMed]
- Beraldi, R.; Li, X.; Fernandez, A.M.; Reyes, S.; Secreto, F.; Terzic, A.; Olson, T.M.; Nelson, T.J. Rbm20-Deficient Cardiogenesis Reveals Early Disruption of RNA Processing and Sarcomere Remodeling Establishing a Developmental Etiology for Dilated Cardiomyopathy. Hum. Mol. Genet. 2014, 23, 3779–3791. [Google Scholar] [CrossRef] [Green Version]
- Ouisse, L.H.; Remy, S.; Lafoux, A.; Larcher, T.; Tesson, L.; Chenouard, V.; Guillonneau, C.; Brusselle, L.; Vimond, N.; Rouger, K.; et al. Immunophenotype of a Rat Model of Duchenne’s Disease and Demonstration of Improved Muscle Strength After Anti-CD45RC Antibody Treatment. Front. Immunol. 2019, 10, 2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, P.L.; Ebner, J.; Koenig, X.; Hamza, O.; Watzinger, S.; Trojanek, S.; Abraham, D.; Todt, H.; Kubista, H.; Schicker, K.; et al. Cardiovascular Phenotype of the Dmdmdx Rat—A Suitable Animal Model for Duchenne Muscular Dystrophy. Dis. Model. Mech. 2021, 14, dmm047704. [Google Scholar] [CrossRef] [PubMed]
- Vakrou, S.; Liu, Y.; Zhu, L.; Greenland, G.V.; Simsek, B.; Hebl, V.B.; Guan, Y.; Woldemichael, K.; Talbot, C.C.; Aon, M.A.; et al. Differences in Molecular Phenotype in Mouse and Human Hypertrophic Cardiomyopathy. Sci. Rep. 2021, 11, 13163. [Google Scholar] [CrossRef] [PubMed]
- Gerbino, A.; Forleo, C.; Milano, S.; Piccapane, F.; Procino, G.; Pepe, M.; Piccolo, M.; Guida, P.; Resta, N.; Favale, S.; et al. Pro-Inflammatory Cytokines as Emerging Molecular Determinants in Cardiolaminopathies. J. Cell. Mol. Med. 2021, 25, 10902–10915. [Google Scholar] [CrossRef]
- Brayson, D.; Frustaci, A.; Verardo, R.; Chimenti, C.; Russo, M.A.; Hayward, R.; Ahmad, S.; Vizcay-Barrena, G.; Protti, A.; Zammit, P.S.; et al. Prelamin A Mediates Myocardial Inflammation in Dilated and HIV-Associated Cardiomyopathies. JCI Insight 2019, 4, 126315. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, K.; Stutzman, A.; Ikegami, S.; Almakki, O.; Liu, S.; Burnicka-turek, O.; Moskowitz, I. Abstract MP230: Nuclear Rupture In A Mouse Model Of Lmna -Related Cardiomyopathy Causes Cytoplasmic Exposure Of The Proinflammatory Signaling Protein Hmgb1. Circ. Res. 2021, 129, AMP230. [Google Scholar] [CrossRef]
- Homma, S.; Iwasaki, M.; Shelton, G.D.; Engvall, E.; Reed, J.C.; Takayama, S. BAG3 Deficiency Results in Fulminant Myopathy and Early Lethality. Am. J. Pathol. 2006, 169, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Lynch, T.L.; Ismahil, M.A.; Jegga, A.G.; Zilliox, M.J.; Troidl, C.; Prabhu, S.D.; Sadayappan, S. Cardiac Inflammation in Genetic Dilated Cardiomyopathy Caused by MYBPC3 Mutation. J. Mol. Cell. Cardiol. 2017, 102, 83–93. [Google Scholar] [CrossRef]
- Maier, H.J.; Schips, T.G.; Wietelmann, A.; Krüger, M.; Brunner, C.; Sauter, M.; Klingel, K.; Böttger, T.; Braun, T.; Wirth, T. Cardiomyocyte-Specific IκB Kinase (IKK)/NF-ΚB Activation Induces Reversible Inflammatory Cardiomyopathy and Heart Failure. Proc. Natl. Acad. Sci. USA 2012, 109, 11794–11799. [Google Scholar] [CrossRef] [Green Version]
