
Mitochondrial Neurodegeneration: Lessons from Drosophila melanogaster Models

Abstract
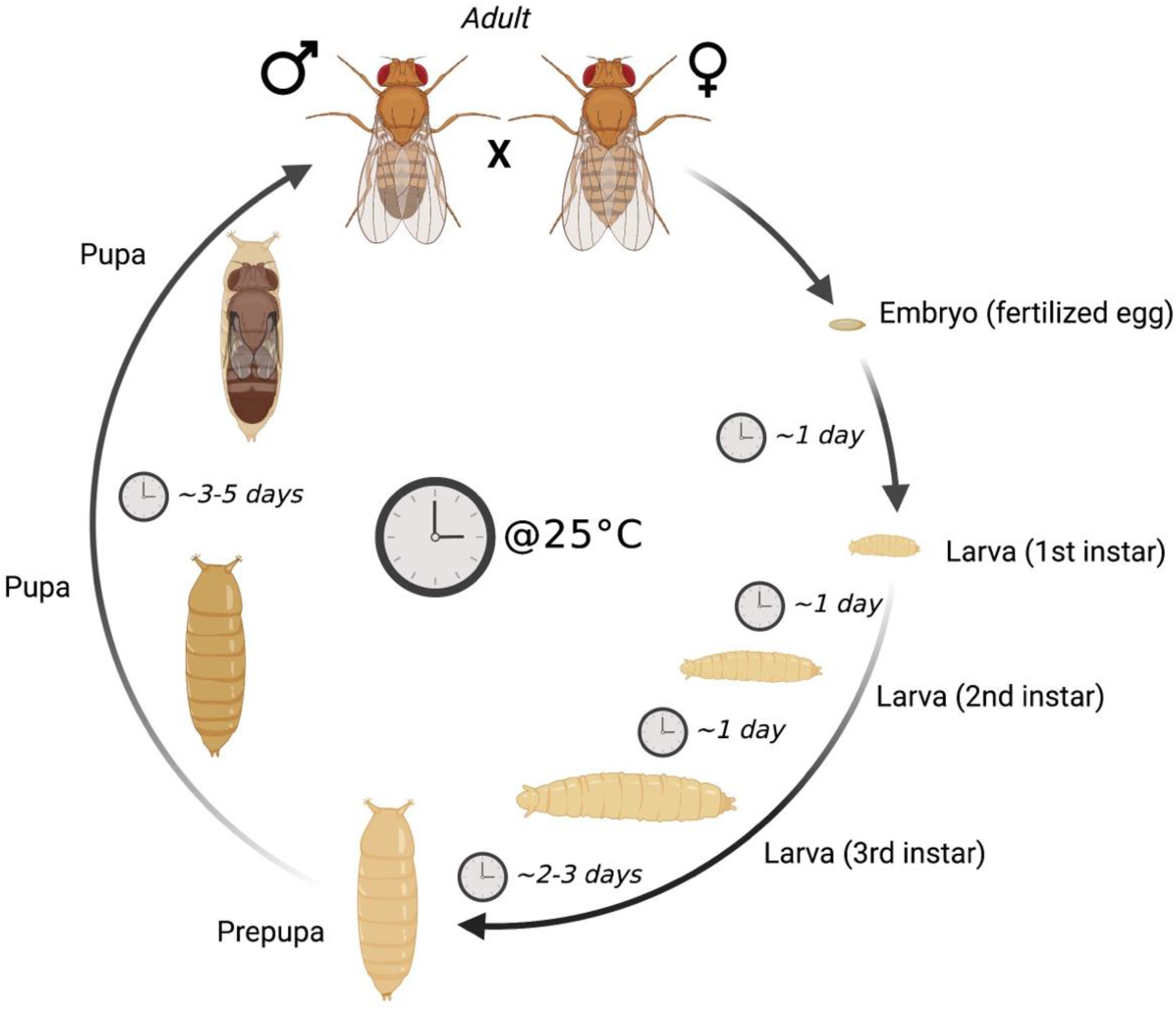
:1. Drosophila melanogaster as a Model Organism to Study Disease
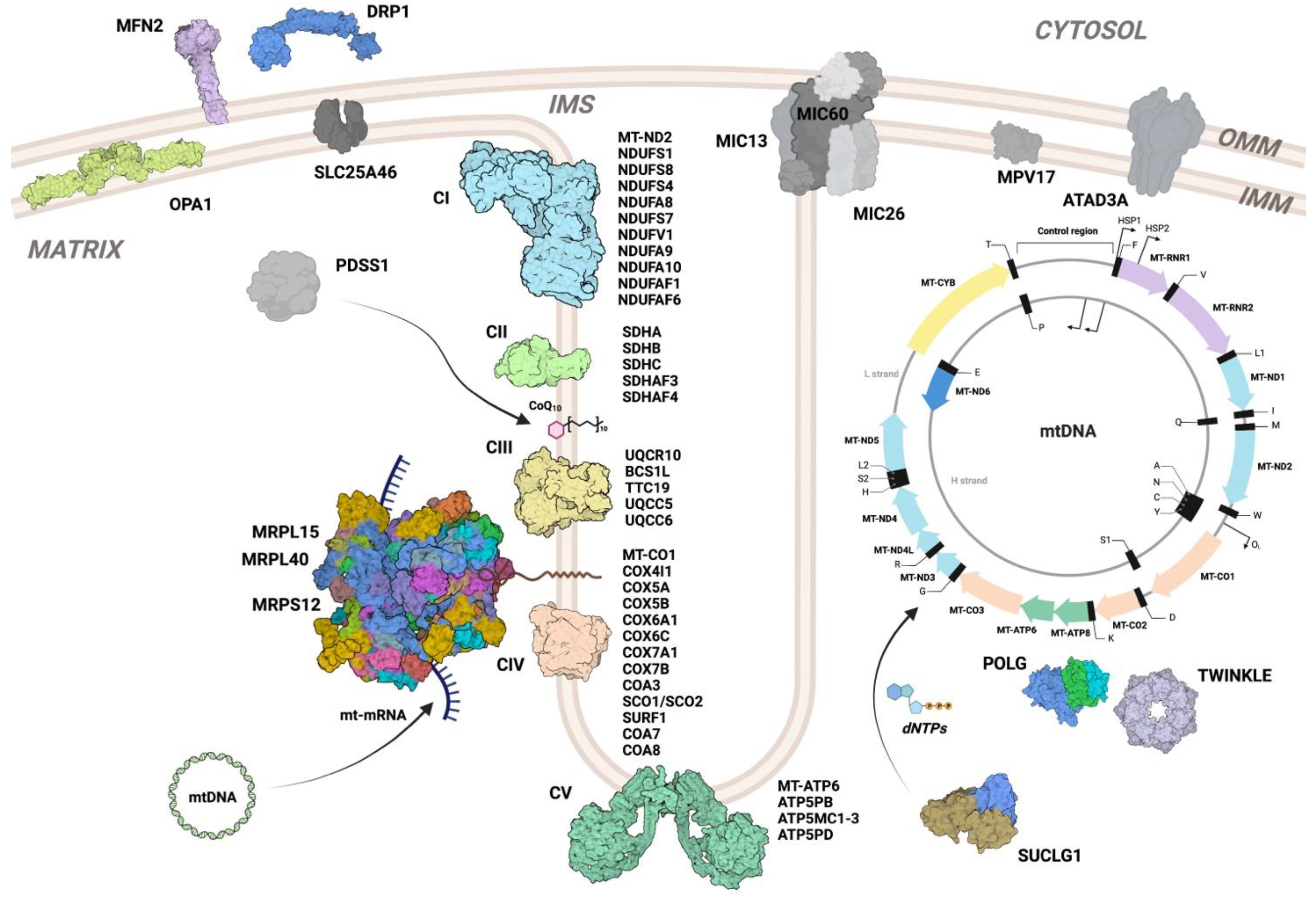
2. Mitochondrial Diseases
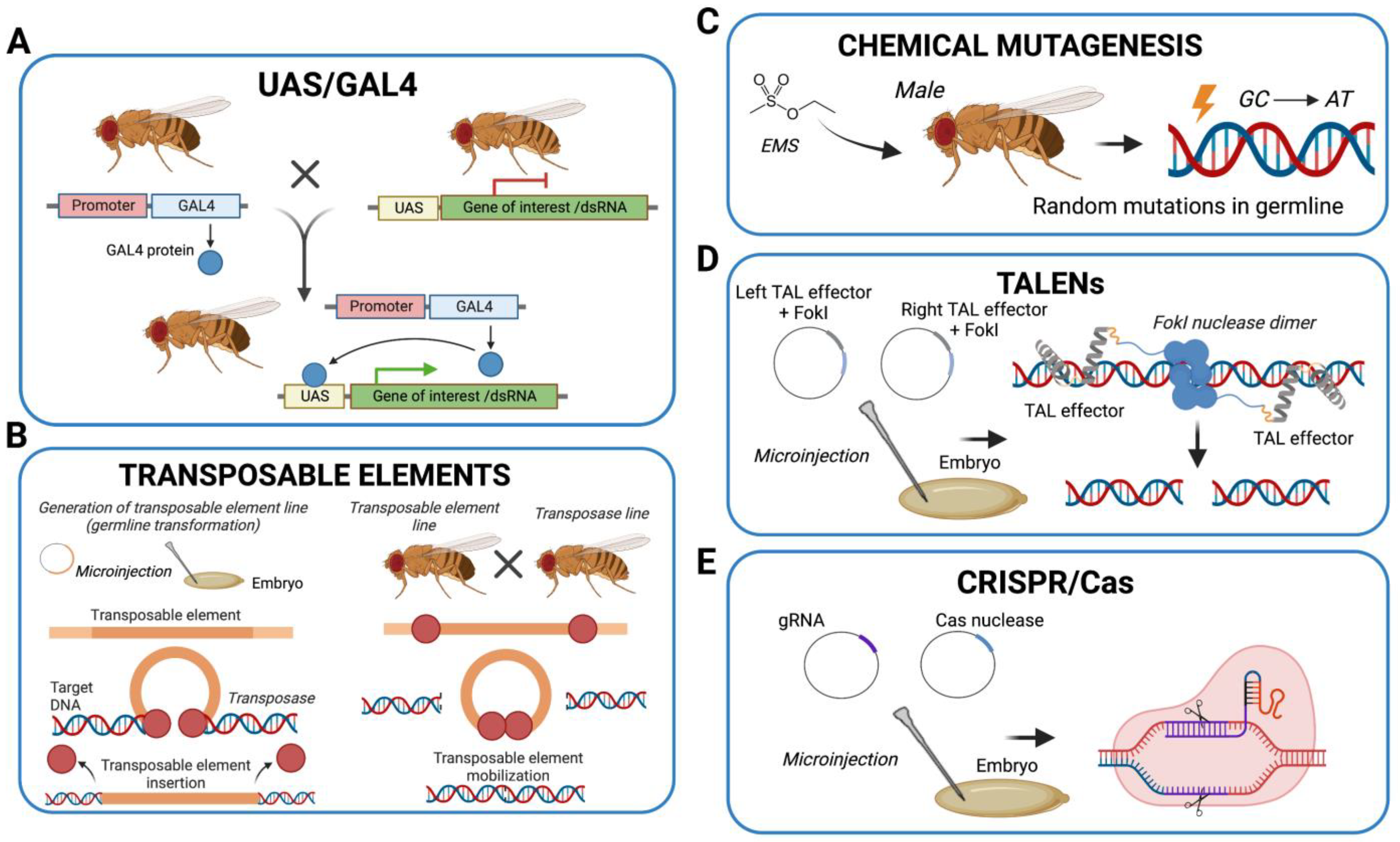
3. Models of Mitochondrial Disease in Drosophila melanogaster
3.1. Fly Models of Complex I Defects
3.2. Fly Models of Complex II Defects
3.3. Fly Models of Complex III Defects
3.4. Fly Models of Complex IV Defects
3.5. Fly Models of Complex V Defects
3.6. Coenzyme Q Deficiency Models

{kind=link}
{kind=link}
{kind=link}
| Fly Gene | Human Ortholog | Function | System | Tissue Specificity | Phenotype | Ref. | |
|---|---|---|---|---|---|---|---|
| Complex I | mt:ND2 | MT-ND2 | Core subunit | Restriction enzymes targeting mtDNA | Ubiquitous | Neuromotor dysfunction, neurodegeneration | [29] |
| ND-75 | NDUFS1 | Core subunit | RNAi | Glia | Neurodegeneration | [28] | |
| RNAi | Ubiquitous | Neurodegeneration | |||||
| RNAi | Neurons | Reduced lifespan | |||||
| ND-23 | NDUFS8 | Core subunit | RNAi | Glia | Neurodegeneration | [30] | |
| RNAi | Ubiquitous | Developmental arrest | |||||
| RNAi | Neurons | Reduced lifespan, neuromotor dysfunction | |||||
| ND-20 | NDUFS7 | Core subunit | RNAi | Ubiquitous | Array of phenotypes depending on RNAi efficiency | [31] | |
| ND-51 | NDUFV1 | Core subunit | RNAi | Ubiquitous | Developmental arrest | ||
| ND-19 | NDUFA8 | Supernumerary subunit | RNAi | Ubiquitous | Developmental arrest | [32] | |
| ND-39 | NDUFA9 | Supernumerary subunit | RNAi | Ubiquitous | Developmental arrest | [32] | |
| ND-42 | NDUFA10 | Supernumerary subunit | RNAi | Ubiquitous | Developmental arrest | [33] | |
| RNAi | Eye | Retinal degeneration | |||||
| Sicily | NDUFAF6 | Assembly factor | FLP/FRT system | Mosaic eye | Retinal degeneration, neurodegeneration | [33] | |
| Transposable elements | Ubiquitous | Developmental arrest | |||||
| ND-18 | NDUFS4 | Supernumerary subunit | RNAi | Ubiquitous | Array of phenotypes depending on RNAi efficiency | [31]; [34] | |
| CIA30 | NDUFAF1 | Assembly factor | Transposable elements | Ubiquitous | Developmental arrest | [38] | |
| RNAi | Ubiquitous | Reduced growth, partial developmental lethality | |||||
| Complex II | SdhA | SDHA | Subunit | FLP/FRT system | Mosaic eye | Retinal degeneration | [41] |
| FLP/FRT system | Ubiquitous | Developmental arrest | |||||
| SdhB | SDHB | Subunit | Transposable elements | Ubiquitous | Reduced lifespan, sensitivity to hyperoxia, age-related neuromotor dysfunction | [43] | |
| SdhC | SDHC | Subunit | Overexpression of dominant negative mutation | Neuronal | Reduced lifespan, oxidative damage | [44] | |
| Sirup/Sdhaf4 | SDHAF4 | Assembly factor | TALENs | Ubiquitous | Reduced lifespan, neurodegeneration, sensitivity to oxidative stress | [45] | |
| Sdhaf3 | SDHAF3 | Assembly factor | Homologous recombination | Ubiquitous | Sensitivity to oxidative stress and hyperoxia, age-related neuromotor dysfunction | [46] | |
| Ttc19 | TTC19 | Assembly factor | Transposable elements | Ubiquitous | Neuromotor dysfunction | [49] | |
| CRISPR/Cas9 KO | Ubiquitous | Neuromotor dysfunction | [50] | ||||
| Bcs1 | BCS1L | Assembly factor | RNAi | Ubiquitous | Developmental arrest, larval neuromotor dysfunction | [51] | |
| RNAi | Neurons | Reduced lifespan, neuromotor dysfunction, paralysis | |||||
| RNAi | Muscle | Developmental arrest | |||||
| RNAi | Fat body | Reduced lifespan | |||||
| Ox | UQCR10 | Supernumerary subunit | Transposable elements | Ubiquitous | Developmental arrest | [52] | |
| sloth1 | SMIM4/ UQCC5 | Assembly factor | RNAi | Ubiquitous | Developmental lethality, neurodegeneration | [56] | |
| CRISPR/Cas9 KO | Ubiquitous (somatic) | Developmental lethality, neurodegeneration | |||||
| CRISPR/Cas9 KO | Ubiquitous (germline) | Developmental lethality, neurodegeneration | |||||
| sloth2 | Brawnin/ UQCC6 | Assembly factor | RNAi | Ubiquitous | Developmental lethality, neurodegeneration | [56] | |
| CRISPR/Cas9 KO | Ubiquitous (somatic) | Developmental lethality, neurodegeneration | |||||
| CRISPR/Cas9 KO | Ubiquitous (germline) | Developmental lethality, neurodegeneration | |||||
| Complex IV | mt:CoI | MT-CO1 | Core subunit | Mitochondrially targeted restriction enzymes | Ubiquitous | Reduced growth, neurodegeneration | [36] |
| COX7B | COX7B | Supernumerary subunit | RNAi | Ubiquitous | Developmental arrest | [64] | |
| cype/ COX6C | COX6C | Supernumerary subunit | FLP/FRT system | Eye | Retinal degeneration | [66] | |
| FLP/FRT system | Germline | Developmental arrest | |||||
| RNAi | Ubiquitous | Developmental arrest | [81] | ||||
| COX5A | COX5A | Supernumerary subunit | FLP/FRT system | Eye | Retinal degeneration | [67] | |
| RNAi | Ubiquitous | Developmental arrest | [70] | ||||
| levy/ COX6A | COX6A1 | Supernumerary subunit | Chemical mutagenesis | Ubiquitous | Temperature-induced paralysis, bang-induced paralysis, neurodegeneration, reduced lifespan | [68] | |
| RNAi | Ubiquitous | Developmental lethality | [70] | ||||
| COX4 | COX4I1 | Supernumerary subunit | RNAi | Ubiquitous | Developmental arrest (strong RNAi), reduced lifespan (mild RNAi) | [69] | |
| COX5B | COX5B | Supernumerary subunit | RNAi | Ubiquitous | Developmental arrest | [69] | |
| RNAi | Ubiquitous | Developmental arrest | [70] | ||||
| COX7A | COX7A1 | Supernumerary subunit | RNAi | Ubiquitous | Developmental arrest | [70] | |
| Ccdc56/Coa3 | COA3 | Assembly factor | Transposable elements | Ubiquitous | Developmental arrest | [71] | |
| Scox | SCO1/ SCO2 | Assembly factor | Transposable elements | Ubiquitous | Developmental arrest | [72] | |
| RNAi | Ubiquitous | Developmental arrest | [73] | ||||
| RNAi | Heart | Reduced lifespan, cardiac dysfunction | [74] | ||||
| RNAi | Glia | Neuromotor dysfunction | [80] | ||||
