
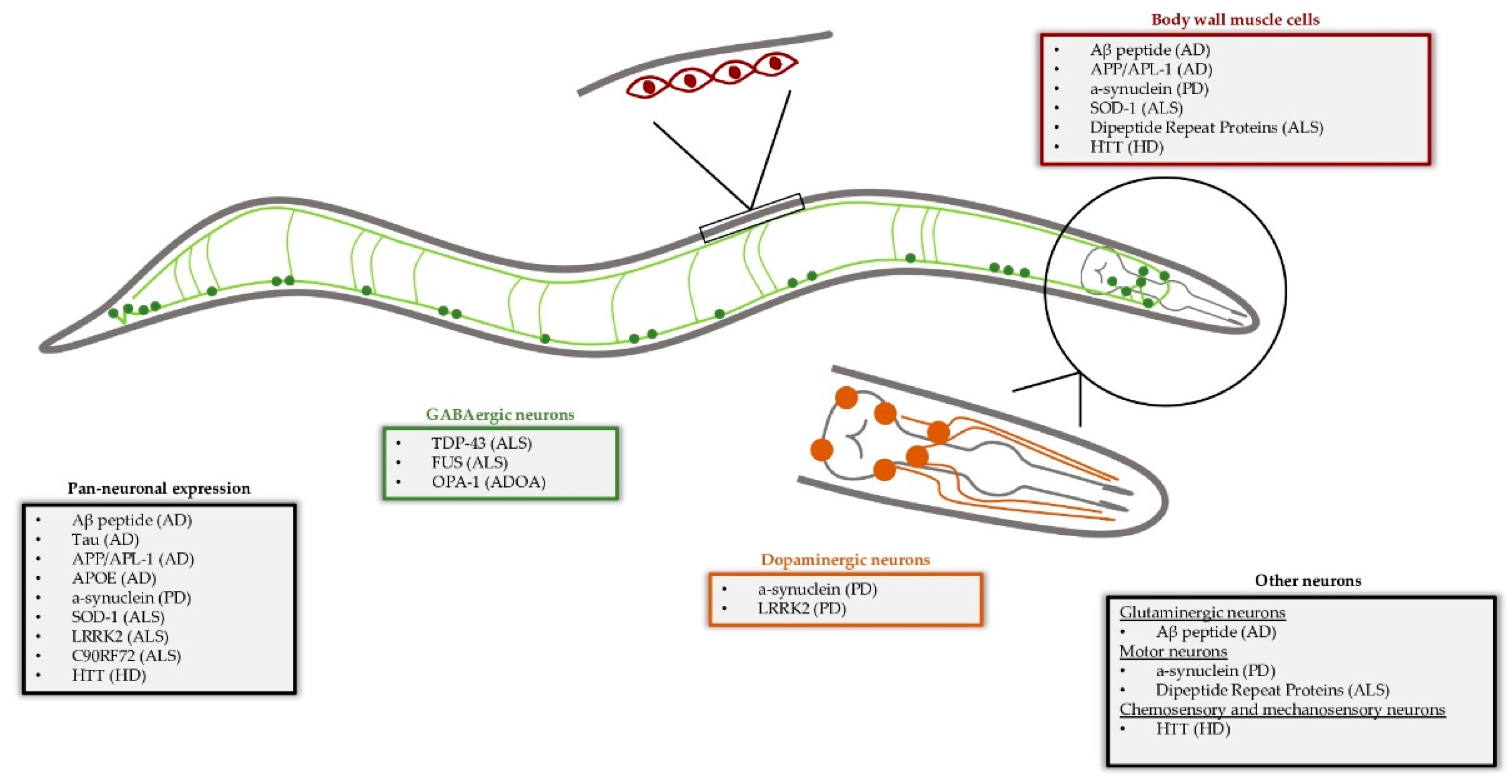
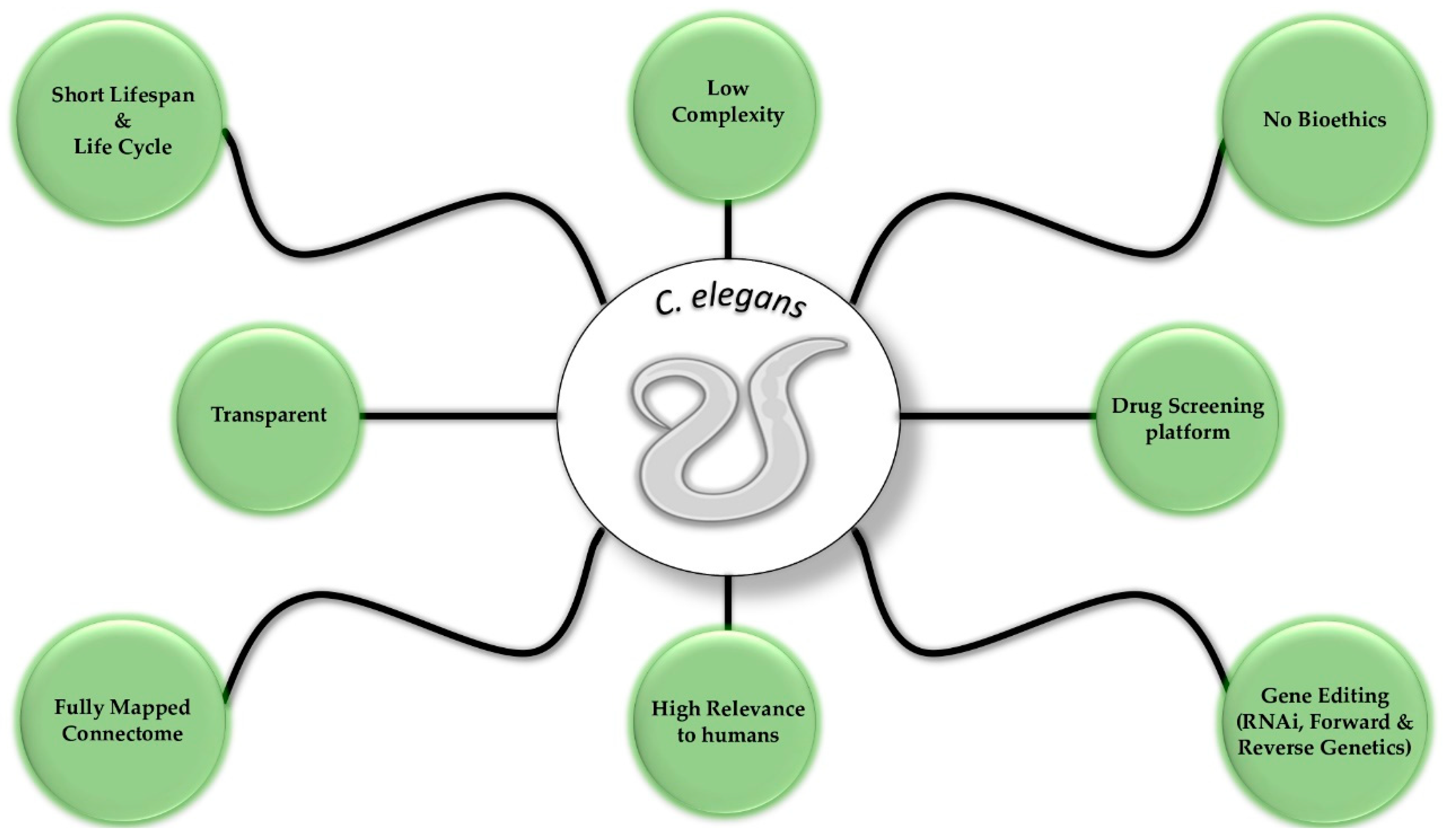
Caenorhabditis elegans as a Model System to Study Human Neurodegenerative Disorders

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Alzheimer’s Disease
1.1.1. Aβ Models
1.1.2. APOE Models
1.1.3. APP Models
1.1.4. Presenilin Models
1.1.5. Tau Models
2. Parkinson’s Disease
2.1. α-Synuclein Models
2.2. LRRK2 Models
3. Amyotrophic Lateral Sclerosis (ALS)
3.1. C9ORF72 Models
3.2. SOD1 Models
3.3. TDP43 Models
3.4. FUS Models
4. Huntington’s Disease
5. Cockayne Syndrome
6. Autosomal Dominant Optic Atrophy (ADOA)
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchyan, G.V.; Abdelmohsen, K.; Andreux, P.; Bagdonaite, I.; Barzilai, N.; Brunak, S.; Cabreiro, F.; de Cabo, R.; Campisi, J.; Cuervo, A.M.; et al. ARDD 2020: From aging mechanisms to interventions. Aging 2020, 12, 24484–24503. [Google Scholar] [CrossRef]
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef] [PubMed]
- Markaki, M.; Tavernarakis, N. Caenorhabditis elegans as a model system for human diseases. Curr. Opin. Biotechnol. 2020, 63, 118–125. [Google Scholar] [CrossRef]
- Frezal, L.; Felix, M.A. C. elegans outside the Petri dish. eLife 2015, 4, e05849. [Google Scholar] [CrossRef]
- Sulston, J. Neuronal Cell Lineages in the Nematode Caenorhabditis elegans. Cold Spring Harb. Symp. Quant. Biol. 1983, 48, 443–452. [Google Scholar] [CrossRef]
- Kaletta, T.; Hengartner, M. Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 2006, 5, 387–399. [Google Scholar] [CrossRef]
- The, C. elegans Sequencing Consortium. Genome Sequence of the Nematode C. elegans: A Platform for Investigating Biology. Science 1998, 282, 2012–2018. [Google Scholar] [CrossRef]
- Culetto, E.; Sattelle, D.B. A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum. Mol. Genet. 2000, 9, 869–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, K.A.; Willicott, C.W.; Caldwell, G.A. Modeling neurodegeneration in Caenorhabditis elegans. Dis. Model. Mech. 2020, 13, dmm046110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, F.; Zhou, T.; Wang, G.; Li, Z. Caenorhabditis elegans as a Useful Model for Studying Aging Mutations. Front. Endocrinol. 2020, 11, 554994. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; Jarrell, T.A.; Brittin, C.A.; Wang, Y.; Bloniarz, A.E.; Yakovlev, M.A.; Nguyen, K.C.Q.; Tang, L.T.-H.; Bayer, E.A.; Duerr, J.S.; et al. Whole-animal connectomes of both Caenorhabditis elegans sexes. Nature 2019, 571, 63–71. [Google Scholar] [CrossRef]
- White, J.G.; Southgate, E.; Thomson, J.N.; Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1986, 314, 1–340. [Google Scholar] [CrossRef]
- Bargmann, C.I.; Marder, E. From the connectome to brain function. Nat. Methods 2013, 10, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Kavalali, E.T.; Jorgensen, E.M. Visualizing presynaptic function. Nat. Neurosci. 2013, 17, 10–16. [Google Scholar] [CrossRef]
- Vashlishan, A.B.; Madison, J.M.; Dybbs, M.; Bai, J.; Sieburth, D.; Ch’Ng, Q.; Tavazoie, M.; Kaplan, J.M. An RNAi Screen Identifies Genes that Regulate GABA Synapses. Neuron 2008, 58, 346–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavell, S.W.; Pokala, N.; Macosko, E.Z.; Albrecht, D.R.; Larsch, J.; Bargmann, C.I. Serotonin and the Neuropeptide PDF Initiate and Extend Opposing Behavioral States in C. elegans. Cell 2013, 154, 1023–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Nakano, S.; Amano, M.; Tsuboi, D.; Nishioka, T.; Ikeda, S.; Yokoyama, G.; Kaibuchi, K.; Mori, I. Single-Cell Memory Regulates a Neural Circuit for Sensory Behavior. Cell Rep. 2016, 14, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Richmond, J.E.; Jorgensen, E.M. One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat. Neurosci. 1999, 2, 791–797. [Google Scholar] [CrossRef]
- Schuske, K.; Beg, A.A.; Jorgensen, E.M. The GABA nervous system in C. elegans. Trends Neurosci. 2004, 27, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Arey, R.N.; Kaletsky, R.; Murphy, C.T. Nervous system-wide profiling of presynaptic mRNAs reveals regulators of associative memory. Sci. Rep. 2019, 9, 20314. [Google Scholar] [CrossRef] [Green Version]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of Genes and Environments for Explaining Alzheimer Disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 17, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-amyloid Peptides and Amyloid Plaques in Alzheimer’s Disease. Neurotherapeutics 2014, 12, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.C. Abeta Plaques. Free Neuropathol. 2020, 1, 31. [Google Scholar] [CrossRef] [PubMed]
