
Cystic Fibrosis Bone Disease: The Interplay between CFTR Dysfunction and Chronic Inflammation

{kind=link}
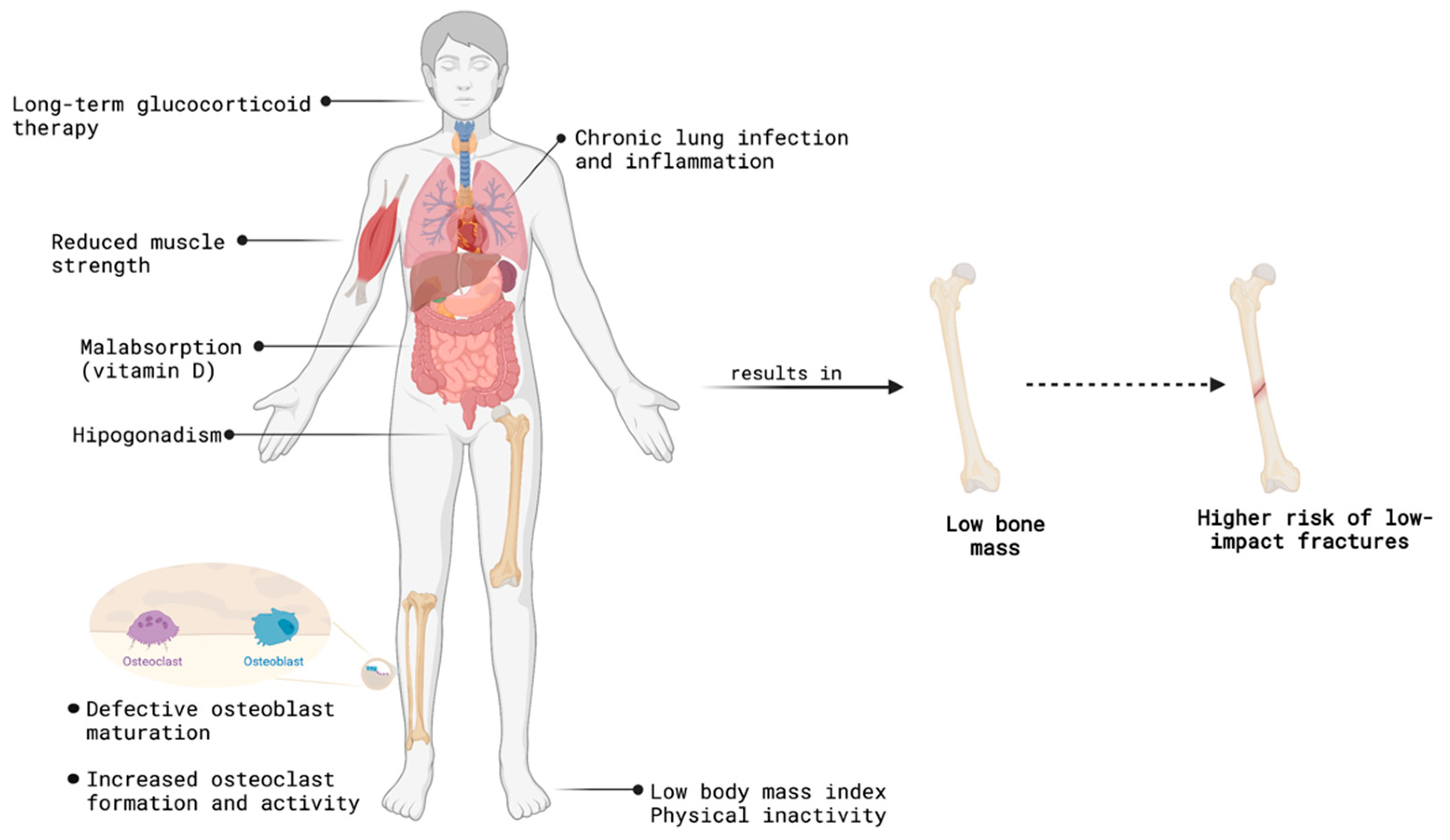{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cystic Fibrosis-Related Bone Disease
Age and Bone Mass in Cystic Fibrosis
3. Cystic Fibrosis-Induced Alterations in Bone Architecture and Turnover
4. CFTR Disfunction in Bone Cells and Their Progenitors
4.1. CFTR Dysfunction on Osteoblasts and Bone Formation
4.2. CFTR Dysfunction and Osteoclastogenesis
5. CFTR-Indirect Effects on Bone Health
5.1. Implications of Chronic Infection
5.2. Dysregulation of Glucose Homeostasis
6. CFTR Modulators and Their Potential Impact in Bone Health
7. Challenges in the Management of Bone Health in Cystic Fibrosis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathologic study. Am. J. Dis. Child. 1938, 56, 344–399. [Google Scholar] [CrossRef]
- Davis, P.B. Cystic fibrosis since 1938. Am. J. Respir. Crit. Care Med. 2006, 173, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Plant, B.J.; Goss, C.H.; Plant, W.D.; Bell, S.C. Management of comorbidities in older patients with cystic fibrosis. Lancet Respir. Med. 2013, 1, 164–174. [Google Scholar] [CrossRef]
- Farrell, P.M. The prevalence of cystic fibrosis in the European Union. J. Cyst. Fibros. 2008, 7, 450–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Liu, K.; Liu, Y.; Situ, Y.; Tian, X.; Xu, K.-F.; Zhang, X. Clinical and genetic characteristics of cystic fibrosis in CHINESE patients: A systemic review of reported cases. Orphanet J. Rare Dis. 2018, 13, 224. [Google Scholar] [CrossRef]
- Stewart, C.; Pepper, M.S. Cystic fibrosis on the African continent. Genet. Med. 2016, 18, 653–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Registry, C.F.P. 2020 Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2021. [Google Scholar]
- Orenti, A.; Jung, Z.A.; van Rens, J.A.; Fox, A.; Krasnyk, M.; Daneau, G.; Hatziagorou, E.; Mei-Zahav, M.; Naehrlich, L.; Storms, V.; et al. ECFSPR Annual Report 2020; European Cystic Fibrosis Society: Karup, Denmark, 2022. [Google Scholar]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Anderson, M.P.; Gregory, R.J.; Thompson, S.; Souza, D.W.; Paul, S.; Mulligan, R.C.; Smith, A.E.; Welsh, M.J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 1991, 253, 202–205. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Zhang, Z.; Csanady, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US CF Foundation, J.H.U.; The Hospital for Sick Children. Clinical and Functional Translation of CFTR. 2011. Available online: https://www.cftr2.org/ (accessed on 15 September 2022).
