

From Genetics to Clinical Implications: A Study of 675 Dutch Osteogenesis Imperfecta Patients
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Methods
2.1. Study Participants
2.2. Data Extraction
2.3. Data Analysis
2.4. Ethical Consideration
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marini, J.C.; Forlino, A.; Bächinger, H.P.; Bishop, N.J.; Byers, P.H.; Paepe, A.; Fassier, F.; Fratzl-Zelman, N.; Kozloff, K.M.; Krakow, D.; et al. Osteogenesis imperfecta. Nat. Rev. Dis. Prim. 2017, 3, 17052. [Google Scholar] [CrossRef]
- Forlino, A.; Marini, J.C. Osteogenesis imperfecta. Lancet 2016, 387, 1657–1671. [Google Scholar] [CrossRef]
- Marom, R.; Rabenhorst, B.M.; Morello, R. Osteogenesis imperfecta: An update on clinical features and therapies. Eur. J. Endocrinol. 2020, 183, R95–R106. [Google Scholar] [CrossRef]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 2019, 179, 2393–2419. [Google Scholar] [CrossRef]
- Zhytnik, L.; Maasalu, K.; Reimand, T.; Duy, B.H.; Kõks, S.; Märtson, A. Inter- and Intrafamilial Phenotypic Variability in Individuals with Collagen-Related Osteogenesis Imperfecta. Clin. Transl. Sci. 2020, 13, 960–971. [Google Scholar] [CrossRef]
- Sillence, D.O.; Senn, A.; Danks, D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979, 16, 101–116. [Google Scholar] [CrossRef]
- van Dijk, F.S.; Cobben, J.M.; Kariminejad, A.; Maugeri, A.; Nikkels, P.G.; van Rijn, R.R.; Pals, G. Osteogenesis Imperfecta: A Review with Clinical Examples. Mol. Syndromol. 2011, 2, 1–20. [Google Scholar] [CrossRef]
- Van Dijk, F.S.; Sillence, D.O. Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am. J. Med. Genet. A 2014, 164, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Byers, P.H.; Wallis, G.A.; Willing, M.C. Osteogenesis imperfecta: Translation of mutation to phenotype. J. Med. Genet. 1991, 28, 433–442. [Google Scholar] [CrossRef]
- Symoens, S.; Hulmes, D.J.; Bourhis, J.M.; Coucke, P.J.; De Paepe, A.; Malfait, F. Type I procollagen C-propeptide defects: Study of genotype-phenotype correlation and predictive role of crystal structure. Hum. Mutat. 2014, 35, 1330–1341. [Google Scholar] [CrossRef]
- Sillence, D.O.; Rimoin, D.L.; Danks, D.M. Clinical variability in osteogenesis imperfecta-variable expressivity or genetic heterogeneity. Birth Defects Orig. Artic. Ser. 1979, 15, 113–129. [Google Scholar]
- Monti, E.; Mottes, M.; Fraschini, P.; Brunelli, P.; Forlino, A.; Venturi, G.; Doro, F.; Perlini, S.; Cavarzere, P.; Antoniazzi, F. Current and emerging treatments for the management of osteogenesis imperfecta. Ther. Clin. Risk Manag. 2010, 6, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N. Molecular structure of collagen. Int. Rev. Connect. Tissue Res. 1963, 1, 127–182. [Google Scholar] [CrossRef]
- Rich, A.; Crick, F.H. The molecular structure of collagen. J. Mol. Biol. 1961, 3, 483–506. [Google Scholar] [CrossRef] [PubMed]
- Claeys, L.; Storoni, S.; Eekhoff, M.; Elting, M.; Wisse, L.; Pals, G.; Bravenboer, N.; Maugeri, A.; Micha, D. Collagen transport and related pathways in Osteogenesis Imperfecta. Hum. Genet. 2021, 140, 1121–1141. [Google Scholar] [CrossRef]
- Leiden Open Variation Database. The Human Varione Project: Disease #05296 Osteogenesis Imperfecta. Available online: https://databases.lovd.nl/shared/diseases/05296 (accessed on 17 November 2022).