- Kabir, T.; Uddin, S.; Setu, J.R.; Ashraf, G.; Bin-Jumah, M.N.; Abdel-Daim, M. Exploring the Role of PSEN Mutations in the Pathogenesis of Alzheimer’s Disease. Neurotox. Res. 2020, 38, 833–849. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Liang, S.-Y.; Wang, Z.-T.; Tan, L.; Yu, J.-T. Tau Toxicity in Neurodegeneration. Mol. Neurobiol. 2022, 59, 3617–3634. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The Role of Tau in Alzheimer’s Disease and Related Disorders. CNS Neurosci. Ther. 2010, 17, 514–524. [Google Scholar] [CrossRef] [PubMed]
- McHatton, M. A theory for timely teaching. Prof. Inferm. 1986, 39, 41–47. [Google Scholar]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Van Pelt, K.M.; Truttmann, M.C. Caenorhabditis elegans as a model system for studying aging-associated neurodegenerative diseases. Transl. Med. Aging 2020, 4, 60–72. [Google Scholar] [CrossRef]
- Alvarez, J.; Alvarez-Illera, P.; Santo-Domingo, J.; Fonteriz, R.I.; Montero, M. Modeling Alzheimer’s Disease in Caenorhabditis elegans. Biomedicines 2022, 10, 288. [Google Scholar] [CrossRef]
- Griffin, E.F.; Caldwell, K.A.; Caldwell, G.A. Genetic and Pharmacological Discovery for Alzheimer’s Disease Using Caenorhabditis elegans. ACS Chem. Neurosci. 2017, 8, 2596–2606. [Google Scholar] [CrossRef]
- Link, C.D. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef] [Green Version]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.S.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional Links Between Aβ Toxicity, Endocytic Trafficking, and Alzheimer’s Disease Risk Factors in Yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, S.; Teo, E.; Ng, L.F.; Chen, C.-B.; Lakshmanan, L.N.; Tsoi, S.Y.; Moore, P.K.; Inoue, T.; Halliwell, B.; Gruber, J. Energy crisis precedes global metabolic failure in a novel Caenorhabditis elegans Alzheimer Disease model. Sci. Rep. 2016, 6, 33781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kulas, J.A.; Wang, C.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118. [Google Scholar] [CrossRef]
- Zhao, J.; Fu, Y.; Yamazaki, Y.; Ren, Y.; Davis, M.D.; Liu, C.-C.; Lu, W.; Wang, X.; Chen, K.; Cherukuri, Y.; et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat. Commun. 2020, 11, 5540. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Asaro, A.; Carlo-Spiewok, A.; Malik, A.R.; Rothe, M.; Schipke, C.G.; Peters, O.; Heeren, J.; Willnow, T.E. Apolipoprotein E4 disrupts the neuroprotective action of sortilin in neuronal lipid metabolism and endocannabinoid signaling. Alzheimer’s Dement. 2020, 16, 1248–1258. [Google Scholar] [CrossRef]
- Spinney, L. Alzheimer’s disease: The forgetting gene. Nature 2014, 510, 26–28. [Google Scholar] [CrossRef] [Green Version]
- Griffin, E.F.; Scopel, S.E.; Stephen, C.A.; Holzhauer, A.C.; Vaji, M.A.; Tuckey, R.A.; Berkowitz, L.A.; Caldwell, K.A.; Caldwell, G.A. ApoE-associated modulation of neuroprotection from Aβ-mediated neurodegeneration in transgenic Caenorhabditis elegans. Dis. Model. Mech. 2019, 12, dmm037218. [Google Scholar] [CrossRef] [Green Version]
- Hornsten, A.; Lieberthal, J.; Fadia, S.; Malins, R.; Ha, L.; Xu, X.; Daigle, I.; Markowitz, M.; O’Connor, G.; Plasterk, R.; et al. APL-1, a Caenorhabditis elegans protein related to the human β-amyloid precursor protein, is essential for viability. Proc. Natl. Acad. Sci. USA 2007, 104, 1971–1976. [Google Scholar] [CrossRef] [Green Version]
- Yi, B.; Sahn, J.J.; Ardestani, P.M.; Evans, A.K.; Scott, L.L.; Chan, J.Z.; Iyer, S.; Crisp, A.; Zuniga, G.; Pierce, J.T.; et al. Small molecule modulator of sigma 2 receptor is neuroprotective and reduces cognitive deficits and neuroinflammation in experimental models of Alzheimer’s disease. J. Neurochem. 2017, 140, 561–575. [Google Scholar] [CrossRef] [Green Version]
- Ewald, C.Y.; Cheng, R.; Tolen, L.; Shah, V.; Gillani, A.; Nasrin, A.; Li, C. Pan-Neuronal Expression of APL-1, an APP-Related Protein, Disrupts Olfactory, Gustatory, and Touch Plasticity in Caenorhabditis elegans. J. Neurosci. 2012, 32, 10156–10169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiese, M.; Antebi, A.; Zheng, H. Intracellular Trafficking and Synaptic Function of APL-1 in Caenorhabditis elegans. PLoS ONE 2010, 5, e12790. [Google Scholar] [CrossRef] [Green Version]
- Ewald, C.Y.; Li, C. Caenorhabditis elegans as a model organism to study APP function. Exp. Brain Res. 2011, 217, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Area-Gomez, E.; de Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins Are Enriched in Endoplasmic Reticulum Membranes Associated with Mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [Green Version]
- Beel, A.J.; Sanders, C.R. Substrate specificity of γ-secretase and other intramembrane proteases. Cell. Mol. Life Sci. 2008, 65, 1311–1334. [Google Scholar] [CrossRef] [Green Version]