| Surf1 | SURF1 | Assembly factor | RNAi | Ubiquitous | Developmental arrest | [75]; [76] | |
| RNAi | Neurons | Mild neuromotor defects | [75] | ||||
| RNAi | Muscle | Developmental arrest | [76] | ||||
| Coa7 | COA7 | Assembly factor | RNAi | Eye | Retinal degeneration | [77] | |
| RNAi | Neurons | Reduced lifespan, neuromotor dysfunction | |||||
| Coa8 | COA8 | Assembly factor | RNAi | Ubiquitous | Sensitivity to oxidative stress, neuromotor dysfunction | [78] | |
| RNAi | Neurons | Sensitivity to oxidative stress, neuromotor dysfunction | |||||
| Complex V | mt:ATPase6 | MT-ATP6 | Core subunit | Isolation of spontaneous mutation | Ubiquitous | Reduced lifespan, progressive neurodegeneration | [84] |
| ATPsynD | ATP5PD | Core subunit | RNAi | Ubiquitous | Developmental arrest | [85] | |
| ATPsynB | ATP5PB | Core subunit | RNAi | Ubiquitous | Developmental arrest | [87] | |
| ATPsynC | ATP5MC1/ATP5MC2/ATP5MC3 | Core subunit | Transposable elements, chemical mutagenesis | Ubiquitous | Range of phenotypes depending on the severity of the genetic lesion | [86] | |
| CoQ | qless | PDSS1 | CoQ biosynthesis | Chemical mutagenesis | Ubiquitous | Developmental arrest | [90] |
| FLP/FRT system | Neurons | Neurodegeneration |
3.7. Defects in Mitochondrial DNA Replication and Maintenance
3.8. Defects in Mitochondrial Gene Expression
3.9. Defects in Mitochondrial Dynamics and Architecture
| Fly Gene | Human Ortholog | Function | System | Tissue Specificity | Phenotype | Ref. | |
|---|---|---|---|---|---|---|---|
| mtDNA replication and maintenance | PolG1/tam | POLG | mtDNA replication | Chemical mutagenesis | Ubiquitous | Developmental arrest, neuromotor dysfunction | [93] |
| Homologous recombination | Ubiquitous | Developmental arrest, reduced growth | [94] | ||||
| RNAi | Ubiquitous | Developmental arrest | [95] | ||||
| KI of PolG1 exo− (mutator) | Ubiquitous | Developmental lethality in homozygosity, increased mtDNA mutation rate in heterozygosity | [94] | ||||
| Transgenic PolG1 exo− (mutator) | Ubiquitous | Reduced lifespan, dose-dependent increase in mtDNA mutation rate | [96] | ||||
| RNAi | Ubiquitous | Partial developmental lethality | [95] | ||||
| mtDNA-helicase | TWNK | mtDNA replication | Transgenic expression of dominant mutations | Ubiquitous | Developmental arrest and mtDNA depletion | [101] | |
| bor | ATAD3A | Component of nucleoids | Transgenic expression of dominant mutation | Ubiquitous | Developmental arrest | [103] | |
| Neurons | Developmental arrest | ||||||
| Muscle-specific | Partial developmental lethality | ||||||
| Transposable elements | Ubiquitous | Developmental arrest | [106] | ||||
| SCSα1 | SUCLG1 | Mitochondrial nucleotide synthesis | CRISPR/Cas9 | Ubiquitous | Developmental delay, altered neuromotor function, and reduced lifespan under starvation | [110] | |
| Mitochondrial translation | tko | MRPS12 | Mitoribosome small subunit | Chemical mutagenesis | Ubiquitous | Bang-induced paralysis, developmental delay, sensitivity to doxycycline | [117]; [118] |
| mRpL15 | MRPL15 | Mitoribosome large subunit | RNAi | Neurons | Disruption of synapse development and function | [119] | |
| mRpL40 | MRPL40 | Mitoribosome large subunit | RNAi | Neurons | Disruption of synapse development and function | [119] | |
| Mitochondrial dynamics/architecture | Drp1 | DRP1 | Mitochondrial fission | Transposable elements | Ubiquitous | Partial developmental lethality, altered neuromotor function | [126] |
| FLP/FRT system | Spermatocytes | Altered spermatogenesis and sperm motility | [127] | ||||
| Opa1 | OPA1 | Mitochondrial fusion | FLP/FRT system | Eye | Retinal degeneration | [128] | |
| Transposable elements | Ubiquitous | Developmental arrest | [128] | ||||
| RNAi | Heart | Cardiomyopathy | [131] | ||||
| Marf | MFN1/2 | Mitochondrial fusion | RNAi | Heart | Cardiomyopathy | [131] | |
| Mic26-27 | MIC26-27 | Cristae architecture | Transposable elements | Ubiquitous | Partial developmental lethality, reduced lifespan, reduced neuromotor function | [138] | |
| Mitofilin | IMMT/ MIC60 | Cristae architecture | Transposable elements | Ubiquitous | Developmental arrest | [139] | |
| RNAi | Muscle | Mild neuromotor defects | |||||
| RNAi | Neurons | Mild neuromotor defects | |||||
| Slc25A46a | SLC25A46 | Mitochondrial dynamics | RNAi | Neurons | Neuromotor dysfunction | [146] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A Systematic Analysis of Human Disease-Associated Gene Sequences in Drosophila Melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, U.B.; Nichols, C.D. Human Disease Models in Drosophila Melanogaster and the Role of the Fly in Therapeutic Drug Discovery. Pharmacol. Rev. 2011, 63, 411–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellen, H.J.; Levis, R.W.; Liao, G.; He, Y.; Carlson, J.W.; Tsang, G.; Evans-Holm, M.; Hiesinger, P.R.; Schulze, K.L.; Rubin, G.M.; et al. The BDGP Gene Disruption Project: Single Transposon Insertions Associated with 40% of Drosophila Genes. Genetics 2004, 167, 761–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellen, H.J.; Levis, R.W.; He, Y.; Carlson, J.W.; Evans-Holm, M.; Bae, E.; Kim, J.; Metaxakis, A.; Savakis, C.; Schulze, K.L.; et al. The Drosophila Gene Disruption Project: Progress Using Transposons with Distinctive Site Specificities. Genetics 2011, 188, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Brand, A.H.; Perrimon, N. Targeted Gene Expression as a Means of Altering Cell Fates and Generating Dominant Phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef]
- Dietzl, G.; Chen, D.; Schnorrer, F.; Su, K.-C.; Barinova, Y.; Fellner, M.; Gasser, B.; Kinsey, K.; Oppel, S.; Scheiblauer, S.; et al. A Genome-Wide Transgenic RNAi Library for Conditional Gene Inactivation in Drosophila. Nature 2007, 448, 151–156. [Google Scholar] [CrossRef]
- Caygill, E.E.; Brand, A.H. The GAL4 System: A Versatile System for the Manipulation and Analysis of Gene Expression. Methods Mol. Biol. 2016, 1478, 33–52. [Google Scholar] [CrossRef]