- Maioli, M.; Gnoli, M.; Boarini, M.; Tremosini, M.; Zambrano, A.; Pedrini, E.; Mordenti, M.; Corsini, S.; D’Eufemia, P.; Versacci, P.; et al. Genotype-phenotype correlation study in 364 osteogenesis imperfecta Italian patients. Eur. J. Hum. Genet. 2019, 27, 1090–1100. [Google Scholar] [CrossRef]
- Byers, P.H. Collagens: Building blocks at the end of the development line. Clin. Genet. 2000, 58, 270–279. [Google Scholar] [CrossRef]
- Bella, J.; Eaton, M.; Brodsky, B.; Berman, H.M. Crystal and molecular structure of a collagen-like peptide at 1.9 A resolution. Science 1994, 266, 75–81. [Google Scholar] [CrossRef]
- Raghunath, M.; Bruckner, P.; Steinmann, B. Delayed triple helix formation of mutant collagen from patients with osteogenesis imperfecta. J. Mol. Biol. 1994, 236, 940–949. [Google Scholar] [CrossRef]
- Marini, J.C.; Forlino, A.; Cabral, W.A.; Barnes, A.M.; San Antonio, J.D.; Milgrom, S.; Hyland, J.C.; Körkkö, J.; Prockop, D.J.; De Paepe, A.; et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007, 28, 209–221. [Google Scholar] [CrossRef]
- Xiao, J.; Cheng, H.; Silva, T.; Baum, J.; Brodsky, B. Osteogenesis imperfecta missense mutations in collagen: Structural consequences of a glycine to alanine replacement at a highly charged site. Biochemistry 2011, 50, 10771–10780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garibaldi, N.; Besio, R.; Dalgleish, R.; Villani, S.; Barnes, A.M.; Marini, J.C.; Forlino, A. Dissecting the phenotypic variability of osteogenesis imperfecta. Dis. Model. Mech. 2022, 15, dmm049398. [Google Scholar] [CrossRef] [PubMed]
- Ben Amor, I.M.; Glorieux, F.H.; Rauch, F. Genotype-phenotype correlations in autosomal dominant osteogenesis imperfecta. J. Osteoporos. 2011, 2011, 540178. [Google Scholar] [CrossRef]
- Yang, L.; Liu, B.; Dong, X.; Wu, J.; Sun, C.; Xi, L.; Cheng, R.; Wu, B.; Wang, H.; Tong, S.; et al. Clinical severity prediction in children with osteogenesis imperfecta caused by COL1A1/2 defects. Osteoporos. Int. 2022, 33, 1373–1384. [Google Scholar] [CrossRef]