- van Opbergen, C.J.M.; Noorman, M.; Pfenniger, A.; Copier, J.S.; Vermij, S.H.; Li, Z.; van der Nagel, R.; Zhang, M.; de Bakker, J.M.T.; Glass, A.M.; et al. Plakophilin-2 Haploinsufficiency Causes Calcium Handling Deficits and Modulates the Cardiac Response Towards Stress. Int. J. Mol. Sci. 2019, 20, 4076. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Hernández, M.; Marrón-Liñares, G.M.; Schlamp, F.; Heguy, A.; van Opbergen, C.J.M.; Mezzano, V.; Zhang, M.; Liang, F.X.; Cerrone, M.; Delmar, M. Transcriptomic Coupling of PKP2 With Inflammatory and Immune Pathways Endogenous to Adult Cardiac Myocytes. Front. Physiol. 2021, 11, 623190. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.B.; de Bakker, J.M.T.; et al. Myocyte Necrosis Underlies Progressive Myocardial Dystrophy in Mouse Dsg2-Related Arrhythmogenic Right Ventricular Cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, K.E.; Delaney, P.J.; Thenet, D.; Murtough, S.; Webb, C.M.; Zaman, N.; Tsisanova, E.; Mastroianni, G.; Walker, S.L.M.; Westaby, J.D.; et al. Early Inflammation Precedes Cardiac Fibrosis and Heart Failure in Desmoglein 2 Murine Model of Arrhythmogenic Cardiomyopathy. Cell Tissue Res. 2021, 386, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Lubos, N.; van der Gaag, S.; Gerçek, M.; Kant, S.; Leube, R.E.; Krusche, C.A. Inflammation Shapes Pathogenesis of Murine Arrhythmogenic Cardiomyopathy. Basic. Res. Cardiol. 2020, 115, 42. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Protonotarios, A.; James, C.A.; Chelko, S.P.; Tichnell, C.; Murray, B.; Tsatsopoulou, A.; Anastasakis, A.; te Riele, A.; Kléber, A.G.; et al. Characterizing the Molecular Pathology of Arrhythmogenic Cardiomyopathy in Patient Buccal Mucosa Cells. Circ. Arrhythm. Electrophysiol. 2016, 9, e003688. [Google Scholar] [CrossRef] [Green Version]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Arndt, A.K.; Adams, E.; Liu, Z.Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a New Modulator of the Intercalated Disc in a Zebrafish Model of Arrhythmogenic Cardiomyopathy. Sci. Transl. Med. 2014, 6, 240ra74. [Google Scholar] [CrossRef] [Green Version]
- Chelko, S.P.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; MacRae, C.A.; Leber, R.; Kleber, A.G.; et al. Central Role for GSK3β in the Pathogenesis of Arrhythmogenic Cardiomyopathy. JCI Insight 2016, 1, e85923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.F.; Haneline, L.S.; Field, L.J.; et al. Restrictive Loss of Plakoglobin in Cardiomyocytes Leads to Arrhythmogenic Cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, R.; da Graca Cabreira-Hansen, M.; Bell, A.; Fromm, R.R.; Willerson, J.T.; Marian, A.J. Nuclear Plakoglobin Is Essential for Differentiation of Cardiac Progenitor Cells to Adipocytes in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Res. 2011, 109, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.X.; Gordon, P.M.K.; Nygren, A.; et al. Transgenic Mice Overexpressing Desmocollin-2 (DSC2) Develop Cardiomyopathy Associated with Myocardial Inflammation and Fibrotic Remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Jiang, C.; Li, Y.; Yang, D.; Ma, Y.; Zhang, B.; Li, X.; Zhang, P.; Hu, X.; Zhao, X.; et al. TMEM43-S358L Mutation Enhances NF-ΚB-TGFβ Signal Cascade in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Protein Cell 2019, 10, 104–119. [Google Scholar] [CrossRef] [Green Version]