- Sarasija, S.; Norman, K.R. A γ-Secretase Independent Role for Presenilin in Calcium Homeostasis Impacts Mitochondrial Function and Morphology in Caenorhabditis elegans. Genetics 2015, 201, 1453–1466. [Google Scholar] [CrossRef] [Green Version]
- Levitan, D.; Greenwald, I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 1995, 377, 351–354. [Google Scholar] [CrossRef]
- Sarasija, S.; Laboy, J.T.; Ashkavand, Z.; Bonner, J.; Tang, Y.; Norman, K.R. Presenilin mutations deregulate mitochondrial Ca2+ homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. eLife 2018, 7, 33052. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Pir, G.J.; Choudhary, B.; Mandelkow, E. Caenorhabditis elegans models of tauopathy. FASEB J. 2017, 31, 5137–5148. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.J.H.; McKinnon, I.A.; Rankin, C.H. The contribution of C. elegans neurogenetics to understanding neurodegenerative diseases. J. Neurogenet. 2020, 34, 527–548. [Google Scholar] [CrossRef] [PubMed]
- Chew, Y.L.; Fan, X.; Götz, J.; Nicholas, H.R. PTL-1 regulates neuronal integrity and lifespan in C. elegans. J. Cell Sci. 2013, 126, 2079–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, B.C.; Zhang, B.; Leverenz, J.B.; Thomas, J.H.; Trojanowski, J.Q.; Schellenberg, G.D. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 9980–9985. [Google Scholar] [CrossRef] [Green Version]
- Brandt, R.; Gergou, A.; Wacker, I.; Fath, T.; Hutter, H. A Caenorhabditis elegans model of tau hyperphosphorylation: Induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 2009, 30, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Fischer, S.; Johnson, G.V.W.; Nehrke, K. Tauopathy-associated tau modifications selectively impact neurodegeneration and mitophagy in a novel C. elegans single-copy transgenic model. Mol. Neurodegener. 2020, 15, 65. [Google Scholar] [CrossRef]
- Miyasaka, T.; Shinzaki, Y.; Yoshimura, S.; Yoshina, S.; Kage-Nakadai, E.; Mitani, S.; Ihara, Y. Imbalanced Expression of Tau and Tubulin Induces Neuronal Dysfunction in C. elegans Models of Tauopathy. Front. Neurosci. 2018, 12, 415. [Google Scholar] [CrossRef] [Green Version]
- Pir, G.J.; Choudhary, B.; Mandelkow, E.; Mandelkow, E.-M. Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol. Neurodegener. 2016, 11, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthrie, C.R.; Greenup, L.; Leverenz, J.B.; Kraemer, B.C. MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum. Mol. Genet. 2011, 20, 1989–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthrie, C.R.; Schellenberg, G.D.; Kraemer, B.C. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum. Mol. Genet. 2009, 18, 1825–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, B.C.; Burgess, J.K.; Chen, J.H.; Thomas, J.H.; Schellenberg, G.D. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum. Mol. Genet. 2006, 15, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Waldherr, S.M.; Strovas, T.J.; Vadset, T.A.; Liachko, N.F.; Kraemer, B.C. Constitutive XBP-1s-mediated activation of the endoplasmic reticulum unfolded protein response protects against pathological tau. Nat. Commun. 2019, 10, 4443. [Google Scholar] [CrossRef] [Green Version]
- Imanikia, S.; Özbey, N.P.; Krueger, C.; Casanueva, M.O.; Taylor, R.C. Neuronal XBP-1 Activates Intestinal Lysosomes to Improve Proteostasis in C. elegans. Curr. Biol. 2019, 29, 2322–2338. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Bai, F. The Association of Tau with Mitochondrial Dysfunction in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef] [Green Version]
- Schulz, K.L.; Eckert, A.; Rhein, V.; Mai, S.; Haase, W.; Reichert, A.S.; Jendrach, M.; Müller, W.E.; Leuner, K. A New Link to Mitochondrial Impairment in Tauopathies. Mol. Neurobiol. 2012, 46, 205–216. [Google Scholar] [CrossRef]
- Palikaras, K.; Achanta, K.; Choi, S.; Akbari, M.; Bohr, V.A. Alteration of mitochondrial homeostasis is an early event in a C. elegans model of human tauopathy. Aging 2021, 13, 23876–23894. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Cooper, J.F.; Van Raamsdonk, J.M. Modeling Parkinson’s Disease in C. elegans. J. Park. Dis. 2018, 8, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene–environment interactions. Neurotoxicology 2015, 46, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Mocko, J.B.; Kern, A.; Moosmann, B.; Behl, C.; Hajieva, P. Phenothiazines interfere with dopaminergic neurodegeneration in Caenorhabditis elegans models of Parkinson’s disease. Neurobiol. Dis. 2010, 40, 120–129. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, Z.; Klaunig, J.E. Caenorhabditis elegans neuron degeneration and mitochondrial suppression caused by selected environmental chemicals. Int. J. Biochem. Mol. Biol. 2013, 4, 191–200. [Google Scholar]