- WHO Collaborating Centre for Drug Statistics Methodology. ATC/DDD Index 2022. Available online: https://www.whocc.no/atc_ddd_index (accessed on 14 November 2022).
- Nijhuis, W.H.; Eastwood, D.M.; Allgrove, J.; Hvid, I.; Weinans, H.H.; Bank, R.A.; Sakkers, R.J. Current concepts in osteogenesis imperfecta: Bone structure, biomechanics and medical management. J. Child. Orthop. 2019, 13, 1–11. [Google Scholar] [CrossRef]
- Makareeva, E.; Sun, G.; Mirigian, L.S.; Mertz, E.L.; Vera, J.C.; Espinoza, N.A.; Yang, K.; Chen, D.; Klein, T.E.; Byers, P.H.; et al. Substitutions for arginine at position 780 in triple helical domain of the α1(I) chain alter folding of the type I procollagen molecule and cause osteogenesis imperfecta. PLoS ONE 2018, 13, e0200264. [Google Scholar] [CrossRef]
- Pollitt, R.; McMahon, R.; Nunn, J.; Bamford, R.; Afifi, A.; Bishop, N.; Dalton, A. Mutation analysis of COL1A1 and COL1A2 in patients diagnosed with osteogenesis imperfecta type I-IV. Hum. Mutat. 2006, 27, 716. [Google Scholar] [CrossRef]
- Lindahl, K.; Åström, E.; Rubin, C.J.; Grigelioniene, G.; Malmgren, B.; Ljunggren, Ö.; Kindmark, A. Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur. J. Hum. Genet. 2015, 23, 1042–1050. [Google Scholar] [CrossRef]
- Starman, B.J.; Eyre, D.; Charbonneau, H.; Harrylock, M.; Weis, M.A.; Weiss, L.; Graham, J.M., Jr.; Byers, P.H. Osteogenesis imperfecta. The position of substitution for glycine by cysteine in the triple helical domain of the pro alpha 1(I) chains of type I collagen determines the clinical phenotype. J. Clin. Investig. 1989, 84, 1206–1214. [Google Scholar] [CrossRef]
- Orwoll, E.S.; Shapiro, J.; Veith, S.; Wang, Y.; Lapidus, J.; Vanek, C.; Reeder, J.L.; Keaveny, T.M.; Lee, D.C.; Mullins, M.A.; et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J. Clin. Investig. 2014, 124, 491–498. [Google Scholar] [CrossRef]
- Rohrbach, M.; Giunta, C. Recessive osteogenesis imperfecta: Clinical, radiological, and molecular findings. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 175–189. [Google Scholar] [CrossRef]