- Shinomiya, H.; Kato, H.; Kuramoto, Y.; Watanabe, N.; Tsuruda, T.; Arimura, T.; Miyashita, Y.; Miyasaka, Y.; Mashimo, T.; Takuwa, A.; et al. Aberrant Accumulation of TMEM43 Accompanied by Perturbed Transmural Gene Expression in Arrhythmogenic Cardiomyopathy. FASEB J. 2021, 35, e21994. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Fox, P.R.; Meurs, K.M.; Towbin, J.A.; Spier, A.W.; Calabrese, F.; Maron, B.J.; Thiene, G. Arrhythmogenic Right Ventricular Cardiomyopathy Causing Sudden Cardiac Death in Boxer Dogs. Circulation 2004, 109, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Pugliese, M.; la Maestra, R.; la Gamba, G.; Lanteri, G.; Passantino, A. Arrhythmogenic Right Ventricular Cardiomyopathy in a Cat. Atti Accad. Peloritana Pericolanti Cl. Sci. Med. Biol. 2021, 109, 1–6. [Google Scholar] [CrossRef]
- Paulus, W.J.; Zile, M.R. From Systemic Inflammation to Myocardial Fibrosis. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.G.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of Titin Causing Dilated Cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Kötter, S.; Gout, L.; von Frieling-Salewsky, M.; Müller, A.E.; Helling, S.; Marcus, K.; dos Remedios, C.; Linke, W.A.; Krüger, M. Differential Changes in Titin Domain Phosphorylation Increase Myofilament Stiffness in Failing Human Hearts. Cardiovasc. Res. 2013, 99, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic Variation in Titin in Arrhythmogenic Right Ventricular Cardiomyopathy-Overlap Syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, M.; Zwinger, L.; Klaassen, S.; Poller, W.; Monserrat Iglesias, L.; Pablo Ochoa, J.; Klingel, K.; Landmesser, U.; Heidecker, B. Severe Heart Failure in the Setting of Inflammatory Cardiomyopathy with Likely Pathogenic Titin Variant. Int. J. Cardiol. Heart Vasc. 2022, 39, 100969. [Google Scholar] [CrossRef]
- Henkens, M.T.H.M.; Stroeks, S.L.V.M.; Raafs, A.G.; Sikking, M.A.; Tromp, J.; Ouwerkerk, W.; Hazebroek, M.R.; Krapels, I.P.C.; Knackstedt, C.; van den Wijngaard, A.; et al. Dynamic Ejection Fraction Trajectory in Patients With Dilated Cardiomyopathy With a Truncating Titin Variant. Circ. Heart Fail. 2022, 15, E009352. [Google Scholar] [CrossRef] [PubMed]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.F.; Varela, J.; David, R.M.; Banks, L.; Huang, C.H.; Li, H.; et al. A Role for Toll-like Receptor 3 Variants in Host Susceptibility to Enteroviral Myocarditis and Dilated Cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar] [CrossRef] [Green Version]
- Ono, R.; Iwai, Y.; Yamazaki, T.; Takahashi, H.; Hori, Y.; Fukushima, K.; Saotome, T. Nivolumab-Induced Myositis and Myocarditis with Positive Anti-Titin Antibody and Anti-Voltage-Gated Potassium Channel Kv1.4 Antibody. Intern. Med. 2022, 61, 2973–2979. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, C.; Lin, S.; Yu, X. Effect of TTN Mutations on Immune Microenvironment and Efficacy of Immunotherapy in Lung Adenocarcinoma Patients. Front. Oncol. 2021, 11, 725292. [Google Scholar] [CrossRef]