- Sherer, T.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of Toxicity in Rotenone Models of Parkinson’s Disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Berman, S.B.; Hastings, T.G. Mic60/mitofilin overexpression alters mitochondrial dynamics and attenuates vulnerability of dopaminergic cells to dopamine and rotenone. Neurobiol. Dis. 2016, 91, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Aliagan, A.D.I.; Ahwazi, M.D.; Tombo, N.; Feng, Y.; Bopassa, J.C. Parkin interacts with Mitofilin to increase dopaminergic neuron death in response to Parkinson’s disease-related stressors. Am. J. Transl. Res. 2020, 12, 7542–7564. [Google Scholar]
- Wu, X.; Nagasawa, S.; Muto, K.; Ueda, M.; Suzuki, C.; Abe, T.; Higashitani, A. Mitochonic Acid 5 Improves Duchenne Muscular Dystrophy and Parkinson’s Disease Model of Caenorhabditis elegans. Int. J. Mol. Sci. 2022, 23, 9572. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamaguchi, H.; Kikusato, M.; Hashizume, O.; Nagatoishi, S.; Matsuo, A.; Sato, T.; Kudo, T.; Matsuhashi, T.; Murayama, K.; et al. Mitochonic Acid 5 Binds Mitochondria and Ameliorates Renal Tubular and Cardiac Myocyte Damage. J. Am. Soc. Nephrol. 2015, 27, 1925–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuhashi, T.; Sato, T.; Kanno, S.-I.; Suzuki, T.; Matsuo, A.; Oba, Y.; Kikusato, M.; Ogasawara, E.; Kudo, T.; Suzuki, K.; et al. Mitochonic Acid 5 (MA-5) Facilitates ATP Synthase Oligomerization and Cell Survival in Various Mitochondrial Diseases. EBioMedicine 2017, 20, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Yamaguchi, H.; Kikusato, M.; Matsuhashi, T.; Matsuo, A.; Sato, T.; Oba, Y.; Watanabe, S.; Minaki, D.; Saigusa, D.; et al. Mitochonic Acid 5 (MA-5), a Derivative of the Plant Hormone Indole-3-Acetic Acid, Improves Survival of Fibroblasts from Patients with Mitochondrial Diseases. Tohoku J. Exp. Med. 2015, 236, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Lakso, M.; Vartiainen, S.; Moilanen, A.-M.; Sirviö, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human α-synuclein. J. Neurochem. 2004, 86, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwahara, T.; Koyama, A.; Gengyo-Ando, K.; Masuda, M.; Kowa, H.; Tsunoda, M.; Mitani, S.; Iwatsubo, T. Familial Parkinson Mutant α-Synuclein Causes Dopamine Neuron Dysfunction in Transgenic Caenorhabditis elegans. J. Biol. Chem. 2006, 281, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.; Plasterk, R.H.A.; Nollen, E.A.A. C. elegans Model Identifies Genetic Modifiers of α-Synuclein Inclusion Formation during Aging. PLoS Genet. 2008, 4, e1000027. [Google Scholar] [CrossRef]
- van der Goot, A.T.; Zhu, W.; Vázquez-Manrique, R.P.; Seinstra, R.I.; Dettmer, K.; Michels, H.; Farina, F.; Krijnen, J.; Melki, R.; Buijsman, R.C.; et al. Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc. Natl. Acad. Sci. USA 2012, 109, 14912–14917. [Google Scholar] [CrossRef] [Green Version]
- Hamamichi, S.; Rivas, R.N.; Knight, A.L.; Cao, S.; Caldwell, K.A.; Caldwell, G.A. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA 2008, 105, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Gelwix, C.C.; Caldwell, K.A.; Caldwell, G.A. Torsin-Mediated Protection from Cellular Stress in the Dopaminergic Neurons of Caenorhabditis elegans. J. Neurosci. 2005, 25, 3801–3812. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtis, N.; Nikoletopoulou, V.; Tavernarakis, N. Small heat-shock proteins protect from heat-stroke-associated neurodegeneration. Nature 2012, 490, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Büttner, S.; Habernig, L.; Broeskamp, F.; Ruli, D.; Vögtle, F.N.; Vlachos, M.; Macchi, F.; Küttner, V.; Carmona-Gutierrez, D.; Eisenberg, T.; et al. Endonuclease G mediates α-synuclein cytotoxicity during Parkinson’s disease. EMBO J. 2013, 32, 3041–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanning, S.; Haque, A.; Imberdis, T.; Baru, V.; Barrasa, M.I.; Nuber, S.; Termine, D.; Ramalingam, N.; Ho, G.P.H.; Noble, T.; et al. Lipidomic Analysis of α-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol. Cell 2019, 73, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson’s Disease. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, P.N.; Chen, S.G.; Wilson-Delfosse, A.L. Leucine-rich repeat kinase 2 (LRRK2): A key player in the pathogenesis of Parkinson’s disease. J. Neurosci. Res. 2009, 87, 1283–1295. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Dawson, T.M.; Dawson, V.L. Models of LRRK2-Associated Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 163–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.; El Khoury, R.; Wang, W.; Byrd, T.A.; Pehek, E.A.; Thacker, C.; Zhu, X.; Smith, M.A.; Wilson-Delfosse, A.L.; Chen, S.G. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol. Dis. 2010, 40, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Fukuzono, T.; Pastuhov, S.I.; Fukushima, O.; Li, C.; Hattori, A.; Iemura, S.