- Baldridge, D.; Schwarze, U.; Morello, R.; Lennington, J.; Bertin, T.K.; Pace, J.M.; Pepin, M.G.; Weis, M.; Eyre, D.R.; Walsh, J.; et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008, 29, 1435–1442. [Google Scholar] [CrossRef]
- Folkestad, L.; Hald, J.D.; Canudas-Romo, V.; Gram, J.; Hermann, A.P.; Langdahl, B.; Abrahamsen, B.; Brixen, K. Mortality and Causes of Death in Patients With Osteogenesis Imperfecta: A Register-Based Nationwide Cohort Study. J. Bone Miner. Res. 2016, 31, 2159–2166. [Google Scholar] [CrossRef]
- Folkestad, L. Mortality and morbidity in patients with osteogenesis imperfecta in Denmark. Dan. Med. J. 2018, 65, B5454. [Google Scholar]
- Storoni, S.; Treurniet, S.; Micha, D.; Celli, M.; Bugiani, M.; van den Aardweg, J.G.; Eekhoff, E.M.W. Pathophysiology of respiratory failure in patients with osteogenesis imperfecta: A systematic review. Ann. Med. 2021, 53, 1676–1687. [Google Scholar] [CrossRef]
- Khan, S.I.; Yonko, E.A.; Carter, E.M.; Dyer, D.; Sandhaus, R.A.; Raggio, C.L. Cardiopulmonary Status in Adults with Osteogenesis Imperfecta: Intrinsic Lung Disease May Contribute More Than Scoliosis. Clin. Orthop. Relat. Res. 2020, 478, 2833–2843. [Google Scholar] [CrossRef]
- McAllion, S.J.; Paterson, C.R. Causes of death in osteogenesis imperfecta. J. Clin. Pathol. 1996, 49, 627–630. [Google Scholar] [CrossRef]
- Ashournia, H.; Johansen, F.T.; Folkestad, L.; Diederichsen, A.C.; Brixen, K. Heart disease in patients with osteogenesis imperfecta—A systematic review. Int. J. Cardiol. 2015, 196, 149–157. [Google Scholar] [CrossRef]
- Rush, E.T.; Li, L.; Goodwin, J.L.; Kreikemeier, R.M.; Craft, M.; Danford, D.A.; Kutty, S. Echocardiographic phenotype in osteogenesis imperfecta varies with disease severity. Heart 2017, 103, 443–448. [Google Scholar] [CrossRef]
- Carré, F.; Achard, S.; Rouillon, I.; Parodi, M.; Loundon, N. Hearing impairment and osteogenesis imperfecta: Literature review. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2019, 136, 379–383. [Google Scholar] [CrossRef]