- Gil-Cruz, C.; Perez-Shibayama, C.; de Martin, A.; Ronchi, F.; van der Borght, K.; Niederer, R.; Onder, L.; Lütge, M.; Novkovic, M.; Nindl, V.; et al. Microbiota-Derived Peptide Mimics Drive Lethal Inflammatory Cardiomyopathy. Science 2019, 366, 881–886. [Google Scholar] [CrossRef]
- Frustaci, A.; Russo, M.A.; Chimenti, C. Randomized Study on the Efficacy of Immunosuppressive Therapy in Patients with Virus-Negative Inflammatory Cardiomyopathy: The TIMIC Study. Eur. Heart J. 2009, 30, 1995–2002. [Google Scholar] [CrossRef]
- de Luca, G.; Campochiaro, C.; Sartorelli, S.; Peretto, G.; Dagna, L. Therapeutic Strategies for Virus-Negative Myocarditis: A Comprehensive Review. Eur. J. Intern. Med. 2020, 77, 9–17. [Google Scholar] [CrossRef]
- de Luca, G.; Campochiaro, C.; Dinarello, C.A.; Dagna, L.; Cavalli, G. Treatment of Dilated Cardiomyopathy With Interleukin-1 Inhibition. Ann. Intern. Med. 2018, 169, 819–820. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, B.; Asimaki, A.; Landstrom, A.P.; Khanji, M.Y.; Odening, K.E.; Cooper, L.T.; Marchlinski, F.E.; Gelzer, A.R.; Semsarian, C.; Reichlin, T.; et al. Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review. Circulation 2021, 144, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Tandri, H.; Duffy, E.R.; Winterfield, J.R.; MacKey-Bojack, S.; Picken, M.M.; Cooper, L.T.; Wilber, D.J.; Marcus, F.I.; Basso, C.; et al. Altered Desmosomal Proteins in Granulomatous Myocarditis and Potential Pathogenic Links to Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2011, 4, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory Mutations in Transforming Growth Factor-Beta3 Gene Cause Arrhythmogenic Right Ventricular Cardiomyopathy Type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Błyszczuk, P. Myocarditis in Humans and in Experimental Animal Models. Front. Cardiovasc. Med. 2019, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W.; et al. A Mutation in the Human Phospholamban Gene, Deleting Arginine 14, Results in Lethal, Hereditary Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef] [Green Version]
- Rusciano, M.R.; Sommariva, E.; Douin-Echinard, V.; Ciccarelli, M.; Poggio, P.; Maione, A.S. CaMKII Activity in the Inflammatory Response of Cardiac Diseases. Int. J. Mol. Sci. 2019, 20, 4374. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.N.; Sbaizero, O.; Taylor, M.R.G.; Mestroni, L. Lamin A/C Cardiomyopathy: Implications for Treatment. Curr. Cardiol. Rep. 2019, 21, 160. [Google Scholar] [CrossRef]
- Bonello-Palot, N.; Simoncini, S.; Robert, S.; Bourgeois, P.; Sabatier, F.; Levy, N.; Dignat-George, F.; Badens, C. Prelamin A Accumulation in Endothelial Cells Induces Premature Senescence and Functional Impairment. Atherosclerosis 2014, 237, 45–52. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular Senescence and the Senescent Secretory Phenotype: Therapeutic Opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Bowles, N.E.; Ni, J.; Marcus, F.; Towbin, J.A. The Detection of Cardiotropic Viruses in the Myocardium of Patients with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J. Am. Coll. Cardiol. 