- Jia, S.; Taylor-Cousar, J.L. Cystic Fibrosis Modulator Therapies. Annu. Rev. Med. 2023, 74, 413–426. [Google Scholar] [CrossRef] [PubMed]
- McGarry, M.E.; McColley, S.A. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr. Pulmonol. 2021, 56, 1496–1503. [Google Scholar] [CrossRef]
- Boucher, R.C. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur. Respir. J. 2004, 23, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esther, C.R., Jr.; Muhlebach, M.S.; Ehre, C.; Hill, D.B.; Wolfgang, M.C.; Kesimer, M.; Ramsey, K.A.; Markovetz, M.R.; Garbarine, I.C.; Forest, M.G.; et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci. Transl. Med. 2019, 11, eaav3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallagi, P.; Hegyi, P.; Rakonczay, Z., Jr. The Physiology and Pathophysiology of Pancreatic Ductal Secretion: The Background for Clinicians. Pancreas 2015, 44, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Novak, I. The cystic fibrosis of exocrine pancreas. Cold Spring Harb. Perspect Med. 2013, 3, a009746. [Google Scholar] [CrossRef]
- Hart, N.J.; Aramandla, R.; Poffenberger, G.; Fayolle, C.; Thames, A.H.; Bautista, A.; Spigelman, A.F.; Babon, J.A.B.; DeNicola, M.E.; Dadi, P.K.; et al. Cystic fibrosis-related diabetes is caused by islet loss and inflammation. JCI Insight 2018, 3, e98240. [Google Scholar] [CrossRef] [Green Version]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Conese, M.; Rejman, J. Stem cells and cystic fibrosis. J. Cyst. Fibros. 2006, 5, 141–143. [Google Scholar] [CrossRef] [Green Version]
- Duchesneau, P.; Waddell, T.; Karoubi, G. Cell-Based Therapeutic Approaches for Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 5219. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front. Pharm. 2016, 7, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2019, 10, 1662. [Google Scholar] [CrossRef] [Green Version]
- Spanò, V.; Barreca, M.; Cilibrasi, V.; Genovese, M.; Renda, M.; Montalbano, A.; Galietta, L.; Barraja, P. Evaluation of Fused Pyrrolothiazole Systems as Correctors of Mutant CFTR Protein. Molecules 2021, 26, 1275. [Google Scholar] [CrossRef]
- Brindani, N.; Gianotti, A.; Giovani, S.; Giacomina, F.; Di Fruscia, P.; Sorana, F.; Bertozzi, S.M.; Ottonello, G.; Goldoni, L.; Penna, I.; et al. Identification, Structure–Activity Relationship, and Biological Characterization of 2,3,4,5-Tetrahydro-1H-pyrido[4,3-b]indoles as a Novel Class of CFTR Potentiators. J. Med. Chem. 2020, 63, 11169–11194. [Google Scholar] [CrossRef]
- Spanò, V.; Venturini, A.; Genovese, M.; Barreca, M.; Raimondi, M.V.; Montalbano, A.; Galietta, L.J.; Barraja, P. Current development of CFTR potentiators in the last decade. Eur. J. Med. Chem. 2020, 204, 112631. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Bertozzi, F.; Caci, E.; Sorana, F.; Di Fruscia, P.; Tomati, V.; Ferrera, L.; Rodríguez-Gimeno, A.; Berti, F.; Pesce, E.; et al. Discovery of a picomolar potency pharmacological corrector of the mutant CFTR chloride channel. Sci. Adv. 2020, 6, eaay9669. [Google Scholar] [CrossRef] [Green Version]
- Costa, E.; Girotti, S.; Pauro, F.; Leufkens, H.G.M.; Cipolli, M. The impact of FDA and EMA regulatory decision-making process on the access to CFTR modulators for the treatment of cystic fibrosis. Orphanet J. Rare Dis. 2022, 17, 188. [Google Scholar] [CrossRef]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.-R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef] [Green Version]
- Bessonova, L.; Volkova, N.; Higgins, M.; Bengtsson, L.; Tian, S.; Simard, C.; Konstan, M.W.; Sawicki, G.S.; Sewall, A.; Nyangoma, S.; et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax 2018, 73, 731–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosz-Griffiths, H.H.; Scambler, T.; Wong, C.H.; Lara-Reyna, S.; Holbrook, J.; Martinon, F.; Savic, S.; Whitaker, P.; Etherington, C.; Spoletini, G.; et al. Different CFTR modulator combinations downregulate inflammation differently in cystic fibrosis. Elife 2020, 9, e54556. [Google Scholar] [CrossRef]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. CFTR-dependent chloride efflux in cystic fibrosis mononuclear cells is increased by ivacaftor therapy. Pediatr. Pulmonol. 2017, 52, 900–908. [Google Scholar]
- Zhang, S.; Shrestha, C.; Kopp, B. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci. Rep. 2018, 8, 17066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeev, V.; Chou, F.Y.; Lam, G.Y.; Hamilton, C.M.; Wilcox, P.G.; Quon, B.S. The Extrapulmonary Effects of Cystic Fibrosis Transmembrane Conductance Regulator Modulators in Cystic Fibrosis. Ann. Am. Thorac. Soc. 2020, 17, 147–154. [Google Scholar] [CrossRef]