- Liu, J.; Li, C.; Yu, Z.; Huang, P.; Wu, H.; Wei, C.; Zhu, N.; Shen, Y.; Chen, Y.; Zhang, B.; et al. Efficient and Specific Modifications of the Drosophila Genome by Means of an Easy TALEN Strategy. J. Genet. Genom. 2012, 39, 209–215. [Google Scholar] [CrossRef]
- Bassett, A.; Liu, J.L. CRISPR/Cas9 Mediated Genome Engineering in Drosophila. Methods 2014, 69, 128–136. [Google Scholar] [CrossRef]
- Housden, B.E.; Lin, S.; Perrimon, N. Cas9-Based Genome Editing in Drosophila. Methods Enzymol. 2014, 546, 415–439. [Google Scholar] [CrossRef]
- Gratz, S.J.; Rubinstein, C.D.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. CRISPR-Cas9 Genome Editing in Drosophila. Curr. Protoc. Mol. Biol. 2015, 2015, 31.2.1–31.2.20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, J.A.; Birchak, G.; Perrimon, N. Precise Genome Engineering in Drosophila Using Prime Editing. Proc. Natl. Acad. Sci. USA 2020, 118, e2021996118. [Google Scholar] [CrossRef]
- Marr, E.; Potter, C.J. Base Editing of Somatic Cells Using CRISPR-Cas9 in Drosophila. CRISPR J. 2021, 4, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Zirin, J.; Bosch, J.; Viswanatha, R.; Mohr, S.E.; Perrimon, N. State-of-the-Art CRISPR for in Vivo and Cell-Based Studies in Drosophila. Trends Genet. 2022, 38, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial Diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Gusic, M.; Prokisch, H. Genetic Basis of Mitochondrial Diseases. FEBS Lett. 2021, 595, 1132–1158. [Google Scholar] [CrossRef]
- Zeviani, M.; Viscomi, C. Mitochondrial Neurodegeneration. Cells 2022, 11, 637. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial Disorders of the OXPHOS System. FEBS Lett. 2021, 595, 1062–1106. [Google Scholar] [CrossRef]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.G.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory Subunits Are Integral for Assembly and Function of Human Mitochondrial Complex i. Nature 2016, 538, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Padavannil, A.; Ayala-Hernandez, M.G.; Castellanos-Silva, E.A.; Letts, J.A. The Mysterious Multitude: Structural Perspective on the Accessory Subunits of Respiratory Complex I. Front. Mol. Biosci. 2022, 8, 1252. [Google Scholar] [CrossRef]
- Rodenburg, R.J. Mitochondrial Complex I-Linked Disease. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.J.; Khajeh, J.; Coulanges, E.; Chen, E.I.-j.; Owusu-Ansah, E. Regulation of Mitochondrial Complex I Biogenesis in Drosophila Flight Muscles. Cell Rep. 2017, 20, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhooms, S.K.; Murari, A.; Goparaju, N.S.V.; Vilanueva, M.; Owusu-Ansah, E. Insights from Drosophila on Mitochondrial Complex I. Cell. Mol. Life Sci. 2020, 77, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Murari, A.; Rhooms, S.K.; Goparaju, N.S.; Villanueva, M.; Owusu-Ansah, E. An Antibody Toolbox to Track Complex I Assembly Defines AIF’s Mitochondrial Function. J. Cell Biol. 2020, 219, e202001071. [Google Scholar] [CrossRef]
- Murari, A.; Rhooms, S.K.; Garcia, C.; Liu, T.; Li, H.; Mishra, B.; Deshong, C.; Owusu-Ansah, E. Dissecting the Concordant and Disparate Roles of NDUFAF3 and NDUFAF4 in Mitochondrial Complex I Biogenesis. iScience 2021, 24, 102869. [Google Scholar] [CrossRef]
- Agip, A.-N.A.; Chung, I.; Sanchez-Martinez, A.; Whitworth, A.J.; Hirst, J. Cryo-EM Structures of Mitochondrial Respiratory Complex I from Drosophila Melanogaster. eLife 2023, 12, e84424. [Google Scholar] [CrossRef]
- Padavannil, A.; Murari, A.; Rhooms, S.-K.; Owusu-Ansah, E.; Letts, J.A. Resting Mitochondrial Complex I from Drosophila Melanogaster Adopts a Helix-Locked State. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hegde, V.R.; Vogel, R.; Feany, M.B. Glia Are Critical for the Neuropathology of Complex I Deficiency in Drosophila. Hum. Mol. Genet. 2014, 23, 4686–4692. [Google Scholar] [CrossRef] [Green Version]
- Burman, J.L.; Itsara, L.S.; Kayser, E.B.; Suthammarak, W.; Wang, A.M.; Kaeberlein, M.; Sedensky, M.M.; Morgan, P.G.; Pallanck, L.J. A Drosophila Model of Mitochondrial Disease Caused by a Complex I Mutation That Uncouples Proton Pumping from Electron Transfer. DMM Dis. Model. Mech. 2014, 7, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Cabirol-Pol, M.J.; Khalil, B.; Rival, T.; Faivre-Sarrailh, C.; Besson, M.T. Glial Lipid Droplets and Neurodegeneration in a Drosophila Model of Complex I Deficiency. Glia 2018, 66, 874–888. [Google Scholar] [CrossRef] [Green Version]
- Foriel, S.; Herma Renkema, G.; Lasarzewski, Y.; Berkhout, J.; Rodenburg, R.J.; Smeitink, J.A.M.; Beyrath, J.; Schenck, A. A Drosophila Mitochondrial Complex i Deficiency Phenotype Array. Front. Genet. 2019, 10, 245. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.; Soikkeli, M.; Portero-Otín, M.; Wilson, A.; Kemppainen, E.; McIlroy, G.; Ellilä, S.; Kemppainen, K.K.; Tuomela, T.; Lakanmaa, M.; et al. Expression of the Yeast NADH Dehydrogenase Ndi1 in Drosophila Confers Increased Lifespan Independently of Dietary Restriction. Proc. Natl. Acad. Sci. USA 2010, 107, 9105–9110. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Li, Z.; Jaiswal, M.; Bayat, V.; Xiong, B.; Sandoval, H.; Charng, W.L.; David, G.; Haueter, C.; Yamamoto, S.; et al. The C8ORF38 Homologue Sicily Is a Cytosolic Chaperone for a Mitochondrial Complex i Subunit. J. Cell Biol. 2013, 200, 807–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foriel, S.; Beyrath, J.; Eidhof, I.; Rodenburg, R.J.; Schenck, A.; Smeitink, J.A.M. Feeding Difficulties, a Key Feature of the Drosophila NDUFS4 Mitochondrial Disease Model. DMM Dis. Model. Mech. 2018, 11, dmm032482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial Dysfunction in Glial Cells: Implications for Neuronal Homeostasis and Survival. Toxicology 2017, 391, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; DeLuca, S.Z.; O’Farrell, P.H. Manipulating the Metazoan Mitochondrial Genome with Targeted Restriction Enzymes. Science 2008, 321, 575–577. [Google Scholar] [CrossRef] [Green Version]