2002, 39, 892–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Q.; Meng, X.; Liu, B.Q.; Li, C.; Gao, Y.Y.; Niu, X.F.; Li, N.; Guan, Y.; Du, Z.X. Involvement of JNK and NF-ΚB Pathways in Lipopolysaccharide (LPS)-Induced BAG3 Expression in Human Monocytic Cells. Exp. Cell Res. 2012, 318, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.P.; de Simone, F.I.; Iorio, V.; de Marco, M.; Khalili, K.; Sariyer, I.K.; Capunzo, M.; Nori, S.L.; Rosati, A. HIV-1 Tat Protein Induces Glial Cell Autophagy through Enhancement of BAG3 Protein Levels. Cell Cycle 2014, 13, 3640–3644. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 Is Required for Transcription of Genes That Control Calcium Cycling and Cardiac Rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampazzo, A.; Beffagna, G.; Nava, A.; Occhi, G.; Bauce, B.; Noiato, M.; Basso, C.; Frigo, G.; Thiene, G.; Towbin, J.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy Type 1 (ARVD1): Confirmation of Locus Assignment and Mutation Screening of Four Candidate Genes. Eur. J. Hum. Genet. 2003, 11, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Dubash, A.D.; Kam, C.Y.; Aguado, B.A.; Patel, D.M.; Delmar, M.; Shea, L.D.; Green, K.J. Plakophilin-2 Loss Promotes TGF-Β1/P38 MAPK-Dependent Fibrotic Gene Expression in Cardiomyocytes. J. Cell Biol. 2016, 212, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Kant, S.; Holthöfer, B.; Magin, T.M.; Krusche, C.A.; Leube, R.E. Desmoglein 2-Dependent Arrhythmogenic Cardiomyopathy Is Caused by a Loss of Adhesive Function. Circ. Cardiovasc. Genet. 2015, 8, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Rimpler, U. Funktionelle Charakterisierung von Desmocollin 2 Während Der Embryonalentwicklung Und Im Adulten Herzen in Der Maus (Functional Characterization of Desmocollin-2 during the Embryonic Development and in the Adult Murine Heart). Ph.D. Thesis, Humboldt-Universität, Berlin, Germany, 2014. [Google Scholar] [CrossRef]
- Beattie, J.; Allan, G.J.; Lochrie, J.D.; Flint, D.J. Insulin-like Growth Factor-Binding Protein-5 (IGFBP-5): A Critical Member of the IGF Axis. Biochem. J. 2006, 395, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of Canonical Wnt/β-Catenin Signaling by Nuclear Plakoglobin Recapitulates Phenotype of Arrhythmogenic Right Ventricular Cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Padrón-Barthe, L.; Villalba-Orero, M.; Gómez-Salinero, J.M.; Domínguez, F.; Román, M.; Larrasa-Alonso, J.; Ortiz-Sánchez, P.; Martínez, F.; López-Olañeta, M.; Bonzón-Kulichenko, E.; et al. Severe Cardiac Dysfunction and Death Caused by Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Are Improved by Inhibition of Glycogen Synthase Kinase-3β. Circulation 2019, 140, 1188–1204. [Google Scholar] [CrossRef]
- Meurs, K.M.; Mauceli, E.; Lahmers, S.; Acland, G.M.; White, S.N.; Lindblad-Toh, K. Genome-Wide Association Identifies a Deletion in the 3 Untranslated Region of Striatin in a Canine Model of Arrhythmogenic Right Ventricular Cardiomyopathy. Hum. Genet. 2010, 128, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Cattanach, B.M.; Dukes-McEwan, J.; Wotton, P.R.; Stephenson, H.M.; Hamilton, R.M. A Pedigree-Based Genetic Appraisal of Boxer ARVC and the Role of the Striatin Mutation. Vet. Rec. 2015, 176, 492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peretto, G.; De Luca, G.; Villatore, A.; Di Resta, C.; Sala, S.; Palmisano, A.; Vignale, D.; Campochiaro, C.; Lazzeroni, D.; De Gaspari, M.; et al. Multimodal detection and targeting of biopsy-proven myocardial inflammation in genetic cardiomyopathies: A pilot report. JACC Basic Transl. Sci. 2023; ahead of print. [Google Scholar]