- Treurniet, S.; Burger, P.; Ghyczy, E.A.E.; Verbraak, F.D.; Curro-Tafili, K.R.; Micha, D.; Bravenboer, N.; Ralston, S.H.; de Vries, R.; Moll, A.C.; et al. Ocular characteristics and complications in patients with osteogenesis imperfecta: A systematic review. Acta Ophthalmol. 2022, 100, e16–e28. [Google Scholar] [CrossRef]
- Storoni, S.; Treurniet, S.; Maugeri, A.; Pals, G.; van den Aardweg, J.G.; van der Pas, S.L.; Elting, M.W.; Kloen, P.; Micha, D.; Eekhoff, E.M.W. Prevalence and Hospital Admissions in Patients With Osteogenesis Imperfecta in The Netherlands: A Nationwide Registry Study. Front. Endocrinol. 2022, 13, 869604. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
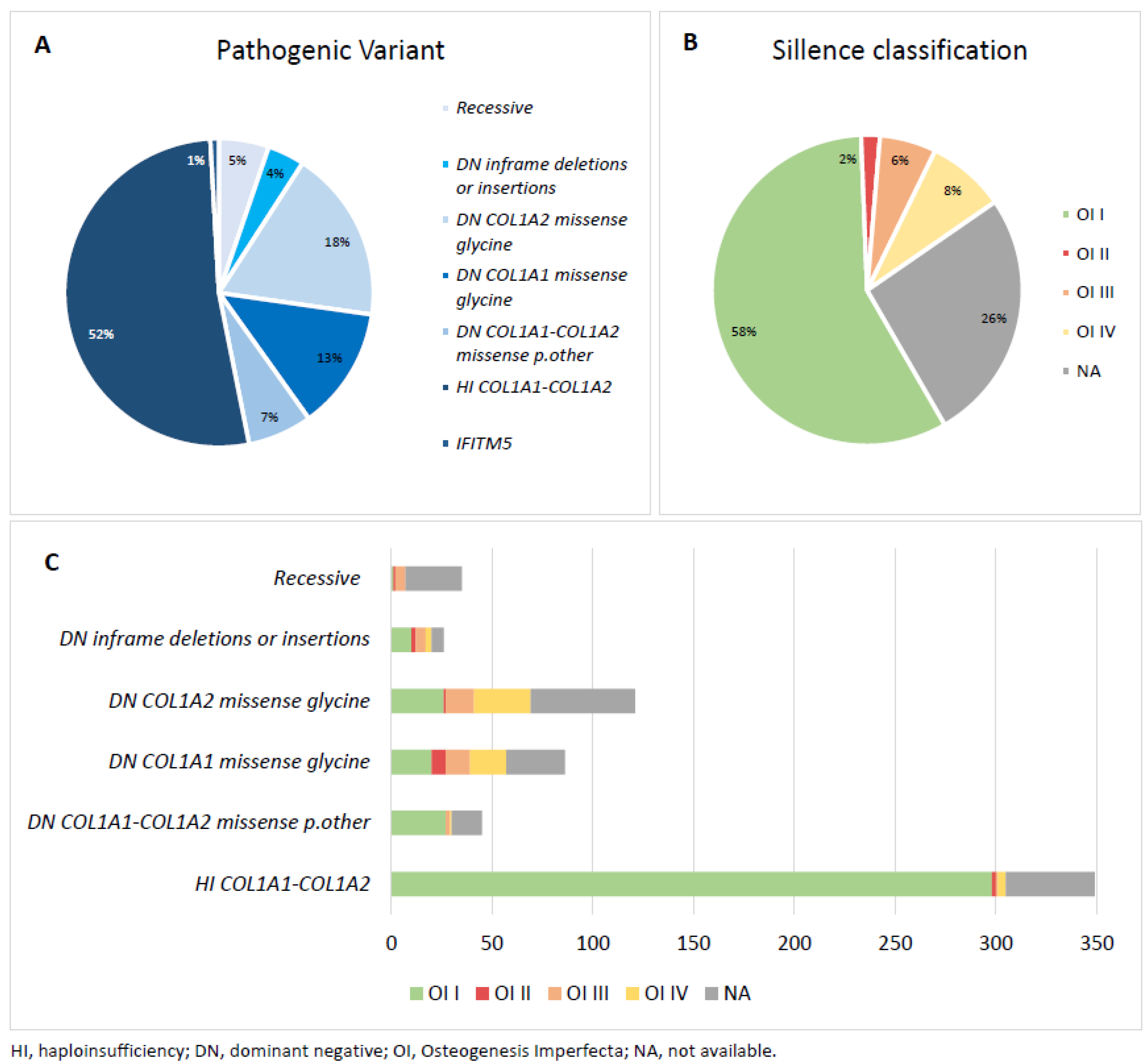
| Pathogenic Variants | Total | ||
|---|---|---|---|
| 675 | |||
| HI COL1A1-COL1A2 | |||
| HI COL1A1 | 349 | ||
| HI COL1A2 | <10 | ||
| DN COL1A1-COL1A2 missense p.other | 45 | ||
| DN COL1A1 missense p.other: | exon 1–5 | <10 | |
| exon 6–10 | <10 | ||
| exon 11–42 | <10 | ||
| exon 43–48 | <10 | ||
| exon 49–51 | <10 | ||
| DN COL1A2 missense p.other: | exon 1–5 | <10 | |
| exon 6–10 | <10 | ||
| exon 11–42 | <10 | ||
| exon 43–48 | <10 | ||
| exon 49–51 | <10 | ||
| DN COL1A1 missense glycine | 86 | ||
| DN COL1A1 missense glycine: | exon 6–10 | <10 | |
| exon 11–42 | 63 | ||
| exon 43–48 | <20 | ||
| DN COL1A2 missense glycine | 121 | ||
| DN COL1A2 missense glycine: | exon 1–5 | <10 | |
| exon 6–10 | <10 | ||
| exon 11–42 | 108 | ||
| exon 43–48 | <10 | ||
| exon 49–51 | <10 | ||
| DN inframe deletions or insertions | 26 | ||
| DN COL1A1 inframe deletion | exon 1–5 | <10 | |
| exon 6–10 | <10 | ||
| exon 11–42 | <10 | ||
| DN COL1A1 inframe insertion: | exon 11–42 | <10 | |
| DN COL1A2 inframe deletion: | exon 6–10 | <10 | |
| exon 11–42 | <10 | ||
| exon 43–48 | <10 | ||
| DN COL1A2 inframe insertion: | exon 11–42 | <10 | |
| exon 43–48 | <10 | ||
| Recessive | 35 | ||
| CRTAP, TMEM38B, CREB3L1, FKBP10, PLOD2, SP7, SERPINF1, P3H1, BMP, PPIB | |||
| IFITM5 | <10 | ||
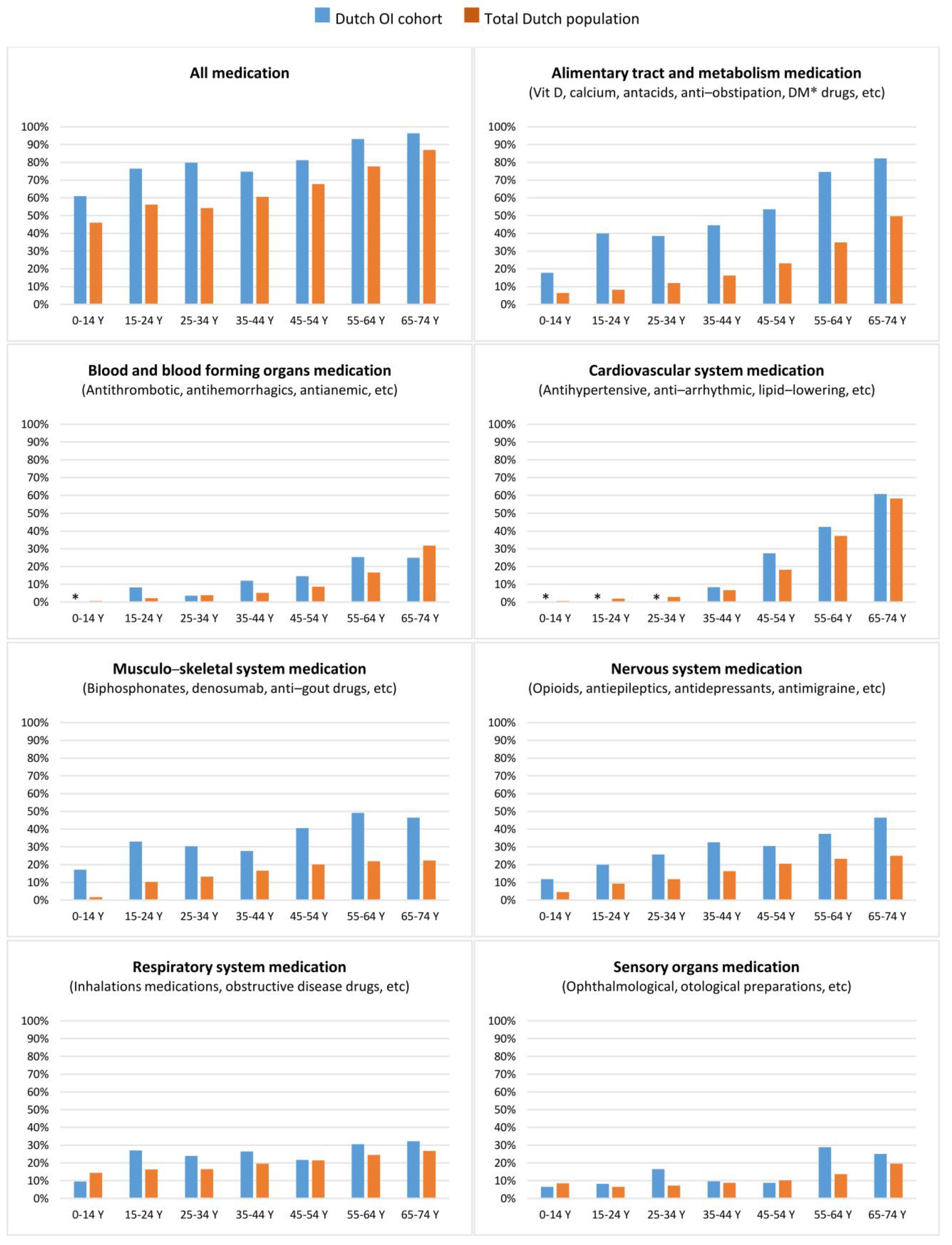