- Hahn, T.J.; Squires, A.E.; Halstead, L.R.; Strominger, D.B. Reduced serum 25-hydroxyvitamin D concentration and disordered mineral metabolism in patients with cystic fibrosis. J. Pediatr. 1979, 94, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Aris, R.M.; Merkel, P.A.; Bachrach, L.K.; Borowitz, D.S.; Boyle, M.; Elkin, S.L.; Guise, T.A.; Hardin, D.S.; Haworth, C.S.; Holick, M.; et al. Guide to bone health and disease in cystic fibrosis. J. Clin. Endocrinol. Metab. 2005, 90, 1888–1896. [Google Scholar] [CrossRef] [Green Version]
- Elkin, S.L.; Fairney, A.; Burnett, S.; Kemp, M.; Kyd, P.; Burgess, J.; Compston, J.E.; Hodson, M.E. Vertebral deformities and low bone mineral density in adults with cystic fibrosis: A cross-sectional study. Osteoporos. Int. 2001, 12, 366–372. [Google Scholar] [CrossRef]
- Haworth, C.S.; Selby, P.L.; Webb, A.K.; Dodd, M.E.; Musson, H.; Niven, R.M.; Economou, G.; Horrocks, A.W.; Freemont, A.J.; Mawer, E.B.; et al. Low bone mineral density in adults with cystic fibrosis. Thorax 1999, 54, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Paccou, J.; Zeboulon, N.; Combescure, C.; Gossec, L.; Cortet, B. The prevalence of osteoporosis, osteopenia, and fractures among adults with cystic fibrosis: A systematic literature review with meta-analysis. Calcif. Tissue Int. 2010, 86, 1–7. [Google Scholar] [CrossRef]
- Garcia, S.; Terroso, G.; Amorim, A.; Redondo, M.; Costa, L. Bone involvement in young adults with cystic fibrosis—A Portuguese cohort. Acta Reumatol. Port 2021, 46, 283–285. [Google Scholar] [PubMed]
- Gensburger, D.; Boutroy, S.; Chapurlat, R.; Nove-Josserand, R.; Roche, S.; Rabilloud, M.; Durieu, I. Reduced bone volumetric density and weak correlation between infection and bone markers in cystic fibrosis adult patients. Osteoporos. Int. 2016, 27, 2803–2813. [Google Scholar] [CrossRef]
- Putman, M.S.; Baker, J.F.; Uluer, A.; Herlyn, K.; Lapey, A.; Sicilian, L.; Tillotson, A.P.; Gordon, C.M.; Merkel, P.A.; Finkelstein, J.S. Trends in bone mineral density in young adults with cystic fibrosis over a 15 year period. J. Cyst. Fibros. 2015, 14, 526–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejero García, S.; Giráldez Sánchez, M.A.; Cejudo, P.; Quintana Gallego, E.; Dapena, J.; García Jiménez, R.; Cano Luis, P.; Gómez de Terreros, I. Bone health, daily physical activity, and exercise tolerance in patients with cystic fibrosis. Chest 2011, 140, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Gur, M.; Bar-Yoseph, R.; Diab, G.; Hanna, M.; Rozen, G.; Daud, F.; Keidar, Z.; Toukan, Y.; Masarweh, K.; Nir, V.; et al. Understanding the interplay between factors that influence bone mineral density in CF. Pediatr. Pulmonol. 2020, 55, 2667–2673. [Google Scholar] [CrossRef]
- Stead, R.J.; Houlder, S.; Agnew, J.; Thomas, M.; Hodson, M.E.; Batten, J.C.; Dandona, P. Vitamin D and parathyroid hormone and bone mineralisation in adults with cystic fibrosis. Thorax 1988, 43, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Cairoli, E.; Eller-Vainicher, C.; Morlacchi, L.; Tarsia, P.; Rossetti, V.; Pappalettera, M.; Arosio, M.; Chiodini, I.; Blasi, F. Bone involvement in young adults with cystic fibrosis awaiting lung transplantation for end-stage respiratory failure. Osteoporos. Int. 2019, 30, 1255–1263. [Google Scholar] [CrossRef]
- Greer, R.M.; Buntain, H.M.; Potter, J.M.; Wainwright, C.E.; Wong, J.C.; O’Rourke, P.K.; Francis, P.W.; Bell, S.C.; Batch, J.A. Abnormalities of the PTH-vitamin D axis and bone turnover markers in children, adolescents and adults with cystic fibrosis: Comparison with healthy controls. Osteoporos. Int. 2003, 14, 404–411. [Google Scholar] [CrossRef]
- Juhász, M.F.; Varannai, O.; Németh, D.; Szakács, Z.; Kiss, S.; Izsák, V.D.; Martonosi, Á.R.; Hegyi, P.; Párniczky, A. Vitamin D supplementation in patients with cystic fibrosis: A systematic review and meta-analysis. J. Cyst. Fibros. 2021, 20, 729–736. [Google Scholar] [CrossRef]
- Durette, G.; Jomphe, V.; Bureau, N.J.; Poirier, C.; Ferraro, P.; Lands, L.C.; Mailhot, G. Long-term bone mineral density changes and fractures in lung transplant recipients with cystic fibrosis. J. Cyst. Fibros. 2021, 20, 525–532. [Google Scholar] [CrossRef]
- Ambroszkiewicz, J.; Sands, D.; Gajewska, J.; Chelchowska, M.; Laskowska-Klita, T. Bone turnover markers, osteoprotegerin and RANKL cytokines in children with cystic fibrosis. Adv. Med. Sci. 2013, 58, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.L.; Colombo, C.; Assael, B.M.; Dubini, A.; Lombardo, M.; Quattrucci, S.; Bella, S.; Collura, M.; Messore, B.; Raia, V.; et al. Treatment of low bone density in young people with cystic fibrosis: A multicentre, prospective, open-label observational study of calcium and calcifediol followed by a randomised placebo-controlled trial of alendronate. Lancet Respir. Med. 2013, 1, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Schall, J.; Stallings, V.A.; Zemel, B.S. Trabecular and cortical bone deficits are present in children and adolescents with cystic fibrosis. Bone 2016, 90, 7–14. [Google Scholar] [CrossRef]