- Reina-Couto, M.; Pereira-Terra, P.; Quelhas-Santos, J.; Silva-Pereira, C.; Albino-Teixeira, A.; Sousa, T. Inflammation in Human Heart Failure: Major Mediators and Therapeutic Targets. Front. Physiol. 2021, 12, 746494. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, F.; Iserin, F.; Raisky, O.; Laux, D.; Bajolle, F.; Boudjemline, Y.; Boddaert, N.; Bonnet, D. Myocardial inflammation on cardiovascular magnetic resonance predicts left ventricular function recovery in children with recent dilated cardiomyopathy. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 756–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peretto, G.; Mazzone, P.; Paglino, G.; Marzi, A.; Tsitsinakis, G.; Rizzo, S.; Basso, C.; Della Bella, P.; Sala, S. Continuous Electrical Monitoring in Patients with Arrhythmic Myocarditis: Insights from a Referral Center. J. Clin. Med. 2021, 10, 5142. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Barzaghi, F.; Cicalese, M.P.; Di Resta, C.; Slavich, M.; Benedetti, S.; Giangiobbe, S.; Rizzo, S.; Palmisano, A.; Esposito, A.; et al. Immunosuppressive therapy in childhood-onset arrhythmogenic inflammatory cardiomyopathy. Pacing Clin. Electrophysiol. 2021, 44, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Kontorovich, A.R. Approaches to Genetic Screening in Cardiomyopathies: Practical Guidance for Clinicians. JACC Heart Fail. 2023, 11, 133–142. [Google Scholar] [CrossRef]
- Lota, A.S.; Hazebroek, M.R.; Theotokis, P.; Wassall, R.; Salmi, S.; Halliday, B.P.; Tayal, U.; Verdonschot, J.; Meena, D.; Owen, R.; et al. Genetic Architecture of Acute Myocarditis and the Overlap with Inherited Cardiomyopathy. Circulation 2022, 146, 1123–1134. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Definition | References |
|---|---|---|
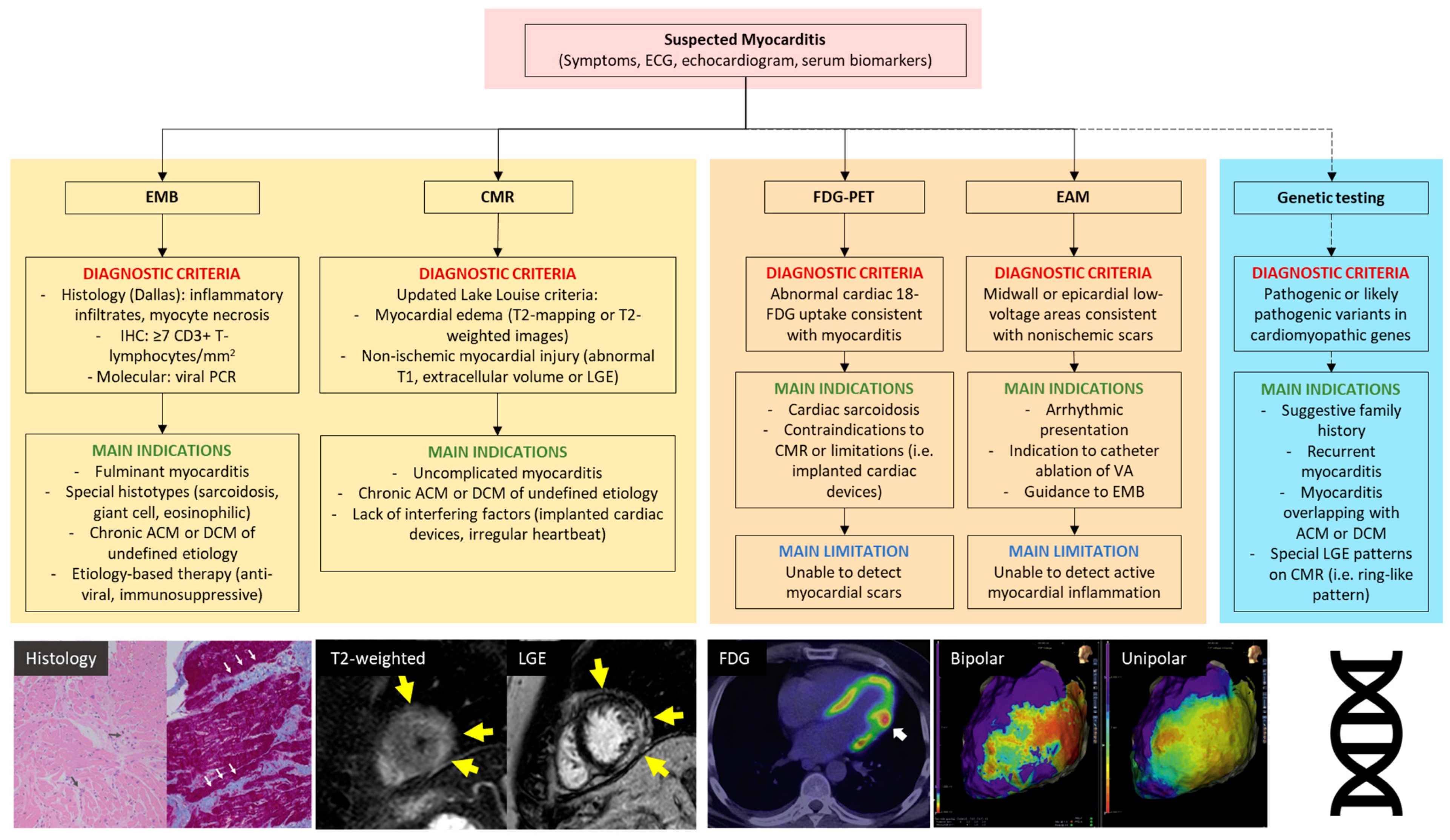
| Myocarditis | Inflammatory disease of the myocardium diagnosed by established Histological, immunological, and immunohistochemical criteria (WHO, ESC). In detail: -Histology: infiltrating inflammatory mononucleated cells with myocyte myocyte degeneration and necrosis of nonischemic origin (Dallas criteria). It is defined borderline myocarditis in the absence of necrosis. It is defined chronic myocarditis in the presence of replacement-type fibrosis. -Immunohistochemistry: ≥14 leucocytes/mm2 including up to 4 monocytes/mm2 with the presence of CD 3 positive T-lymphocytes ≥7 cells/mm2. Clinical classification based on symptom onset: -Acute (<1 month) -Subacute (1–3 months) -Chronic (>3 months) | [6,7,8] |
| Cardiomyopathy | Myocardial disorders in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular disease and congenital heart disease sufficient to cause the observed myocardial abnormality (ESC). It may also include electrical diseases prone to life-threatening arrhythmias (AHA). | [9,10,11] |