- Silva-Pinheiro, P.; Minczuk, M. The Potential of Mitochondrial Genome Engineering. Nat. Rev. Genet. 2022, 23, 199–214. [Google Scholar] [CrossRef]
- Cho, J.; Hur, J.H.; Graniel, J.; Benzer, S.; Walker, D.W. Expression of Yeast NDI1 Rescues a Drosophila Complex I Assembly Defect. PLoS ONE 2012, 7, e50644. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, D.; Goffrini, P.; Uziel, G.; Horvath, R.; Klopstock, T.; Lochmüller, H.; D’Adamo, P.; Gasparini, P.; Strom, T.M.; Prokisch, H.; et al. SDHAF1, Encoding a LYR Complex-II Specific Assembly Factor, Is Mutated in SDH-Defective Infantile Leukoencephalopathy. Nat. Genet. 2009, 41, 654–656. [Google Scholar] [CrossRef]
- Hao, H.X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.P.; Kunst, H.; Devilee, P.; Cremers, C.W.R.J.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a Gene Required for Flavination of Succinate Dehydrogenase, Is Mutated in Paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [Green Version]
- Mast, J.D.; Tomalty, K.M.H.; Vogel, H.; Clandinin, T.R. Reactive Oxygen Species Act Remotely to Cause Synapese Loss in a Drosophila Model of Developmental Mitochondrial Encephalopathy. Development 2008, 135, 2669–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, F.; Sam, R.; Ryan, E.; Alvarado, K.; Villa-Cuesta, E. Rapamycin as a Potential Treatment for Succinate Dehydrogenase Deficiency. Heliyon 2019, 5, e01217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, D.W.; Hájek, P.; Muffat, J.; Knoepfle, D.; Cornelison, S.; Attardi, G.; Benzer, S. Hypersensitivity to Oxygen and Shortened Lifespan in a Drosophila Mitochondrial Complex II Mutant. Proc. Natl. Acad. Sci. USA 2006, 103, 16382–16387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, M.; Sugiura, T.; Ishii, T.; Ishii, N.; Aigaki, T. A Mev-1-like Dominant-Negative SdhC Increases Oxidative Stress and Reduces Lifespan in Drosophila. Biochem. Biophys. Res. Commun. 2007, 363, 342–346. [Google Scholar] [CrossRef]
- Van Vranken, J.G.; Bricker, D.K.; Dephoure, N.; Gygi, S.P.; Cox, J.E.; Thummel, C.S.; Rutter, J. SDHAF4 Promotes Mitochondrial Succinate Dehydrogenase Activity and Prevents Neurodegeneration. Cell Metab. 2014, 20, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Na, U.; Yu, W.; Cox, J.; Bricker, D.K.; Brockmann, K.; Rutter, J.; Thummel, C.S.; Winge, D.R. The LYR Factors Sdhaf1 and SDHAF3 Mediate Maturation of the Iron-Sulfur Subunit of Succinate Dehydrogenase. Cell Metab. 2014, 20, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Bayley, J.P.; Devilee, P. Hypothesis: Why Different Types of SDH Gene Variants Cause Divergent Tumor Phenotypes. Genes 2022, 13, 1025. [Google Scholar] [CrossRef]
- Fernández-Vizarra, E.; Zeviani, M. Nuclear Gene Mutations as the Cause of Mitochondrial Complex III Deficiency. Front. Genet. 2015, 6, 134. [Google Scholar] [CrossRef]
- Ghezzi, D.; Arzuffi, P.; Zordan, M.; Da Re, C.; Lamperti, C.; Benna, C.; D’Adamo, P.; Diodato, D.; Costa, R.; Mariotti, C.; et al. Mutations in TTC19 Cause Mitochondrial Complex III Deficiency and Neurological Impairment in Humans and Flies. Nat. Genet. 2011, 43, 259–263. [Google Scholar] [CrossRef]
- Peruzzo, R.; Corrà, S.; Costa, R.; Brischigliaro, M.; Varanita, T.; Biasutto, L.; Rampazzo, C.; Ghezzi, D.; Leanza, L.; Zoratti, M.; et al. Exploiting Pyocyanin to Treat Mitochondrial Disease Due to Respiratory Complex III Dysfunction. Nat. Commun. 2021, 12, 2103. [Google Scholar] [CrossRef]
- Brischigliaro, M.; Frigo, E.; Corrà, S.; De Pittà, C.; Szabò, I.; Zeviani, M.; Costa, R. Modelling of BCS1L-Related Human Mitochondrial Disease in Drosophila Melanogaster. J. Mol. Med. 2021, 99, 1471–1485. [Google Scholar] [CrossRef] [PubMed]
- Frolov, M.V.; Benevolenskaya, E.V.; Birchler, J.A. The Oxen Gene of Drosophila Encodes a Homolog of Subunit 9 of Yeast Ubiquinol-Cytochrome c Oxidoreductase Complex: Evidence for Modulation of Gene Expression in Response to Mitochondrial Activity. Genetics 2000, 156, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Visapää, I.; Fellman, V.; Vesa, J.; Dasvarma, A.; Hutton, J.L.; Kumar, V.; Payne, G.S.; Makarow, M.; Van Coster, R.; Taylor, R.W.; et al. GRACILE Syndrome, a Lethal Metabolic Disorder with Iron Overload, Is Caused by a Point Mutation in BCS1L. Am. J. Hum. Genet. 2002, 71, 863–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, R.A.; Priestley, J.R.C.; Wilstermann, A.M.; Reese, K.J.; Mark, P.R. Clinical Spectrum of BCS1L Mitopathies and Their Underlying Structural Relationships. Am. J. Med. Genet. Part A 2019, 179, 373–380. [Google Scholar] [CrossRef]
- Calvo, S.E.; Compton, A.G.; Hershman, S.G.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular Diagnosis of Infantile Mitochondrial Disease with Targeted Next-Generation Sequencing. Sci. Transl. Med. 2012, 4, 118ra10. [Google Scholar] [CrossRef] [Green Version]
- Bosch, J.A.; Ugur, B.; Pichardo-Casas, I.; Rabasco, J.; Escobedo, F.; Zuo, Z.; Brown, B.; Celniker, S.; Sinclair, D.A.; Bellen, H.J.; et al. Two Neuronal Peptides Encoded from a Single Transcript Regulate Mitochondrial Complex III in Drosophila. eLife 2022, 11, e82709. [Google Scholar] [CrossRef]
- Zhang, S.; Reljić, B.; Liang, C.; Kerouanton, B.; Francisco, J.C.; Peh, J.H.; Mary, C.; Jagannathan, N.S.; Olexiouk, V.; Tang, C.; et al. Mitochondrial Peptide BRAWNIN Is Essential for Vertebrate Respiratory Complex III Assembly. Nat. Commun. 2020, 11, 1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennerlein, S.; Poerschke, S.; Oeljeklaus, S.; Wang, C.; Richter-Dennerlein, R.; Sattmann, J.; Bauermeister, D.; Hanitsch, E.; Stoldt, S.; Langer, T.; et al. Defining the Interactome of the Human Mitochondrial Ribosome Identifies SMIM4 and TMEM223 as Respiratory Chain Assembly Factors. eLife 2021, 10, e68213. [Google Scholar] [CrossRef]