- Lucidi, V.; Bizzarri, C.; Alghisi, F.; Bella, S.; Russo, B.; Ubertini, G.; Cappa, M. Bone and body composition analyzed by Dual-energy X-ray Absorptiometry (DXA) in clinical and nutritional evaluation of young patients with Cystic Fibrosis: A cross-sectional study. BMC Pediatr. 2009, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Jaksic, M.; Fenwick, S.; Byrnes, C.; Cundy, T. Accrual of Bone Mass in Children and Adolescents With Cystic Fibrosis. J. Clin. Endocrinol. Metab. 2017, 102, 1734–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, R.M.; Zemel, B.S.; Stallings, V.A.; Leonard, M.B.; Tsao, J.; Kelly, A. Bone accrual and structural changes over one year in youth with cystic fibrosis. J. Clin. Transl. Endocrinol. 2022, 28, 100297. [Google Scholar] [CrossRef]
- Baker, J.F.; Putman, M.S.; Herlyn, K.; Tillotson, A.P.; Finkelstein, J.S.; Merkel, P.A. Body composition, lung function, and prevalent and progressive bone deficits among adults with cystic fibrosis. Jt. Bone Spine 2016, 83, 207–211. [Google Scholar] [CrossRef]
- Atlas, G.; Yap, M.; Lim, A.; Vidmar, S.; Smith, N.; King, L.; Jones, A.; Hong, J.; Ranganathan, S.; Simm, P.J. The clinical features that contribute to poor bone health in young Australians living with cystic fibrosis: A recommendation for BMD screening. Pediatr. Pulmonol. 2021, 56, 2014–2022. [Google Scholar] [CrossRef]
- Smith, N.; Lim, A.; Yap, M.; King, L.; James, S.; Jones, A.; Ranganathan, S.; Simm, P. Bone mineral density is related to lung function outcomes in young people with cystic fibrosis-A retrospective study. Pediatr. Pulmonol. 2017, 52, 1558–1564. [Google Scholar] [CrossRef]
- Alicandro, G.; Bisogno, A.; Battezzati, A.; Bianchi, M.L.; Corti, F.; Colombo, C. Recurrent pulmonary exacerbations are associated with low fat free mass and low bone mineral density in young adults with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Aris, R.M.; Stephens, A.R.; Ontjes, D.A.; Blackwood, A.D.; Lark, R.K.; Hensler, M.B.; Neuringer, I.P.; Lester, G.E. Adverse alterations in bone metabolism are associated with lung infection in adults with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2000, 162, 1674–1678. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Bolívar, V.; Olveira, C.; Ruiz-García, I.; Porras, N.; García-Olivares, M.; Sánchez-Torralvo, F.J.; Girón, M.V.; Alonso-Gallardo, S.P.; Olveira, G. Handgrip Strength: Associations with Clinical Variables, Body Composition, and Bone Mineral Density in Adults with Cystic Fibrosis. Nutrients 2021, 13, 4107. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.S.; Gemma; Patel, A. Factors associated with low bone mineral density in patients with cystic fibrosis. J. Bone Miner. Metab. 2015, 33, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, I.H.; Pressler, T.; Oturai, P.; Katzenstein, T.L.; Skov, M.; Frikke-Schmidt, R.; Hitz, M.F. Osteoporosis Is Associated with Deteriorating Clinical Status in Adults with Cystic Fibrosis. Int. J. Endocrinol. 2018, 2018, 4803974. [Google Scholar] [CrossRef] [Green Version]
- Aris, R.M.; Renner, J.B.; Winders, A.D.; Buell, H.E.; Riggs, D.B.; Lester, G.E.; Ontjes, D.A. Increased rate of fractures and severe kyphosis: Sequelae of living into adulthood with cystic fibrosis. Ann. Intern. Med. 1998, 128, 186–193. [Google Scholar] [CrossRef]
- Anabtawi, A.; Holyoak, M.; He, J.; Cristiano, E.; Polineni, D.; Graves, L. 3rd. Trabecular bone score in people with cystic fibrosis. Osteoporos. Int. 2022, 33, 1137–1145. [Google Scholar] [CrossRef]
- Latzin, P.; Griese, M.; Hermanns, V.; Kammer, B. Sternal fracture with fatal outcome in cystic fibrosis. Thorax 2005, 60, 616. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.M.; Darukhanavala, A.; Hicks, R.; Kelly, A. An update on methods for assessing bone quality and health in Cystic fibrosis. J. Clin. Transl. Endocrinol. 2022, 27, 100281. [Google Scholar] [CrossRef]
- Kovacs, C.S.; Chaussain, C.; Osdoby, P.; Brandi, M.L.; Clarke, B.; Thakker, R.V. The role of biomineralization in disorders of skeletal development and tooth formation. Nat. Rev. Endocrinol. 2021, 17, 336–349. [Google Scholar] [CrossRef]
- Bolamperti, S.; Villa, I.; Rubinacci, A. Bone remodeling: An operational process ensuring survival and bone mechanical competence. Bone Res. 2022, 10, 48. [Google Scholar] [CrossRef]
- Oliveira, T.C.; Gomes, M.; Gomes, A. The Crossroads between Infection and Bone Loss. Microorganisms 2020, 8, 1765. [Google Scholar] [CrossRef] [PubMed]
- Weitzmann, M.N.; Ofotokun, I. Physiological and pathophysiological bone turnover—role of the immune system. Nat. Rev. Endocrinol. 2016, 12, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiao, Z.; Quarles, L.D.; Li, W. Osteoporosis: Mechanism, Molecular Target and Current Status on Drug Development. Curr. Med. Chem. 2021, 28, 1489–1507. [Google Scholar] [CrossRef] [PubMed]