| Dilated cardiomyopathy (DCM) | Dilation and impaired contraction of the left or both ventricles that is not explained by abnormal loading conditions or coronary artery disease. Phenotype classification:-Hypokinetic nondilated cardiomyopathy (LV systolic dysfunction with no dilation). -Overt DCM (LV dilation and systolic dysfunction). | [6,12] |
| Arrhythmogenic cardiomyopathy (ACM) | Arrhythmogenic heart muscle disorder not explained by ischemic, hypertensive, or valvular heart disease. Phenotype classification: -Classic right ventricular ACM (modified Task Force Criteria). -Biventricular ACM. -Left-dominant ACM (Padua criteria). | [13,14] |
| Inflammatory cardiomyopathy | Myocarditis in association with cardiac dysfunction (i.e., LV ejection fraction <50%) | [6,7] |
| Myocardial inflammation (M-Infl) | Evidence of myocardial inflammation fulfilling the histological (±myocyte necrosis, i.e., borderline myocarditis) and immunohistochemical criteria for myocarditis, in a patient with clinical diagnosis of ACM or DCM. | [3,5] |
| Gene | Model | Main Findings | References |
|---|---|---|---|
| DSP, PKP2 | Clinical setting | Patients with DSP variant cardiomyopathy including 16/105 (15%) who had “acute myocardial injury episodes” akin to clinical myocarditis. | [18] |
| DSP | Clinical setting | Acute myocarditis reflects an active phase of ACM that leads to changes in phenotype and abrupt progression of ACM. | [19] |
| DSP | Clinical setting | Cohort of patients initially presenting with a classic myocarditis syndrome (chest pain, troponin elevation) who were subsequently diagnosed with ACM. Most patients had a DSP genetic variant. | [20,21,22] |
| DSP, LAMA4, LDB3, MYBPC3 DSC2, RYR2, SOS1, SCN5A, SGCD, LPL, PKP2, MYH1, GATA6, and DSG2 | Human heart specimens from autopsy cases | Minimal inflammatory foci may be an early sign of inherited cardiomyopathy. | [23] |
| DSP, FLNC, PKP2, TMPO, TTN | Human EMB | Retrospective multicenter study on patients with undefined LV ACM and extensive overlap between EMB-proven myocardial inflammation and rare genetic variants of the DCM/ACM spectrum. | [5] |
| Human EMB | Most asymptomatic relatives of dilated cardiomyopathy patients with mild left ventricular enlargement already showed infiltration of inflammatory cells, at levels that were similar to those of patients with established disease. | [24] | |
| SCN5A | Case report | Young SCN5A variant carrier with recurrent ventricular fibrillation and massive myocardial inflammation | [25] |
| Models of Genetic Etiologies Linked to DCM | |||
|---|---|---|---|
| Gene | Model | Phenotype | References |
| TTN | C57BL6/J mice | Cardiac inflammation | [82] |
| RBM20 | shRbm20 iPSCs | Dysregulation of genes involved in inflammation | [83] |
| DMD | Dmdmdx rat | Infiltration of leukocytes | [84] |
| DMD | Dmdmdx rat | Infiltration of macrophages | [85] |
| MYH | MyH-mutant mouse | Upregulation of inflammasome pathways | [86] |
| LMNA | HEK293 cells expressing LMNA-p. Leu140_Ala146dup | Upregulation of Hsp70 | [87] |
| LMNA | csPLA transgenic mouse | Inflammatory cardiomyopathy; activation of NF-κB | [88] |
| LMNA | LmnaCMKO mouse | Upregulation of pro-inflammatory gene expression programs | [89] |