| A The Number of Diagnosis Treatment Combinations per Person in the OI Cohort Compared to the Total Dutch Population on Average during 2013–2017 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Medical Specialty | DN Missense p.Other | HI | DN COL1A2 Missense Glycine | DN COL1A1 Missense Glycine | Recessive | DN Inframe Deletions or Insertions | Total | |
| Total | 1.8 | 2.4 | 2.8 | 3.1 | 3.3 | 4.1 | 2.6 | |
| Ophthalmology | 0.9 | 1.1 | 1.0 | 0.8 | 1.3 | 1.9 | 1.1 | |
| Otolaryngology | 1.7 | 2.3 | 2.5 | 1.4 | 1.3 | 3.3 | 2.2 | |
| Surgery | 1.9 | 3.2 | 2.3 | 3.4 | 1.7 | 2.0 | 2.9 | |
| Surgery (plastics) | 0.9 | 1.7 | 1.4 | 0.7 | 10.2 | 6.5 | 1.7 | |
| Orthopedic surgery | 4.6 | 6.3 | 8.2 | 9.3 | 9.2 | 13.2 | 7.4 | |
| Urology | 0.1 | 0.9 | 0.7 | 1.1 | 1.3 | 0.2 | 0.8 | |
| Gynecology | 2.1 | 1.5 | 2.3 | 1.4 | 0.6 | 1.0 | 1.6 | |
| Dermatology | 0.9 | 0.8 | 0.5 | 0.9 | 2.0 | 0.7 | 0.9 | |
| Internal medicine | 1.1 | 1.8 | 1.5 | 2.3 | 1.8 | 2.6 | 1.7 | |
| Pediatrics | 6.6 | 8.4 | 12.7 | 11.2 | 13.4 | 13.2 | 9.9 | |
| Gastroenterology | 1.3 | 0.8 | 0.6 | 0.5 | 0 | 0 | 0.7 | |
| Cardiology | 0.6 | 0.7 | 0.4 | 0.4 | 1.0 | 0.3 | 0.6 | |
| Pulmonology | 1.6 | 1.4 | 1.8 | 5.2 | 5.4 | 3.6 | 2.3 | |
| Rheumatology | 0.5 | 0.9 | 0.6 | 0.4 | 4.4 | 2.4 | 0.8 | |
| Rehabilitation | 11.5 | 15.3 | 25.4 | 29.2 | 28.0 | 54.0 | 20.9 | |
| Geriatrics | 0 | 2.0 | 0 | 1.4 | 1.3 | 0 | 1.2 | |
| Anesthesiology | 0.3 | 1.3 | 1.9 | 3.0 | 2.6 | 2.7 | 1.6 | |
| Clinical genetics | 17.6 | 22.8 | 31.2 | 24.4 | 34.6 | 45.8 | 25.8 | |
| Audiology | 3.7 | 7.5 | 8.7 | 9.0 | 0 | 13.9 | 7.5 | |
| B The number of hospital admissions per 100 people on average per year in the dutch OI cohort during 2013–2019 | ||||||||
| Medical specialty | DN missense p.other | HI | DN COL1A2 missense glycine | DN COL1A1 missense glycine | recessive | DN inframe deletions or insertions | Total | |
| Total | 36.0 | 59.8 | 85.4 | 91.5 | 112.4 | 124.0 | 71.5 | per 100 people |
| Pediatrics | 12.0 | 20.9 | 41.6 | 42.2 | 61.4 | 49.6 | 29.6 | per 100 people |