- Putman, M.S.; Milliren, C.E.; Derrico, N.; Uluer, A.; Sicilian, L.; Lapey, A.; Sawicki, G.; Gordon, C.M.; Bouxsein, M.L.; Finkelstein, J.S. Compromised bone microarchitecture and estimated bone strength in young adults with cystic fibrosis. J. Clin. Endocrinol. Metab. 2014, 99, 3399–3407. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, K.K.; Agarwal, S.; Kepley, A.; Rosete, F.; Hu, Y.; Guo, X.E.; Keating, C.L.; DiMango, E.A.; Shane, E. Adults with cystic fibrosis have deficits in bone structure and strength at the distal tibia despite similar size and measuring standard and relative sites. Bone 2018, 107, 181–187. [Google Scholar] [CrossRef]
- Mailhot, G.; Dion, N.; Farlay, D.; Rizzo, S.; Bureau, N.J.; Jomphe, V.; Sankhe, S.; Boivin, G.; Lands, L.C.; Ferraro, P.; et al. Impaired rib bone mass and quality in end-stage cystic fibrosis patients. Bone 2017, 98, 9–17. [Google Scholar] [CrossRef]
- Elkin, S.L.; Vedi, S.; Bord, S.; Garrahan, N.J.; Hodson, M.E.; Compston, J.E. Histomorphometric analysis of bone biopsies from the iliac crest of adults with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2002, 166, 1470–1474. [Google Scholar] [CrossRef]
- Haworth, C.S.; Webb, A.K.; Egan, J.J.; Selby, P.L.; Hasleton, P.S.; Bishop, P.W.; Freemont, T.J. Bone histomorphometry in adult patients with cystic fibrosis. Chest 2000, 118, 434–439. [Google Scholar] [CrossRef]
- Dif, F.; Marty, C.; Baudoin, C.; de Vernejoul, M.C.; Levi, G. Severe osteopenia in CFTR-null mice. Bone 2004, 35, 595–603. [Google Scholar] [CrossRef]
- Mora Vallellano, J.; Delgado Pecellín, C.; Delgado Pecellín, I.; Quintana Gallego, E.; López-Campos, J.L. Evaluation of bone metabolism in children with cystic fibrosis. Bone 2021, 147, 115929. [Google Scholar] [CrossRef]
- Rossini, M.; Del Marco, A.; Santo, F.D.; Gatti, D.; Braggion, C.; James, G.; Adami, S. Prevalence and correlates of vertebral fractures in adults with cystic fibrosis. Bone 2004, 35, 771–776. [Google Scholar] [CrossRef]
- Sermet-Gaudelus, I.; Souberbielle, J.C.; Ruiz, J.C.; Vrielynck, S.; Heuillon, B.; Azhar, I.; Cazenave, A.; Lawson-Body, E.; Chedevergne, F.; Lenoir, G. Low bone mineral density in young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 951–957. [Google Scholar] [CrossRef]
- Bianchi, M.L.; Romano, G.; Saraifoger, S.; Costantini, D.; Limonta, C.; Colombo, C. BMD and body composition in children and young patients affected by cystic fibrosis. J. Bone Miner. Res. 2006, 21, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, J.; Delion, M.; Gangloff, S.; Braux, J.; Velard, F. Bone disease in cystic fibrosis: New pathogenic insights opening novel therapies. Osteoporos. Int. 2016, 27, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Shead, E.F.; Haworth, C.S.; Condliffe, A.M.; McKeon, D.J.; Scott, M.A.; Compston, J.E. Cystic fibrosis transmembrane conductance regulator (CFTR) is expressed in human bone. Thorax 2007, 62, 650–651. [Google Scholar] [CrossRef] [Green Version]
- Bronckers, A.; Kalogeraki, L.; Jorna, H.J.; Wilke, M.; Bervoets, T.J.; Lyaruu, D.M.; Zandieh-Doulabi, B.; DenBesten, P.; de Jonge, H. The cystic fibrosis transmembrane conductance regulator (CFTR) is expressed in maturation stage ameloblasts, odontoblasts and bone cells. Bone 2010, 46, 1188–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Yang, L.; Ma, T.; Zhao, Y. Functional expression of cystic fibrosis transmembrane conductance regulator in mouse chondrocytes. Clin. Exp. Pharmacol. Physiol. 2010, 37, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Orlando, V.; Morin, G.; Laffont, A.; Lénart, D.; Barrera, C.S.; Mustafy, T.; Sankhe, S.; Villemure, I.; Mailhot, G. CFTR deletion affects mouse osteoblasts in a gender-specific manner. J Cell Physiol. 2020, 235, 6736–6753. [Google Scholar] [CrossRef]
- Pashuck, T.D.; Franz, S.E.; Altman, M.K.; Wasserfall, C.H.; Atkinson, M.A.; Wronski, T.J.; Flotte, T.R.; Stalvey, M.S. Murine model for cystic fibrosis bone disease demonstrates osteopenia and sex-related differences in bone formation. Pediatr. Res. 2009, 65, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Haston, C.K.; Li, W.; Li, A.; Lafleur, M.; Henderson, J.E. Persistent osteopenia in adult cystic fibrosis transmembrane conductance regulator-deficient mice. Am. J. Respir. Crit. Care Med. 2008, 177, 309–315. [Google Scholar] [CrossRef]
- Le Henaff, C.; Gimenez, A.; Hay, E.; Marty, C.; Marie, P.; Jacquot, J. The F508del mutation in cystic fibrosis transmembrane conductance regulator gene impacts bone formation. Am. J. Pathol. 2012, 180, 2068–2075. [Google Scholar] [CrossRef] [PubMed]
- Le Henaff, C.; Haÿ, E.; Velard, F.; Marty, C.; Tabary, O.; Marie, P.J.; Jacquot, J.P. Enhanced F508del-CFTR channel activity ameliorates bone pathology in murine cystic fibrosis. Am. J. Pathol. 2014, 184, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Stalvey, M.S.; Clines, K.L.; Havasi, V.; McKibbin, C.R.; Dunn, L.K.; Chung, W.J.; Clines, G.A. Osteoblast CFTR inactivation reduces differentiation and osteoprotegerin expression in a mouse model of cystic fibrosis-related bone disease. PLoS ONE 2013, 8, e80098. [Google Scholar] [CrossRef] [PubMed]