- Brischigliaro, M.; Zeviani, M. Cytochrome c Oxidase Deficiency. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148335. [Google Scholar] [CrossRef]
- Massa, V.; Fernandez-Vizarra, E.; Alshahwan, S.; Bakhsh, E.; Goffrini, P.; Ferrero, I.; Mereghetti, P.; D’Adamo, P.; Gasparini, P.; Zeviani, M. Severe Infantile Encephalomyopathy Caused by a Mutation in COX6B1, a Nucleus-Encoded Subunit of Cytochrome C Oxidase. Am. J. Hum. Genet. 2008, 82, 1281–1289. [Google Scholar] [CrossRef] [Green Version]
- Shoubridge, E.A. Cytochrome c Oxidase Deficiency. Am. J. Med. Genet. Semin. Med. Genet. 2001, 106, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Darin, N.; Moslemi, A.R.; Lebon, S.; Rustin, P.; Holme, E.; Oldfors, A.; Tulinius, M. Genotypes and Clinical Phenotypes in Children with Cytochrome-c Oxidase Deficiency. Neuropediatrics 2003, 34, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Timón-Gómez, A.; Nývltová, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial Cytochrome c Oxidase Biogenesis: Recent Developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Brischigliaro, M.; Cabrera-Orefice, A.; Sturlese, M.; Elurbe, D.M.; Frigo, E.; Fernandez-Vizarra, E.; Moro, S.; Huynen, M.A.; Arnold, S.; Viscomi, C.; et al. CG7630 Is the Drosophila Melanogaster Homolog of the Cytochrome c Oxidase Subunit COX7B. EMBO Rep. 2022, 23, e54825. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Miriyala, G.K.; Littleton, A.J.; Yang, H.; Trinh, K.; Young, J.M.; Kennedy, S.R.; Yamashita, Y.M.; Pallanck, L.J.; Malik, H.S. A Mitochondrial DNA Hypomorph of Cytochrome Oxidase Specifically Impairs Male Fertility in Drosophila Melanogaster. eLife 2016, 5, e16923. [Google Scholar] [CrossRef]
- Szuplewski, S.; Terracol, R. The Cyclope Gene of Drosophila Encodes a Cytochrome c Oxidase Subunit VIc Homolog. Genetics 2001, 158, 1629–1643. [Google Scholar] [CrossRef]
- Mandal, S.; Guptan, P.; Owusu-Ansah, E.; Banerjee, U. Mitochondrial Regulation of Cell Cycle Progression during Development as Revealed by the Tenured Mutation in Drosophila. Dev. Cell 2005, 9, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Gnanasambandam, R.; Benjamin, J.; Kaur, G.; Getman, P.B.; Siegel, A.J.; Shortridge, R.D.; Singh, S. Mutations in Cytochrome c Oxidase Subunit VIa Cause Neurodegeneration and Motor Dysfunction in Drosophila. Genetics 2007, 176, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Klichko, V.; Sohal, B.H.; Radyuk, S.N.; Orr, W.C.; Sohal, R.S. Decrease in Cytochrome c Oxidase Reserve Capacity Diminishes Robustness of Drosophila Melanogaster and Shortens Lifespan. Biochem. J. 2014, 459, 127–135. [Google Scholar] [CrossRef]
- Kemppainen, K.K.; Rinne, J.; Sriram, A.; Lakanmaa, M.; Zeb, A.; Tuomela, T.; Popplestone, A.; Singh, S.; Sanz, A.; Rustin, P.; et al. Expression of Alternative Oxidase in Drosophila Ameliorates Diverse Phenotypes Due to Cytochrome Oxidase Deficiency. Hum. Mol. Genet. 2014, 23, 2078–2093. [Google Scholar] [CrossRef] [Green Version]
- Peralta, S.; Clemente, P.; Sánchez-Martínez, Á.; Calleja, M.; Hernández-Sierra, R.; Matsushima, Y.; Adán, C.; Ugalde, C.; Fernández-Moreno, M.Á.; Kaguni, L.S.; et al. Coiled Coil Domain-Containing Protein 56 (CCDC56) Is a Novel Mitochondrial Protein Essential for Cytochrome c Oxidase Function. J. Biol. Chem. 2012, 287, 24174–24185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, D.; Oliva, M.; Duchi, S.; Latorre, D.; Cavaliere, V.; Barsanti, P.; Villani, G.; Gargiulo, G.; Caggese, C. Genetic, Functional and Evolutionary Characterization of Scox, the Drosophila Melanogaster Ortholog of the Human SCO1 Gene. Mitochondrion 2010, 10, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Ida, H.; Shimamura, M.; Kitazawa, D.; Akao, S.; Yoshida, H.; Inoue, Y.H.; Yamaguchi, M. Role of SCOX in Determination of Drosophila Melanogaster Lifespan. Am. J. Cancer Res. 2014, 4, 325–336. [Google Scholar] [PubMed]
- Martínez-Morentin, L.; Martínez, L.; Piloto, S.; Yang, H.; Schon, E.A.; Garesse, R.; Bodmer, R.; Ocorr, K.; Cervera, M.; Arredondo, J.J. Cardiac Deficiency of Single Cytochrome Oxidase Assembly Factor Scox Induces P53-Dependent Apoptosis in a Drosophila Cardiomyopathy Model. Hum. Mol. Genet. 2015, 24, 3608–3622. [Google Scholar] [CrossRef] [Green Version]
- Zordan, M.A.; Cisotto, P.; Benna, C.; Agostino, A.; Rizzo, G.; Piccin, A.; Pegoraro, M.; Sandrelli, F.; Perini, G.; Tognon, G.; et al. Post-Transcriptional Silencing and Functional Characterization of the Drosophila Melanogaster Homolog of Human Surf1. Genetics 2006, 172, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Da-Rè, C.; von Stockum, S.; Biscontin, A.; Millino, C.; Cisotto, P.; Zordan, M.A.; Zeviani, M.; Bernardi, P.; De Pittà, C.; Costa, R. Leigh Syndrome in Drosophila Melanogaster. J. Biol. Chem. 2014, 289, 29235–29246. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, Y.; Okunushi, R.; Hara, T.; Hashiguchi, A.; Yuan, J.; Yoshimura, A.; Murayama, K.; Ohtake, A.; Ando, M.; Hiramatsu, Y.; et al. Mutations in COA7 Cause Spinocerebellar Ataxia with Axonal Neuropathy. Brain 2018, 141, 1622–1636. [Google Scholar] [CrossRef]
- Brischigliaro, M.; Corrà, S.; Tregnago, C.; Fernandez-Vizarra, E.; Zeviani, M.; Costa, R.; De Pittà, C. Knockdown of APOPT1/COA8 Causes Cytochrome c Oxidase Deficiency, Neuromuscular Impairment, and Reduced Resistance to Oxidative Stress in Drosophila Melanogaster. Front. Physiol. 2019, 10, 1143. [Google Scholar] [CrossRef]
- Ostergaard, E.; Weraarpachai, W.; Ravn, K.; Born, A.P.; Jønson, L.; Duno, M.; Wibrand, F.; Shoubridge, E.A.; Vissing, J. Mutations in COA3 Cause Isolated Complex IV Deficiency Associated with Neuropathy, Exercise Intolerance, Obesity, and Short Stature. J. Med. Genet. 2015, 52, 203–207. [Google Scholar] [CrossRef]
- Kowada, R.; Kodani, A.; Ida, H.; Yamaguchi, M.; Lee, I.S.; Okada, Y.; Yoshida, H. The Function of Scox in Glial Cells Is Essential for Locomotive Ability in Drosophila. Sci. Rep. 2021, 11, 21207. [Google Scholar] [CrossRef]