| BAG3 | BAG3-deficient mice | Minimal cardiac muscle inflammation | [90] |
| MYBPC3 | cMyBP-C(t/t) mouse | Macrophage infiltration; upregulation of inflammatory pathways | [91] |
| - | IKKMyHC mouse | Excessive inflammatory response and myocyte atrophy | [92] |
| Models of Genetic Etiologies Linked to ACM | |||
| Gene | Model | Phenotype | References |
| PKP2 | PKP2-Hz mouse | More sensitivity to experimental autoimmune myocarditis | [93] |
| PKP2 | C57BL/6 RiboTagflox mice | Abundance of transcripts involved in the inflammatory/immune response | [94] |
| PKP2 | hiPSC line from an ACM patient with a c.2013delC (p.Lys672Argfs*12) variant in plakophilin-2 (PKP2) | Secretion of inflammatory cytokines | [95] |
| DSG2 | N271Sdsg2 transgenic mouse | Presence of massive inflammatory infiltrates | [96] |
| DSG2 | Dsg2+/+ mouse, Dsg2+/− mouse, Dsg2−/− mouse | Activation of inflammatory pathways; upregulation of genes linked to specific macrophage populations | [97] |
| DSG2 | Dsg2MT mouse, Dsg2cKO mouse | Presence of inflammatory response, recruitment of immune cell | [98] |
| DSG2 | Dsg2mut/mut mouse | Activation of NFκB; increased levels of inflammatory cytokines and chemotactic molecules | [95] |
| DSC2 | DSC2 transgenic mouse | Upregulation of inflammatory and fibrotic remodeling pathways | [99] |
| JUP | Neonatal rat ventricular myocytes expressing 2057del2 plakoglobin | Release of inflammatory mediators, reduced by SB216763 treatment | [100] |
| JUP | Dsg2mut/mut mouse, JUP2157del2 mouse | Extensive inflammation, improved by SB216763 treatment | [101] |
| JUP | Neonatal rat ventricular myocytes expressing JUP2157del2 | Activation of NFκB | [95] |
| JUP | JUP mutant mouse | Expression of pro-inflammatory cytokines | [102] |
| JUP | PGTR mouse | Overexpression of IGFBP5 | [103] |
| DSP | Cardiac-specific Dsp knockout mouse | Fibro-fatty substitution of the myocardium; cardiomyocyte death | [104] |
| TMEM43 | Tmem43-S358L mutant mice | Activation of NFκB | [105] |
| TMEM43 | Tmem43-S358L KI | Infiltration of inflammatory cells | [106] |
| - | Boxer right ventricular ACM | Myocarditis | [107] |
| - | Cat right ventricular ACM | Signs of inflammatory infiltrates | [108] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peretto, G.; Sommariva, E.; Di Resta, C.; Rabino, M.; Villatore, A.; Lazzeroni, D.; Sala, S.; Pompilio, G.; Cooper, L.T. Myocardial Inflammation as a Manifestation of Genetic Cardiomyopathies: From Bedside to the Bench. Biomolecules 2023, 13, 646. https://doi.org/10.3390/biom13040646
Peretto G, Sommariva E, Di Resta C, Rabino M, Villatore A, Lazzeroni D, Sala S, Pompilio G, Cooper LT. Myocardial Inflammation as a Manifestation of Genetic Cardiomyopathies: From Bedside to the Bench. Biomolecules. 2023; 13(4):646. https://doi.org/10.3390/biom13040646
Chicago/Turabian StylePeretto, Giovanni, Elena Sommariva, Chiara Di Resta, Martina Rabino, Andrea Villatore, Davide Lazzeroni, Simone Sala, Giulio Pompilio, and Leslie T. Cooper. 2023. "Myocardial Inflammation as a Manifestation of Genetic Cardiomyopathies: From Bedside to the Bench" Biomolecules 13, no. 4: 646. https://doi.org/10.3390/biom13040646