- Paradis, J.; Wilke, M.; Haston, C.K. Osteopenia in Cftr-deltaF508 mice. J. Cyst. Fibros. 2010, 9, 239–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, C.; Bacchetta, J.; Braillon, P.; Chapurlat, R.; Drai, J.; Reix, P. Children and adolescents with cystic fibrosis display moderate bone microarchitecture abnormalities: Data from high-resolution peripheral quantitative computed tomography. Osteoporos. Int. 2017, 28, 3179–3188. [Google Scholar] [CrossRef] [PubMed]
- King, S.J.; Topliss, D.J.; Kotsimbos, T.; Nyulasi, I.B.; Bailey, M.; Ebeling, P.R.; Wilson, J.W. Reduced bone density in cystic fibrosis: DeltaF508 mutation is an independent risk factor. Eur. Respir. J. 2005, 25, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Braux, J.; Jourdain, M.-L.; Guillaume, C.; Untereiner, V.; Piot, O.; Baehr, A.; Klymiuk, N.; Winter, N.; Berri, M.; Buzoni-Gatel, D.; et al. CFTR-deficient pigs display alterations of bone microarchitecture and composition at birth. J. Cyst. Fibros. 2020, 19, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Stalvey, M.S.; Havasi, V.; Tuggle, K.L.; Wang, D.; Birket, S.; Rowe, S.M.; Sorscher, E.J. Reduced bone length, growth plate thickness, bone content, and IGF-I as a model for poor growth in the CFTR-deficient rat. PLoS ONE 2017, 12, e0188497. [Google Scholar] [CrossRef] [Green Version]
- Velard, F.; Delion, M.; Le Henaff, C.; Guillaume, C.; Gangloff, S.; Jacquot, J.; Tabary, O.; Touqui, L.; Barthes, F.; Sermet-Gaudelus, I. Cystic fibrosis and bone disease: Defective osteoblast maturation with the F508del mutation in cystic fibrosis transmembrane conductance regulator. Am. J. Respir. Crit. Care Med. 2014, 189, 746–748. [Google Scholar] [CrossRef]
- Gimenez-Maitre, A.; Le Henaff, C.; Norez, C.; Guillaumé, C.; Ravoninjatovo, B.; Laurent-Maquin, D.; Becq, F.; Jacquot, J. Deficit of osteoprotegerin release by osteoblasts from a patient with cystic fibrosis. Eur. Respir. J. 2012, 39, 780–781. [Google Scholar] [CrossRef] [Green Version]
- Le Henaff, C.; Mansouri, R.; Modrowski, D.; Zarka, M.; Geoffroy, V.; Marty, C.; Tarantino, N.; Laplantine, E.; Marie, P.J. Increased NF-kappaB Activity and Decreased Wnt/beta-Catenin Signaling Mediate Reduced Osteoblast Differentiation and Function in DeltaF508 Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Mice. J. Biol. Chem. 2015, 290, 18009–18017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Gault, W.J.; Gotthardt, D.; Rohatgi, R.; Klein, T.J.; Shao, Y.; Lee, H.J.; Wu, A.L.; Fang, Y.; Satlin, L.M.; et al. Electrochemical cues regulate assembly of the Frizzled/Dishevelled complex at the plasma membrane during planar epithelial polarization. Nat. Cell Biol. 2009, 11, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, Y.; Zhang, J.; Chen, Y.; Liu, Y.; Lin, Z.; Liu, M.; Sheng, K.; Liao, H.; Tsang, K.S.; et al. CFTR mutation enhances Dishevelled degradation and results in impairment of Wnt-dependent hematopoiesis. Cell Death Dis. 2018, 9, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Henaff, C.; Faria Da Cunha, M.; Hatton, A.; Tondelier, D.; Marty, C.; Collet, C.; Zarka, M.; Geoffroy, V.; Zatloukal, K.; Laplantine, E.; et al. Genetic deletion of keratin 8 corrects the altered bone formation and osteopenia in a mouse model of cystic fibrosis. Hum. Mol. Genet. 2016, 25, 1281–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Heron, L.; Guillaume, C.; Velard, F.; Braux, J.; Touqui, L.; Moriceau, S.; Sermet-Gaudelus, I.; Laurent-Maquin, D.; Jacquot, J. Cystic fibrosis transmembrane conductance regulator (CFTR) regulates the production of osteoprotegerin (OPG) and prostaglandin (PG) E2 in human bone. J. Cyst. Fibros. 2010, 9, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Baudouin-Legros, M.; Colas, J.; Moriceau, S.; Kelly, M.; Planelles, G.; Edelman, A.; Ollero, M. Long-term CFTR inhibition modulates 15d-prostaglandin J2 in human pulmonary cells. Int. J. Biochem. Cell Biol. 2012, 44, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Goessling, W.; North, T.E.; Loewer, S.; Lord, A.M.; Lee, S.; Stoick-Cooper, C.L.; Weidinger, G.; Puder, M.; Daley, G.Q.; Moon, R.T.; et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 2009, 136, 1136–1147. [Google Scholar] [CrossRef] [Green Version]
- Napimoga, M.H.; Demasi, A.P.; Bossonaro, J.P.; de Araujo, V.C.; Clemente-Napimoga, J.T.; Martinez, E.F. Low doses of 15d-PGJ2 induce osteoblast activity in a PPAR-gamma independent manner. Int. Immunopharmacol. 2013, 16, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, K.A.; Raisz, L.; Pilbeam, C. Prostaglandins in bone: Bad cop, good cop? Trends Endocrinol. Metab. 2010, 21, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Lam, G.Y.; Desai, S.; Fu, J.; Hu, X.Y.; Jang, J.; Goshtasebi, A.; Kalyan, S.; Quon, B.S. IL-8 correlates with reduced baseline femoral neck bone mineral density in adults with cystic fibrosis: A single center retrospective study. Sci. Rep. 2021, 11, 15405. [Google Scholar] [CrossRef]