- Brischigliaro, M.; Badocco, D.; Costa, R.; Viscomi, C.; Zeviani, M.; Pastore, P.; Fernández-Vizarra, E. Mitochondrial Cytochrome c Oxidase Defects Alter Cellular Homeostasis of Transition Metals. Front. Cell Dev. Biol. 2022, 10, 1090. [Google Scholar] [CrossRef]
- Sánchez-Caballero, L.; Elurbe, D.M.; Baertling, F.; Guerrero-Castillo, S.; van den Brand, M.; van Strien, J.; van Dam, T.J.P.; Rodenburg, R.; Brandt, U.; Huynen, M.A.; et al. TMEM70 Functions in the Assembly of Complexes I and V. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148202. [Google Scholar] [CrossRef]
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. TMEM70 and TMEM242 Help to Assemble the Rotor Ring of Human ATP Synthase and Interact with Assembly Factors for Complex I. Proc. Natl. Acad. Sci. USA 2021, 118, e2100558118. [Google Scholar] [CrossRef] [PubMed]
- Celotto, A.M.; Frank, A.C.; McGrath, S.W.; Fergestad, T.; Van Voorhies, W.A.; Buttle, K.F.; Mannella, C.A.; Palladino, M.J. Mitochondrial Encephalomyopathy in Drosophila. J. Neurosci. 2006, 26, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wheeler, C.T.; Yolitz, J.; Laslo, M.; Alberico, T.; Sun, Y.; Song, Q.; Zou, S. A Mitochondrial ATP Synthase Subunit Interacts with TOR Signaling to Modulate Protein Homeostasis and Lifespan in Drosophila. Cell Rep. 2014, 8, 1781–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovero, D.; Giordano, L.; Marsano, R.M.; Sanchez-Martinez, A.; Boukhatmi, H.; Drechsler, M.; Oliva, M.; Whitworth, A.J.; Porcelli, D.; Caggese, C. Characterization of Drosophila ATPsynC Mutants as a New Model of Mitochondrial ATP Synthase Disorders. PLoS ONE 2018, 13, e0201811. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.N.; Wu, C.H.; Zheng, Y.; Li, J.J.; Wang, J.L.; Wang, Y.F. Knockdown of ATPsyn-b Caused Larval Growth Defect and Male Infertility in Drosophila. Arch. Insect Biochem. Physiol. 2015, 88, 144–154. [Google Scholar] [CrossRef]
- Teixeira, F.K.; Sanchez, C.G.; Hurd, T.R.; Seifert, J.R.K.; Czech, B.; Preall, J.B.; Hannon, G.J.; Lehmann, R. ATP Synthase Promotes Germ Cell Differentiation Independent of Oxidative Phosphorylation. Nat. Cell Biol. 2015, 17, 689–696. [Google Scholar] [CrossRef] [Green Version]
- Alcázar-Fabra, M.; Rodríguez-Sánchez, F.; Trevisson, E.; Brea-Calvo, G. Primary Coenzyme Q Deficiencies: A Literature Review and Online Platform of Clinical Features to Uncover Genotype-Phenotype Correlations. Free Radic. Biol. Med. 2021, 167, 141–180. [Google Scholar] [CrossRef]
- Grant, J.; Saldanha, J.W.; Gould, A.P. A Drosophila Model for Primary Coenzyme Q Deficiency and Dietary Rescue in the Developing Nervous System. DMM Dis. Model. Mech. 2010, 3, 799–806. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA Replication: Clinical Syndromes. Essays Biochem. 2018, 62, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.; Ng, Y.S.; Nicholls, T.J. The Maintenance of Mitochondrial DNA Integrity and Dynamics by Mitochondrial Membranes. Life 2020, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, B.; Roote, J.; Campos, A.R. The Tamas Gene, Identified as a Mutation That Disrupts Larval Behavior in Drosophila Melanogaster, Codes for the Mitochondrial DNA Polymerase Catalytic Subunit (DNApol-Γ125). Genetics 1999, 153, 1809–1824. [Google Scholar] [CrossRef]
- Bratic, A.; Kauppila, T.E.S.; Macao, B.; Grönke, S.; Siibak, T.; Stewart, J.B.; Baggio, F.; Dols, J.; Partridge, L.; Falkenberg, M.; et al. Complementation between Polymerase- and Exonuclease-Deficient Mitochondrial DNA Polymerase Mutants in Genomically Engineered Flies. Nat. Commun. 2015, 6, 8808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.P.C.; Camargo, A.F.; Andjelković, A.; Jacobs, H.T.; Oliveira, M.T. Developmental Arrest in Drosophila Melanogaster Caused by Mitochondrial DNA Replication Defects Cannot Be Rescued by the Alternative Oxidase. Sci. Rep. 2018, 8, 10882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstag, C.L.; Hoekstra, J.G.; Huang, C.H.; Chaisson, M.J.; Youle, R.J.; Kennedy, S.R.; Pallanck, L.J. Deleterious Mitochondrial DNA Point Mutations Are Overrepresented in Drosophila Expressing a Proofreading-Defective DNA Polymerase γ. PLoS Genet. 2018, 14, e1007805. [Google Scholar] [CrossRef] [PubMed]
- Vermulst, M.; Bielas, J.H.; Kujoth, G.C.; Ladiges, W.C.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. Mitochondrial Point Mutations Do Not Limit the Natural Lifespan of Mice. Nat. Genet. 2007, 39, 540–543. [Google Scholar] [CrossRef]
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA Deletions and Clonal Mutations Drive Premature Aging in Mitochondrial Mutator Mice. Nat. Genet. 2008, 40, 392–394. [Google Scholar] [CrossRef]
- Edgar, D.; Shabalina, I.; Camara, Y.; Wredenberg, A.; Calvaruso, M.A.; Nijtmans, L.; Nedergaard, J.; Cannon, B.; Larsson, N.G.; Trifunovic, A. Random Point Mutations with Major Effects on Protein-Coding Genes Are the Driving Force behind Premature Aging in MtDNA Mutator Mice. Cell Metab. 2009, 10, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Andreazza, S.; Samstag, C.L.; Sanchez-Martinez, A.; Fernandez-Vizarra, E.; Gomez-Duran, A.; Lee, J.J.; Tufi, R.; Hipp, M.J.; Schmidt, E.K.; Nicholls, T.J.; et al. Mitochondrially-Targeted APOBEC1 Is a Potent MtDNA Mutator Affecting Mitochondrial Function and Organismal Fitness in Drosophila. Nat. Commun. 2019, 10, 3280. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martinez, A.; Calleja, M.; Peralta, S.; Matsushima, Y.; Hernandez-Sierra, R.; Whitworth, A.J.; Kaguni, L.S.; Garesse, R. Modeling Pathogenic Mutations of Human Twinkle in Drosophila Suggests an Apoptosis Role in Response to Mitochondrial Defects. PLoS ONE 2012, 7, e43954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Cooper, H.M.; Reyes, A.; Di Re, M.; Sembongi, H.; Gao, J.; Neuman, K.C.; Fearnley, I.M.; Spinazzola, A.; Walker, J.E.; et al. Mitochondrial Nucleoid Interacting Proteins Support Mitochondrial Protein Synthesis. Nucleic Acids Res. 2012, 40, 6109–6121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harel, T.; Yoon, W.H.; Garone, C.; Gu, S.; Coban-Akdemir, Z.; Eldomery, M.K.; Posey, J.E.; Jhangiani, S.N.; Rosenfeld, J.A.; Cho, M.T.; et al. Recurrent De Novo and Biallelic Variation of ATAD3A, Encoding a Mitochondrial Membrane Protein, Results in Distinct Neurological Syndromes. Am. J. Hum. Genet. 2016, 99, 831–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazier, A.E.; Compton, A.G.; Kishita, Y.; Hock, D.H.; Welch, A.M.E.; Amarasekera, S.S.C.; Rius, R.; Formosa, L.E.; Imai-Okazaki, A.; Francis, D.; et al. Fatal Perinatal Mitochondrial Cardiac Failure Caused by Recurrent De Novo Duplications in the ATAD3 Locus. Med 2021, 2, 49–73.e10. [Google Scholar] [CrossRef] [PubMed]