-I.; Natsume, T.; Shibuya, H.; Hanafusa, H.; Matsumoto, K.; et al. Chaperone complex BAG2-HSC70 regulates localization of Caenorhabditis elegans leucine-rich repeat kinase LRK-1 to the Golgi. Genes Cells 2016, 21, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi-Nakashima, A.; Meir, J.Y.; Jin, Y.; Matsumoto, K.; Hisamoto, N. LRK-1, a C. elegans PARK8-Related Kinase, Regulates Axonal-Dendritic Polarity of SV Proteins. Curr. Biol. 2007, 17, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, B.; Kamak, M.; Ratnakaran, N.; Kumar, J.; Awasthi, A.; Li, C.; Nguyen, K.; Matsumoto, K.; Hisamoto, N.; Koushika, S.P. UNC-16/JIP3 regulates early events in synaptic vesicle protein trafficking via LRK-1/LRRK2 and AP complexes. PLoS Genet. 2017, 13, e1007100. [Google Scholar] [CrossRef] [Green Version]
- Sämann, J.; Hegermann, J.; von Gromoff, E.; Eimer, S.; Baumeister, R.; Schmidt, E. Caenorhabditits elegans LRK-1 and PINK-1 Act Antagonistically in Stress Response and Neurite Outgrowth. J. Biol. Chem. 2009, 284, 16482–16491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Guillily, M.D.; Ferree, A.; Lanceta, J.; Chan, D.; Ghosh, J.; Hsu, C.H.; Segal, L.; Raghavan, K.; Matsumoto, K.; et al. LRRK2 Modulates Vulnerability to Mitochondrial Dysfunction in Caenorhabditis elegans. J. Neurosci. 2009, 29, 9210–9218. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Hamamichi, S.; Lee, B.D.; Yang, D.; Ray, A.; Caldwell, G.A.; Caldwell, K.A.; Dawson, T.M.; Smith, W.W.; Dawson, V.L. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum. Mol. Genet. 2011, 20, 3933–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longinetti, E.; Fang, F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Saánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.D.; Ranum, L.P. New developments in RAN translation: Insights from multiple diseases. Curr. Opin. Genet. Dev. 2017, 44, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Rudich, P.; Snoznik, C.; Watkins, S.C.; Monaghan, J.; Pandey, U.B.; Lamitina, S.T. Nuclear localized C9orf72-associated arginine-containing dipeptides exhibit age-dependent toxicity in C. elegans. Hum. Mol. Genet. 2017, 26, 4916–4928. [Google Scholar] [CrossRef] [Green Version]
- van Blitterswijk, M.; DeJesus-Hernandez, M.; Niemantsverdriet, E.; Murray, M.E.; Heckman, M.G.; Diehl, N.N.; Brown, P.H.; Baker, M.C.; Finch, N.A.; Bauer, P.O.; et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): A cross-sectional cohort study. Lancet Neurol. 2013, 12, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Therrien, M.; Rouleau, G.A.; Dion, P.A.; Parker, J.A. Deletion of C9ORF72 Results in Motor Neuron Degeneration and Stress Sensitivity in C. elegans. PLoS ONE 2013, 8, e83450. [Google Scholar] [CrossRef] [Green Version]
- Corrionero, A.; Horvitz, H.R. A C9orf72 ALS/FTD Ortholog Acts in Endolysosomal Degradation and Lysosomal Homeostasis. Curr. Biol. 2018, 28, 1522–1535. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hao, L.; Saur, T.; Joyal, K.; Zhao, Y.; Zhai, D.; Li, J.; Pribadi, M.; Coppola, G.; Cohen, B.M.; et al. Forward Genetic Screen in Caenorhabditis elegans Suggests F57A10.2 and acp-4 As Suppressors of C9ORF72 Related Phenotypes. Front. Mol. Neurosci. 2016, 9, 113. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Model. Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxidative Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef]
- Landis, G.N.; Tower, J. Superoxide dismutase evolution and life span regulation. Mech. Ageing Dev. 2005, 126, 365–379. [Google Scholar] [CrossRef]
- Oeda, T.; Shimohama, S.; Kitagawa, N.; Kohno, R.; Imura, T.; Shibasaki, H.; Ishii, N. Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum. Mol. Genet. 2001, 10, 2013–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Farr, G.W.; Hall, D.H.; Li, F.; Furtak, K.; Dreier, L.; Horwich, A.L. An ALS-Linked Mutant SOD1 Produces a Locomotor Defect Associated with Aggregation and Synaptic Dysfunction When Expressed in Neurons of Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Huang, K.-X.; Le, W.-D. Establishing a novel C. elegans model to investigate the role of autophagy in amyotrophic lateral sclerosis. Acta Pharmacol. Sin. 2013, 34, 644–650. [Google Scholar] [CrossRef] [Green Version]