- Delion, M.; Braux, J.; Jourdain, M.-L.; Guillaume, C.; Bour, C.; Gangloff, S.; Le Pimpec-Barthes, F.; Sermet-Gaudelus, I.; Jacquot, J.; Velard, F. Overexpression of RANKL in osteoblasts: A possible mechanism of susceptibility to bone disease in cystic fibrosis. J. Pathol. 2016, 240, 50–60. [Google Scholar] [CrossRef]
- Shead, E.F.; Haworth, C.S.; Gunn, E.; Bilton, D.; Scott, M.A.; Compston, J.E. Osteoclastogenesis during infective exacerbations in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 306–311. [Google Scholar] [CrossRef]
- Jourdain, M.-L.; Sergheraert, J.; Braux, J.; Guillaume, C.; Gangloff, S.C.; Hubert, D.; Velard, F.; Jacquot, J. Osteoclastogenesis and sphingosine-1-phosphate secretion from human osteoclast precursor monocytes are modulated by the cystic fibrosis transmembrane conductance regulator. Biochim. Biophys. Acta. Mol. Basis. Dis. 2021, 1867, 166010. [Google Scholar] [CrossRef]
- Jourdain, M.-L.; Sergheraert, J.; Braux, J.; Guillaume, C.; Gangloff, S.C.; Hubert, D.; Velard, F.; Jacquot, J. Impaired interleukin-8 chemokine secretion by staphylococcus aureus-activated epithelium and T-cell chemotaxis in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 644–650. [Google Scholar]
- Augarten, A.; Paret, G.; Avneri, I.; Akons, H.; Aviram, M.; Bentur, L.; Blau, H.; Efrati, O.; Szeinberg, A.; Barak, A.; et al. Systemic inflammatory mediators and cystic fibrosis genotype. Clin. Exp. Med. 2004, 4, 99–102. [Google Scholar] [CrossRef]
- Augarten, A.; Paret, G.; Avneri, I.; Akons, H.; Aviram, M.; Bentur, L.; Blau, H.; Efrati, O.; Szeinberg, A.; Barak, A.; et al. Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis. J. Immunol. 2011, 186, 6990–6998. [Google Scholar]
- Augarten, A.; Paret, G.; Avneri, I.; Akons, H.; Aviram, M.; Bentur, L.; Blau, H.; Efrati, O.; Szeinberg, A.; Barak, A.; et al. Connections between genetics and clinical data: Role of MCP-1, CFTR, and SPINK-1 in the setting of acute, acute recurrent, and chronic pancreatitis. Am. J. Gastroenterol. 2010, 105, 199–206. [Google Scholar]
- Augarten, A.; Paret, G.; Avneri, I.; Akons, H.; Aviram, M.; Bentur, L.; Blau, H.; Efrati, O.; Szeinberg, A.; Barak, A.; et al. Genistein inhibits constitutive and inducible NFkappaB activation and decreases IL-8 production by human cystic fibrosis bronchial gland cells. Am. J. Pathol. 1999, 155, 473–481. [Google Scholar]
- Zaman, M.M.; Gelrud, A.; Junaidi, O.; Regan, M.M.; Warny, M.; Shea, J.C.; Kelly, C.; O’Sullivan, B.P.; Freedman, S.D. Interleukin 8 secretion from monocytes of subjects heterozygous for the deltaF508 cystic fibrosis transmembrane conductance regulator gene mutation is altered. Clin. Diagn. Lab. Immunol. 2004, 11, 819–824. [Google Scholar]
- Kim, M.S.; Day, C.; Morrison, N. MCP-1 is induced by receptor activator of nuclear factor-{kappa}B ligand, promotes human osteoclast fusion, and rescues granulocyte macrophage colony-stimulating factor suppression of osteoclast formation. J. Biol. Chem. 2005, 280, 16163–16169. [Google Scholar] [CrossRef] [Green Version]
- Shead, E.F.; Haworth, C.S.; Barker, H.; Bilton, D.; Compston, J.E. Osteoclast function, bone turnover and inflammatory cytokines during infective exacerbations of cystic fibrosis. J. Cyst. Fibros. 2010, 9, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarique, A.A.; Sly, P.D.; Cardenas, D.G.; Luo, L.; Stow, J.L.; Bell, S.C.; Wainwright, C.E.; Fantino, E. Differential expression of genes and receptors in monocytes from patients with cystic fibrosis. J. Cyst. Fibros. 2019, 18, 342–348. [Google Scholar] [CrossRef]
- Bruscia, E.M.; Bonfield, T.L. Update on Innate and Adaptive Immunity in Cystic Fibrosis. Clin. Chest Med. 2022, 43, 603–615. [Google Scholar] [CrossRef]
- Averna, M.; Melotti, P.; Sorio, C. Revisiting the Role of Leukocytes in Cystic Fibrosis. Cells 2021, 10, 3380. [Google Scholar] [CrossRef]
- Morales-Nebreda, L.; Misharin, A.V.; Perlman, H.; Budinger, G.R. The heterogeneity of lung macrophages in the susceptibility to disease. Eur. Respir. Rev. 2015, 24, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Hubeau, C.; Puchelle, E.; Gaillard, D. Distinct pattern of immune cell population in the lung of human fetuses with cystic fibrosis. J. Allergy Clin. Immunol. 2001, 108, 524–529. [Google Scholar] [CrossRef]