- Yap, Z.Y.; Park, Y.H.; Wortmann, S.B.; Gunning, A.C.; Ezer, S.; Lee, S.; Duraine, L.; Wilichowski, E.; Wilson, K.; Mayr, J.A.; et al. Functional Interpretation of ATAD3A Variants in Neuro-Mitochondrial Phenotypes. Genome Med. 2021, 13, 55. [Google Scholar] [CrossRef]
- Gilquin, B.; Taillebourg, E.; Cherradi, N.; Hubstenberger, A.; Gay, O.; Merle, N.; Assard, N.; Fauvarque, M.-O.; Tomohiro, S.; Kuge, O.; et al. The AAA + ATPase ATAD3A Controls Mitochondrial Dynamics at the Interface of the Inner and Outer Membranes. Mol. Cell. Biol. 2010, 30, 1984–1996. [Google Scholar] [CrossRef] [Green Version]
- Ostergaard, E.; Christensen, E.; Kristensen, E.; Mogensen, B.; Duno, M.; Shoubridge, E.A.; Wibrand, F. Deficiency of the α Subunit of Succinate-Coenzyme A Ligase Causes Fatal Infantile Lactic Acidosis with Mitochondrial DNA Depletion. Am. J. Hum. Genet. 2007, 81, 383–387. [Google Scholar] [CrossRef] [Green Version]
- Carrozzo, R.; Verrigni, D.; Rasmussen, M.; de Coo, R.; Amartino, H.; Bianchi, M.; Buhas, D.; Mesli, S.; Naess, K.; Born, A.P.; et al. Succinate-CoA Ligase Deficiency Due to Mutations in SUCLA2 and SUCLG1: Phenotype and Genotype Correlations in 71 Patients. J. Inherit. Metab. Dis. 2016, 39, 243–252. [Google Scholar] [CrossRef]
- Demirbas, D.; Harris, D.J.; Arn, P.H.; Huang, X.; Waisbren, S.E.; Anselm, I.; Lerner-Ellis, J.P.; Wong, L.J.; Levy, H.L.; Berry, G.T. Phenotypic Variability in Deficiency of the α Subunit of Succinate-CoA Ligase. JIMD Rep. 2019, 46, 63–69. [Google Scholar] [CrossRef]
- Quan, X.; Sato-Miyata, Y.; Tsuda, M.; Muramatsu, K.; Asano, T.; Takeo, S.; Aigaki, T. Deficiency of Succinyl-CoA Synthetase α Subunit Delays Development, Impairs Locomotor Activity and Reduces Survival under Starvation in Drosophila. Biochem. Biophys. Res. Commun. 2017, 483, 566–571. [Google Scholar] [CrossRef]
- Kodani, A.; Yamaguchi, M.; Itoh, R.; Huynh, M.A.; Yoshida, H. A Drosophila Model of the Neurological Symptoms in Mpv17-Related Diseases. Sci. Rep. 2022, 12, 22632. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Viscomi, C.; Fernandez-Vizarra, E.; Carrara, F.; D’Adamo, P.; Calvo, S.; Marsano, R.M.; Donnini, C.; Weiher, H.; Strisciuglio, P.; et al. MPV17 Encodes an Inner Mitochondrial Membrane Protein and Is Mutated in Infantile Hepatic Mitochondrial DNA Depletion. Nat. Genet. 2006, 38, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, B.A.; Mehta, N.; Hameed, B.; Pekmezci, M.; Packman, S.; Ralph, J. Adult-Onset Fatal Neurohepatopathy in a Woman Caused by MPV17 Mutation. JIMD Rep. 2014, 13, 37–41. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, A.R.; Minczuk, M. Mitochondrial Transcription and Translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boczonadi, V.; Horvath, R. Mitochondria: Impaired Mitochondrial Translation in Human Disease. Int. J. Biochem. Cell Biol. 2014, 48, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.; Del’Olio, S.; Barrientos, A. The Diseased Mitoribosome. FEBS Lett. 2021, 595, 1025–1061. [Google Scholar] [CrossRef]
- Royden, C.S.; Pirrotta, V.; Jan, L.Y. The Tko Locus, Site of a Behavioral Mutation in D. Melanogaster, Codes for a Protein Homologous to Prokaryotic Ribosomal Protein S12. Cell 1987, 51, 165–173. [Google Scholar] [CrossRef]
- Toivonen, J.M.; O’Dell, K.M.C.; Petit, N.; Irvine, S.C.; Knight, G.K.; Lehtonen, M.; Longmuir, M.; Luoto, K.; Touraille, S.; Wang, Z.; et al. Technical Knockout, a Drosophila Model of Mitochondrial Deafness. Genetics 2001, 159, 241–254. [Google Scholar] [CrossRef]
- Gokhale, A.; Lee, C.E.; Zlatic, S.A.; Freeman, A.A.H.; Shearing, N.; Hartwig, C.; Ogunbona, O.; Bassell, J.L.; Wynne, M.E.; Werner, E.; et al. Mitochondrial Proteostasis Requires Genes Encoded in a Neurodevelopmental Syndrome Locus. J. Neurosci. 2021, 41, 6596–6616. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Hales, K.G.; Fuller, M.T. Developmentally Regulated Mitochondrial Fusion Mediated by a Conserved, Novel, Predicted GTPase. Cell 1997, 90, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.I.; et al. Mitochondrial Fission Factor Drp1 Is Essential for Embryonic Development and Synapse Formation in Mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The Dynamin-Related GTPase Drp1 Is Required for Embryonic and Brain Development in Mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [Green Version]
- Verstreken, P.; Ly, C.V.; Venken, K.J.T.; Koh, T.W.; Zhou, Y.; Bellen, H.J. Synaptic Mitochondria Are Critical for Mobilization of Reserve Pool Vesicles at Drosophila Neuromuscular Junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Aldridge, A.C.; Benson, L.P.; Siegenthaler, M.M.; Whigham, B.T.; Stowers, R.S.; Hales, K.G. Roles for Drp1, a Dynamin-Related Protein, and Milton, a Kinesin-Associated Protein, in Mitochondrial Segregation, Unfurling and Elongation during Drosophila Spermatogenesis. Fly 2007, 1, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Yarosh, W.; Monserrate, J.; Tong, J.J.; Tse, S.; Le, P.K.; Nguyen, K.; Brachmann, C.B.; Wallace, D.C.; Huang, T. The Molecular Mechanisms of OPA1-Mediated Optic Atrophy in Drosophila Model and Prospects for Antioxidant Treatment. PLoS Genet. 2008, 4, 0062–0071. [Google Scholar] [CrossRef] [Green Version]