| Surgery | 4.0 | 9.8 | 6.4 | 10.5 | 11.0 | 10.4 | 9.0 | per 100 people |
| Orthopedic surgery | 8.4 | 10.3 | 18.5 | 23.1 | 22.8 | 28.0 | 14.4 | per 100 people |
| Internal medicine | 0.9 | 2.5 | 4.3 | 3.4 | 1.4 | 24.8 | 3.7 | per 100 people |
| Neurology | 0 | 1.7 | 1.7 | 3.4 | 2.8 | 3.2 | 1.9 | per 100 people |
| Otolaryngology | 2.7 | 3.1 | 1.5 | 2.0 | 4.1 | 1.6 | 2.6 | per 100 people |
| Ophthalmology | 0 | 1.1 | 0.4 | 0.3 | 2.1 | 1.6 | 0.9 | per 100 people |
| Gynecology | 3.1 | 2.7 | 5.7 | 2.6 | 0.7 | 0.8 | 3.1 | per 100 people |
| Gastroenterology | 4.0 | 2.5 | 1.3 | 0.6 | 0.7 | 0.8 | 2.0 | per 100 people |
| Cardiology | 0.4 | 1.4 | 0.8 | 0.6 | 1.4 | 0 | 1.0 | per 100 people |
| Pulmonology | 0 | 1.4 | 0.8 | 0.9 | 0 | 0.8 | 1.0 | per 100 people |
| Urology | 0 | 0.9 | 0.8 | 0.9 | 0.7 | 0.8 | 0.8 | per 100 people |
| Dentistry | 0.4 | 0.6 | 0.9 | 0.9 | 2.8 | 0.8 | 0.8 | per 100 people |
| Other | 0 | 0.9 | 0.8 | 0.3 | 0.7 | 0.8 | 0.7 | per 100 people |
| The number of hospital admissions per person in the OI cohort compared to the total dutch population on average between during 2013–2019 | ||||||||
| Mean yearly admission rate in total Dutch population | 0.19 | |||||||
| Mean yearly admission rate in the OI cohort | 0.26 (median: 0.1, std. dev: 0.4) | 0.43 (median: 0.3, std. dev: 0.5) | 0.61 (median: 0.3, std. dev: 0.9) | 0.65 (median: 0.3, std. dev: 0.8) | 0.8 (median: 0.4, std. dev: 1.1) | 0.89 (median: 0.6, std. dev: 1.0) | 0.51 (median: 0.3, std. dev: 0.7) | |
| Incidence rate ratio OI cohort vs. total Dutch population | 1.35 | 2.25 | 3.21 | 3.44 | 4.23 | 4.66 | 2.69 | |
| Hospital Admissions (2013–2019) | Exp (Estimate) | 95% CI | ||
|---|---|---|---|---|
| HI | 0.41 | 0.33–0.50 | ⱡ | |
| DN missense p.other | 0.24 | 0.13–0.45 | ꬸ | |
| DN COL1A1 missense glycine | 0.58 | 0.42–0.81 | ||
| DN COL1A2 missense glycine | 0.53 | 0.40–0.71 | ||
| DN inframe deletions or insertions | 0.89 | 0.56–1.41 | ⱡ,ꬸ | |
| Recessive | 0.66 | 0.43–1.00 | ||
| Diagnosis treatment combinations (DTCs) (2013–2017) | Exp (estimate) | 95% CI | ||