- Baskoylu, S.N.; Yersak, J.; O’Hern, P.; Grosser, S.; Simon, J.; Kim, S.; Schuch, K.; Dimitriadi, M.; Yanagi, K.S.; Lins, J.; et al. Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 2018, 14, e1007682. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Jia, C.; Cheng, C.; Wu, H.; Cai, H.; Le, W. Activation of autophagy attenuates motor deficits and extends lifespan in a C. elegans model of ALS. Free. Radic. Biol. Med. 2022, 181, 52–61. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Meneses, A.; Koga, S.; O’Leary, J.; Dickson, D.W.; Bu, G.; Na Zhao, N. TDP-43 Pathology in Alzheimer’s Disease. Mol. Neurodegener. 2021, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hwang, H.-Y.; Hao, H.; Talbot, C., Jr.; Wang, J. Caenorhabditis elegans RNA-processing Protein TDP-1 Regulates Protein Homeostasis and Life Span. J. Biol. Chem. 2012, 287, 8371–8382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaccaro, A.; Tauffenberger, A.; Ash, P.E.A.; Carlomagno, Y.; Petrucelli, L.; Parker, J.A. TDP-1/TDP-43 Regulates Stress Signaling and Age-Dependent Proteotoxicity in Caenorhabditis elegans. PLoS Genet. 2012, 8, e1002806. [Google Scholar] [CrossRef] [Green Version]
- Vaccaro, A.; Tauffenberger, A.; Aggad, D.; Rouleau, G.; Drapeau, P.; Parker, J.A. Mutant TDP-43 and FUS Cause Age-Dependent Paralysis and Neurodegeneration in C. elegans. PLoS ONE 2012, 7, e31321. [Google Scholar] [CrossRef]
- Ash, P.E.; Zhang, Y.-J.; Roberts, C.M.; Saldi, T.; Hutter, H.; Buratti, E.; Petrucelli, L.; Link, C.D. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 2010, 19, 3206–3218. [Google Scholar] [CrossRef]
- Liachko, N.F.; Guthrie, C.R.; Kraemer, B.C. Phosphorylation Promotes Neurotoxicity in a Caenorhabditis elegans Model of TDP-43 Proteinopathy. J. Neurosci. 2010, 30, 16208–16219. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.S.; Michel, C.H.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef] [Green Version]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef]
- Yang, L.; Gal, J.; Chen, J.; Zhu, H. Self-assembled FUS binds active chromatin and regulates gene transcription. Proc. Natl. Acad. Sci. USA 2014, 111, 17809–17814. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Blechingberg, J.; Fernandes, A.M.; Li, S.; Fryland, T.; Børglum, A.D.; Bolund, L.; Nielsen, A.L. EWS and FUS bind a subset of transcribed genes encoding proteins enriched in RNA regulatory functions. BMC Genom. 2015, 16, 929. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, J.C.; Ebmeier, C.C.; Podell, E.R.; Heimiller, J.; Taatjes, D.J.; Cech, T.R. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012, 26, 2690–2695. [Google Scholar] [CrossRef] [Green Version]
- Therrien, M.; Rouleau, G.A.; Dion, P.A.; Parker, J.A. FET proteins regulate lifespan and neuronal integrity. Sci. Rep. 2016, 6, 25159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Yang, S.-P.; Xie, L.; Kawano, T.; Fu, D.; Mukai, A.; Bohm, C.; Chen, F.; Robertson, J.; Suzuki, H.; et al. ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum. Mol. Genet. 2011, 21, 1–9. [Google Scholar] [CrossRef]
- Jorgensen, E.M. Gaba. WormBook 2005, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Labarre, A.; Tossing, G.; Maios, C.; Doyle, J.J.; Parker, J.A. A single copy transgenic mutant FUS strain reproduces age-dependent ALS phenotypes in C. elegans. Micropublication Biol. 2021, 2021. [Google Scholar] [CrossRef]
- Markert, S.M.; Skoruppa, M.; Yu, B.; Mulcahy, B.; Zhen, M.; Gao, S.; Sendtner, M.; Stigloher, C. Overexpression of an ALS-associated FUS mutation in C. elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol. Open 2020, 9, bio055129. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and Neuropathology of Huntington’s Disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar] [CrossRef] [Green Version]
- Duyao, M.; Ambrose, C.; Myers, R.; Novelletto, A.; Persichetti, F.; Frontali, M.; Folstein, S.; Ross, C.; Franz, M.; Abbott, M.; et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Genet. 1993, 4, 387–392. [Google Scholar] [CrossRef]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [Green Version]
- Faber, P.W.; Alter, J.R.; MacDonald, M.E.; Hart, A.C. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc. Natl. Acad. Sci. USA 1999, 96, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.A.; Connolly, J.B.; Wellington, C.; Hayden, M.; Dausset, J.; Neri, C. Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc. Natl. Acad. Sci. USA 2001, 98, 13318–13323. [Google Scholar] [CrossRef] [Green Version]
- Cordeiro, L.M.; Machado, M.L.; da Silva, A.F.; Baptista, F.B.O.; da Silveira, T.L.; Soares, F.A.A.; Arantes, L.P. Rutin protects Huntington’s disease through the insulin/IGF1 (IIS) signaling pathway and autophagy activity: Study in Caenorhabditis elegans model. Food Chem. Toxicol. 2020, 141, 111323. [Google Scholar] [CrossRef]
- Cordeiro, L.M.; Soares, M.V.; da Silva, A.F.; Machado, M.L.; Baptista, F.B.O.; da Silveira, T.L.; Arantes, L.P.; Soares, F.A.A. Neuroprotective effects of rutin on ASH neurons in Caenorhabditis elegans model of Huntington’s disease. Nutr. Neurosci. 2022, 25, 2288–2301. [Google Scholar] [CrossRef]