- Shahrestani, P.; Leung, H.T.; Le, P.K.; Pak, W.L.; Tse, S.; Ocorr, K.; Huang, T. Heterozygous Mutation of Drosophila Opa1 Causes the Development of Multiple Organ Abnormalities in an Age-Dependent and Organ-Specific Manner. PLoS ONE 2009, 4, e6867. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Le, P.K.; Tse, S.; Wallace, D.C.; Huang, T. Heterozygous Mutation of Opa1 in Drosophila Shortens Lifespan Mediated through Increased Reactive Oxygen Species Production. PLoS ONE 2009, 4, e4492. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W.; Clark, C.F.; Eschenbacher, W.H.; Kang, M.Y.; Engelhard, J.T.; Warner, S.J.; Matkovich, S.J.; Jowdy, C.C. MARF and Opa1 Control Mitochondrial and Cardiac Function in Drosophila. Circ. Res. 2011, 108, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.M.; Anselmi, C.; Wittig, I.; Faraldo-Gómez, J.D.; Kühlbrandt, W. Structure of the Yeast F 1F O-ATP Synthase Dimer and Its Role in Shaping the Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, T.B.; Hahn, A.; Meier, T.; Davies, K.M.; Kühlbrandt, W. Dimers of Mitochondrial ATP Synthase Induce Membrane Curvature and Self-Assemble into Rows. Proc. Natl. Acad. Sci. USA 2019, 116, 4250–4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, I.; Ghosh, M.; Meinecke, M. MICOS and the Mitochondrial Inner Membrane Morphology—When Things Get out of Shape. FEBS Lett. 2021, 595, 1159–1183. [Google Scholar] [CrossRef] [PubMed]
- Guarani, V.; Jardel, C.; Chrétien, D.; Lombès, A.; Bénit, P.; Labasse, C.; Lacène, E.; Bourillon, A.; Imbard, A.; Benoist, J.F.; et al. QIL1 Mutation Causes MICOS Disassembly and Early Onset Fatal Mitochondrial Encephalopathy with Liver Disease. eLife 2016, 5, e17163. [Google Scholar] [CrossRef]
- Zeharia, A.; Friedman, J.R.; Tobar, A.; Saada, A.; Konen, O.; Fellig, Y.; Shaag, A.; Nunnari, J.; Elpeleg, O. Mitochondrial Hepato-Encephalopathy Due to Deficiency of QIL1/MIC13 (C19orf70), a MICOS Complex Subunit. Eur. J. Hum. Genet. 2016, 24, 1778–1782. [Google Scholar] [CrossRef] [Green Version]
- Russell, B.E.; Whaley, K.G.; Bove, K.E.; Labilloy, A.; Lombardo, R.C.; Hopkin, R.J.; Leslie, N.D.; Prada, C.; Assouline, Z.; Barcia, G.; et al. Expanding and Underscoring the Hepato-Encephalopathic Phenotype of QIL1/MIC13. Hepatology 2019, 70, 1066–1070. [Google Scholar] [CrossRef]
- Benincá, C.; Zanette, V.; Brischigliaro, M.; Johnson, M.; Reyes, A.; Valle, D.A.D.; Robinson, A.J.; Degiorgi, A.; Yeates, A.; Telles, B.A.; et al. Mutation in the MICOS Subunit Gene APOO (MIC26) Associated with an X-Linked Recessive Mitochondrial Myopathy, Lactic Acidosis, Cognitive Impairment and Autistic Features. J. Med. Genet. 2021, 58, 155–167. [Google Scholar] [CrossRef]
- Tsai, P.I.; Lin, C.H.; Hsieh, C.H.; Papakyrikos, A.M.; Kim, M.J.; Napolioni, V.; Schoor, C.; Couthouis, J.; Wu, R.M.; Wszolek, Z.K.; et al. PINK1 Phosphorylates MIC60/Mitofilin to Control Structural Plasticity of Mitochondrial Crista Junctions. Mol. Cell 2018, 69, 744–756.e6. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.I.; Papakyrikos, A.M.; Hsieh, C.H.; Wang, X. Drosophila MIC60/Mitoflin Conducts Dual Roles in Mitochondrial Motility and Crista Structure. Mol. Biol. Cell 2017, 28, 3471–3479. [Google Scholar] [CrossRef]
- Guarani, V.; McNeill, E.M.; Paulo, J.A.; Huttlin, E.L.; Fröhlich, F.; Gygi, S.P.; Vactor, D.V.; Wade Harper, J. QIL1 Is a Novel Mitochondrial Protein Required for MICOS Complex Stability and Cristae Morphology. eLife 2015, 4, e06265. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, G.; Balint, B.; Mencacci, N.E.; Carr, L.; Wood, N.W.; Bhatia, K.P. SLC25A46 Mutations Underlie Progressive Myoclonic Ataxia with Optic Atrophy and Neuropathy. Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 1249–1251. [Google Scholar] [CrossRef] [PubMed]
- Janer, A.; Prudent, J.; Paupe, V.; Fahiminiya, S.; Majewski, J.; Sgarioto, N.; Des Rosiers, C.; Forest, A.; Lin, Z.; Gingras, A.; et al. SLC 25A46 Is Required for Mitochondrial Lipid Homeostasis and Cristae Maintenance and Is Responsible for Leigh Syndrome. EMBO Mol. Med. 2016, 8, 1019–1038. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Steffen, J.; Yourshaw, M.; Mamsa, H.; Andersen, E.; Rudnik-Schöneborn, S.; Pope, K.; Howell, K.B.; McLean, C.A.; Kornberg, A.J.; et al. Loss of Function of SLC25A46 Causes Lethal Congenital Pontocerebellar Hypoplasia. Brain 2016, 139, 2877–2890. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.; Boesten, I.; Hellebrekers, D.M.E.I.; Mulder-den Hartog, N.M.; de Coo, I.F.M.; Smeets, H.J.M.; Gerards, M. Novel Pathogenic SLC25A46 Splice-Site Mutation Causes an Optic Atrophy Spectrum Disorder. Clin. Genet. 2017, 91, 121–125. [Google Scholar] [CrossRef]
- Ali, M.S.; Suda, K.; Kowada, R.; Ueoka, I.; Yoshida, H.; Yamaguchi, M. Neuron-Specific Knockdown of Solute Carrier Protein SLC25A46a Induces Locomotive Defects, an Abnormal Neuron Terminal Morphology, Learning Disability, and Shortened Lifespan. IBRO Rep. 2020, 8, 65–75. [Google Scholar] [CrossRef]
- Garone, C.; Viscomi, C. Towards a Therapy for Mitochondrial Disease: An Update. Biochem. Soc. Trans. 2018, 46, 1247–1261. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brischigliaro, M.; Fernandez-Vizarra, E.; Viscomi, C. Mitochondrial Neurodegeneration: Lessons from Drosophila melanogaster Models. Biomolecules 2023, 13, 378. https://doi.org/10.3390/biom13020378
Brischigliaro M, Fernandez-Vizarra E, Viscomi C. Mitochondrial Neurodegeneration: Lessons from Drosophila melanogaster Models. Biomolecules. 2023; 13(2):378. https://doi.org/10.3390/biom13020378
Chicago/Turabian StyleBrischigliaro, Michele, Erika Fernandez-Vizarra, and Carlo Viscomi. 2023. "Mitochondrial Neurodegeneration: Lessons from Drosophila melanogaster Models" Biomolecules 13, no. 2: 378. https://doi.org/10.3390/biom13020378