| HI | 2.68 | 2.32–3.08 | ⱡ | |
| DN missense p.other | 2.04 | 1.52–2.72 | ꬸ,ꬷ,† | |
| DN COL1A1 missense glycine | 3.41 | 2.76–4.21 | ꬷ | |
| DN COL1A2 missense glycine | 3.06 | 2.54–3.67 | ||
| DN inframe deletions or insertions | 4.59 | 3.34–6.30 | ⱡ,ꬸ | |
| Recessive | 3.76 | 2.78–5.08 | † | |
| Outpatient clinic visits (2013–2017) | Exp (estimate) | 95% CI | ||
| HI | 4.66 | 4.01–5.41 | ||
| DN missense p.other | 3.73 | 2.79–4.99 | ||
| DN COL1A1 missense glycine | 5.51 | 4.39–6.91 | ||
| DN COL1A2 missense glycine | 4.80 | 3.95–5.83 | ||
| DN inframe deletions or insertions | 6.37 | 4.47–9.08 | ||
| Recessive | 5.74 | 4.13–7.98 | ||
| X-rays (2013–2017) | Exp (estimate) | 95% CI | ||
| HI | 1.37 | 1.13–1.67 | ||
| DN missense p.other | 0.91 | 0.60–1.40 | ꬸ,ꬷ,† | |
| DN COL1A1 missense glycine | 1.93 | 1.44–2.59 | ꬷ | |
| DN COL1A2 missense glycine | 1.66 | 1.29–2.14 | ||
| DN inframe deletions or insertions | 2.30 | 1.46–3.61 | ꬸ | |
| Recessive | 2.09 | 1.38–3.16 | † | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Storoni, S.; Verdonk, S.J.E.; Zhytnik, L.; Pals, G.; Treurniet, S.; Elting, M.W.; Sakkers, R.J.B.; van den Aardweg, J.G.; Eekhoff, E.M.W.; Micha, D. From Genetics to Clinical Implications: A Study of 675 Dutch Osteogenesis Imperfecta Patients. Biomolecules 2023, 13, 281. https://doi.org/10.3390/biom13020281
Storoni S, Verdonk SJE, Zhytnik L, Pals G, Treurniet S, Elting MW, Sakkers RJB, van den Aardweg JG, Eekhoff EMW, Micha D. From Genetics to Clinical Implications: A Study of 675 Dutch Osteogenesis Imperfecta Patients. Biomolecules. 2023; 13(2):281. https://doi.org/10.3390/biom13020281
Chicago/Turabian StyleStoroni, Silvia, Sara J. E. Verdonk, Lidiia Zhytnik, Gerard Pals, Sanne Treurniet, Mariet W. Elting, Ralph J. B. Sakkers, Joost G. van den Aardweg, Elisabeth M. W. Eekhoff, and Dimitra Micha. 2023. "From Genetics to Clinical Implications: A Study of 675 Dutch Osteogenesis Imperfecta Patients" Biomolecules 13, no. 2: 281. https://doi.org/10.3390/biom13020281