- Jia, K.; Hart, A.C.; Levine, B. Autophagy Genes Protect Against Disease Caused by Polyglutamine Expansion Proteins in Caenorhabditis elegans. Autophagy 2007, 3, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Baptista, F.B.O.; Arantes, L.P.; Machado, M.L.; da Silva, A.F.; Cordeiro, L.M.; da Silveira, T.L.; Soares, F.A.A. Diphenyl diselenide protects a Caenorhabditis elegans model for Huntington’s disease by activation of the antioxidant pathway and a decrease in protein aggregation. Metallomics 2020, 12, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Tauffenberger, A.; Vaccaro, A.; Aulas, A.; Velde, C.V.; Parker, J.A. Glucose delays age-dependent proteotoxicity. Aging Cell 2012, 11, 856–866. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.L.; Ung, H.M.; Sands, L.P.; Kikis, E.A. A new Caenorhabditis elegans model of human huntingtin 513 aggregation and toxicity in body wall muscles. PLoS ONE 2017, 12, e0173644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiela, E.; Rudich, P.D.; Traa, A.; Anglas, U.; Soo, S.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Targeting Mitochondrial Network Disorganization is Protective in C. elegans Models of Huntington’s Disease. Aging Dis. 2021, 12, 1753. [Google Scholar] [CrossRef]
- Traa, A.; Machiela, E.; Rudich, P.D.; Soo, S.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Identification of Novel Therapeutic Targets for Polyglutamine Diseases That Target Mitochondrial Fragmentation. Int. J. Mol. Sci. 2021, 22, 13447. [Google Scholar] [CrossRef]
- Fan, H.-C.; Ho, L.-I.; Chi, C.-S.; Chen, S.-J.; Peng, G.-S.; Chan, T.-M.; Lin, S.-Z.; Harn, H.-J. Polyglutamine (PolyQ) Diseases: Genetics to Treatments. Cell Transplant. 2014, 23, 441–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikkineth, A.C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017, 33, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. Mitochondrial deficiency in Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 275–283. [Google Scholar] [CrossRef] [Green Version]
- van der Horst, G.T.; Meira, L.; Gorgels, T.G.; de Wit, J.; Velasco-Miguel, S.; Richardson, J.A.; Kamp, Y.; Vreeswijk, M.P.; Smit, B.; Bootsma, D.; et al. UVB radiation-induced cancer predisposition in Cockayne syndrome group A (Csa) mutant mice. DNA Repair 2002, 1, 143–157. [Google Scholar] [CrossRef]
- Liakos, A.; Lavigne, M.D.; Fousteri, M. Nucleotide Excision Repair: From Neurodegeneration to Cancer. Adv. Exp. Med. Biol. 2017, 1007, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Ahn, B.; Choi, I.S.; Koo, H.-S. The gene expression and deficiency phenotypes of Cockayne syndrome B protein in Caenorhabditis elegans. FEBS Lett. 2002, 522, 47–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, V.; Hofmann, K.; Schumacher, B. A C. elegans homolog of the Cockayne syndrome complementation group A gene. DNA Repair 2014, 24, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, M.M.; Castells-Roca, L.; Babu, V.; Ermolaeva, M.A.; Müller, R.-U.; Frommolt, P.; Williams, A.B.; Greiss, S.; Schneider, J.I.; Benzing, T.; et al. DAF-16/FOXO and EGL-27/GATA promote developmental growth in response to persistent somatic DNA damage. Nature 2014, 16, 1168–1179. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.F.C.; Bozek, K.; Herholz, M.; Trifunovic, A.; Rieckher, M.; Schumacher, B. A C. elegans model for neurodegeneration in Cockayne syndrome. Nucleic Acids Res. 2020, 48, 10973–10985. [Google Scholar] [CrossRef]
- Liakos, A.; Konstantopoulos, D.; Lavigne, M.D.; Fousteri, M. Continuous transcription initiation guarantees robust repair of all transcribed genes and regulatory regions. Nat. Commun. 2020, 11, 916. [Google Scholar] [CrossRef] [Green Version]
- Babu, V.; Schumacher, B. A C. elegans homolog for the UV-hypersensitivity syndrome disease gene UVSSA. DNA Repair 2016, 41, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Zaninello, M.; Palikaras, K.; Naon, D.; Iwata, K.; Herkenne, S.; Quintana-Cabrera, R.; Semenzato, M.; Grespi, F.; Ross-Cisneros, F.N.; Carelli, V.; et al. Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat. Commun. 2020, 11, 4029. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Votruba, M.; Pesch, U.E.A.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Griffoin, J.-M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Zaninello, M.; Palikaras, K.; Sotiriou, A.; Tavernarakis, N.; Scorrano, L. Sustained intracellular calcium rise mediates neuronal mitophagy in models of autosomal dominant optic atrophy. Cell Death Differ. 2021, 29, 167–177. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitophagy in neurodegeneration and aging. Front. Genet. 2012, 3, 297. [Google Scholar] [CrossRef] [Green Version]
- Weinhouse, C.; Truong, L.; Meyer, J.N.; Allard, P. Caenorhabditis elegans as an emerging model system in environmental epigenetics. Environ. Mol. Mutagen. 2018, 59, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Maloy, S.R.; Hughes, K.T. Brenner’s Encyclopedia of Genetics, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- O’Reilly, L.P.; Luke, C.J.; Perlmutter, D.H.; Silverman, G.A.; Pak, S.C. C. elegans in high-throughput drug discovery. Adv. Drug Deliv. Rev. 2013, 69–70, 247–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Zhao, Y.; Chen, Y.; Cheng, B.; Peng, A.; Huang, K. Caenorhabditis elegans as a model system for target identification and drug screening against neurodegenerative diseases. Eur. J. Pharmacol. 2018, 819, 169–180. [Google Scholar] [CrossRef]
- McIntyre, R.L.; Molenaars, M.; Schomakers, B.V.; Gao, A.W.; Kamble, R.; Jongejan, A.; van Weeghel, M.; van Kuilenburg, A.B.; Possemato, R.; Houtkooper, R.H.; et al. Anti-retroviral treatment with zidovudine alters pyrimidine metabolism, reduces translation, and extends healthy longevity via ATF-4. Cell Rep. 2022, 42, 111928. [Google Scholar] [CrossRef]
- Chen, X.; Barclay, J.W.; Burgoyne, R.D.; Morgan, A. Using C. elegans to discover therapeutic compounds for ageing-associated neurodegenerative diseases. Chem. Central J. 2015, 9, 65. [Google Scholar] [CrossRef] [Green Version]
- Patten, S.A.; Aggad, D.; Martinez, J.; Tremblay, E.; Petrillo, J.; Armstrong, G.A.; La Fontaine, A.; Maios, C.; Liao, M.; Ciura, S.; et al. Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight 2017, 2, e97152. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.Q.; Pontifex, M.G.; Phelan, M.M.; Pidathala, C.; Kraemer, B.C.; Barclay, J.W.; Berry, N.G.; O’Neill, P.M.; Burgoyne, R.D.; Morgan, A. α-Methyl-α-phenylsuccinimide ameliorates neurodegeneration in a C. elegans model of TDP-43 proteinopathy. Neurobiol. Dis. 2018, 118, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Matlack, K.E.S.; Tardiff, D.F.; Narayan, P.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Clioquinol promotes the degradation of metal-dependent amyloid-β (Aβ) oligomers to restore endocytosis and ameliorate Aβ toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 4013–4018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujols, J.; Peña-Díaz, S.; Lázaro, D.F.; Peccati, F.; Pinheiro, F.; González, D.; Carija, A.; Navarro, S.; Conde-Giménez, M.; García, J.; et al. Small molecule inhibits α-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, K.A.; Hu, C.J.; Pan, H.; Lu, J.; Abskharon, R.; Bowler, J.T.; Rosenberg, G.M.; Williams, C.K.; Elezi, G.; Balbirnie, M.; et al. Small molecules disaggregate alpha-synuclein and prevent seeding from patient brain-derived fibrils. Proc. Natl. Acad. Sci. USA 2023, 120, e2217835120. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Hegarty, E.; Martin, C.; Gökçe, S.K.; Ghorashian, N.; Ben-Yakar, A. Large-scale microfluidics providing high-resolution and high-throughput screening of Caenorhabditis elegans poly-glutamine aggregation model. Nat. Commun. 2016, 7, 13023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohrabi, S.; Mor, D.E.; Kaletsky, R.; Keyes, W.; Murphy, C.T. High-throughput behavioral screen in C. elegans reveals Parkinson’s disease drug candidates. Commun. Biol. 2021, 4, 203. [Google Scholar] [CrossRef]
- Xie, C.; Zhuang, X.-X.; Niu, Z.; Ai, R.; Lautrup, S.; Zheng, S.; Jiang, Y.; Han, R.; Gupta, T.S.; Cao, S.; et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat. Biomed. Eng. 2022, 6, 76–93. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Schellenberg, G.D. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum. Mol. Genet. 2007, 16, 1959–1971. [Google Scholar] [CrossRef]
- Knight, A.L.; Yan, X.; Hamamichi, S.; Ajjuri, R.R.; Mazzulli, J.R.; Zhang, M.W.; Daigle, J.G.; Zhang, S.; Borom, A.R.; Roberts, L.R.; et al. The Glycolytic Enzyme, GPI, Is a Functionally Conserved Modifier of Dopaminergic Neurodegeneration in Parkinson’s Models. Cell Metab. 2014, 20, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.-C.; et al. α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Yacoubian, T.A.; Wilson, S.; Xie, Z.-L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol. Brain 2008, 1, 17–18. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roussos, A.; Kitopoulou, K.; Borbolis, F.; Palikaras, K. Caenorhabditis elegans as a Model System to Study Human Neurodegenerative Disorders. Biomolecules 2023, 13, 478. https://doi.org/10.3390/biom13030478
Roussos A, Kitopoulou K, Borbolis F, Palikaras K. Caenorhabditis elegans as a Model System to Study Human Neurodegenerative Disorders. Biomolecules. 2023; 13(3):478. https://doi.org/10.3390/biom13030478
Chicago/Turabian StyleRoussos, Antonis, Katerina Kitopoulou, Fivos Borbolis, and Konstantinos Palikaras. 2023. "Caenorhabditis elegans as a Model System to Study Human Neurodegenerative Disorders" Biomolecules 13, no. 3: 478. https://doi.org/10.3390/biom13030478