- Brennan, S.; Sly, P.D.; Gangell, C.L.; Sturges, N.; Winfield, K.; Wikstrom, M.; Gard, S.; Upham, J.W.; Cf, O.B.O.A. Alveolar macrophages and CC chemokines are increased in children with cystic fibrosis. Eur. Respir. J. 2009, 34, 655–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elizur, A.; Cannon, C.; Ferkol, T. Airway inflammation in cystic fibrosis. Chest 2008, 133, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.A.; Brennan, S.; Gard, S.; Berry, L.; Gangell, C.; Stick, S.M.; Clements, B.S.; Sly, P.D. Acquisition and eradication of P. aeruginosa in young children with cystic fibrosis. Eur. Respir. J. 2009, 33, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Lévêque, M.; Le Trionnaire, S.; Del Porto, P.; Martin-Chouly, C. The impact of impaired macrophage functions in cystic fibrosis disease progression. J. Cyst. Fibros. 2017, 16, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Bonfield, T.L.; Hodges, C.A.; Cotton, C.U.; Drumm, M.L. Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J. Leukoc. Biol. 2012, 92, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Shmarina, G.; Pukhalsky, A.; Petrova, N.; Zakharova, E.; Avakian, L.; Kapranov, N.; Alioshkin, V. TNF gene polymorphisms in cystic fibrosis patients: Contribution to the disease progression. J. Transl. Med. 2013, 11, 19. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, A.; Strait, K.; Zhang, H.; Nanes, M.S.; Weitzmann, M.N. Endogenous TNFalpha lowers maximum peak bone mass and inhibits osteoblastic Smad activation through NF-kappaB. J. Bone Miner. Res. 2007, 22, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.C.; Sousa, D.M.; Oliveira, T.C. Serum Amyloid A proteins reduce bone mass during mycobacterial infections. bioRxiv 2022, 2022, 10.24.513637. [Google Scholar]
- Bruscia, E.M.; Zhang, P.X.; Ferreira, E.; Caputo, C.; Emerson, J.W.; Tuck, D.; Krause, D.S.; Egan, M.E. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/- mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Ode, K.L.; Moran, A. New insights into cystic fibrosis-related diabetes in children. Lancet Diabetes Endocrinol. 2013, 1, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Wei, J.; Yoshizawa, T.; Del Fattore, A.; DePinho, R.A.; Teti, A.; Ducy, P.; Karsenty, G. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 2010, 142, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Guo, Q.; Xiao, Y.; Guo, Q.; Huang, Y.; Li, C.; Luo, X. Endocrine role of bone in the regulation of energy metabolism. Bone Res. 2021, 9, 25. [Google Scholar] [CrossRef]
- Karsenty, G.; Ferron, M. The contribution of bone to whole-organism physiology. Nature 2012, 481, 314–320. [Google Scholar] [CrossRef]
- Wei, J.; Ferron, M.; Clarke, C.J.; Hannun, Y.A.; Jiang, H.; Blaner, W.S.; Karsenty, G. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J. Clin. Investig. 2014, 124, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Nguyen, A.; Enriquez, R.F.; Luzuriaga, J.; Bensellam, M.; Laybutt, R.; Baldock, P.A.; Herzog, H. NPY signalling in early osteoblasts controls glucose homeostasis. Mol. Metab. 2015, 4, 164–174. [Google Scholar] [CrossRef]
- Napoli, N.; Chandran, M.; Pierroz, D.D.; Abrahamsen, B.; Schwartz, A.V.; Ferrari, S.L. Mechanisms of diabetes mellitus-induced bone fragility. Nat. Rev. Endocrinol. 2017, 13, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Napoli, N.; Chandran, M.; Pierroz, D.D.; Abrahamsen, B.; Schwartz, A.V.; Ferrari, S.L. The impact of dysglycaemia on bone mineral accrual in young people with cystic fibrosis. Clin. Endocrinol. 2013, 78, 36–42. [Google Scholar]
- Mathiesen, I.H.; Hitz, M.F.; Katzenstein, T.L.; Oturai, P.; Skov, M.; Jørgensen, N.R.; Jensen, P.Ø.; Mikkelsen, C.R.; Krogh-Madsen, R.; Pressler, T.; et al. Markers of bone turnover are reduced in patients with CF related diabetes; the role of glucose. J. Cyst. Fibros. 2019, 18, 436–441. [Google Scholar] [CrossRef]
- Putman, M.S.; Anabtawi, A.; Le, T.; Tangpricha, V.; Sermet-Gaudelus, I. Cystic fibrosis bone disease treatment: Current knowledge and future directions. J. Cyst. Fibros. 2019, 18 (Suppl. 2), S56–S65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conwell, L.S.; Chang, A. Bisphosphonates for osteoporosis in people with cystic fibrosis. Cochrane Database Syst. Rev. 2014, 2014, CD002010. [Google Scholar] [CrossRef] [Green Version]
- Keown, K.; Brown, R.; Doherty, D.F.; Houston, C.; McKelvey, M.C.; Creane, S.; Linden, D.; McAuley, D.F.; Kidney, J.C.; Weldon, S.; et al. Airway Inflammation and Host Responses in the Era of CFTR Modulators. Int. J. Mol. Sci. 2020, 21, 6379. [Google Scholar] [CrossRef] [PubMed]
- Keown, K.; Brown, R.; Doherty, D.F.; Houston, C.; McKelvey, M.C.; Creane, S.; Linden, D.; McAuley, D.F.; Kidney, J.C.; Weldon, S.; et al. Ivacaftor decreases monocyte sensitivity to interferon-γ in people with cystic fibrosis. ERJ Open Res. 2020, 6, 00318–02019. [Google Scholar]
- Kopp, B.T.; Fitch, J.; Jaramillo, L.; Shrestha, C.L.; Robledo-Avila, F.; Zhang, S.; Palacios, S.; Woodley, F.; Hayes, D.; Partida-Sanchez, S.; et al. Whole-blood transcriptomic responses to lumacaftor/ivacaftor therapy in cystic fibrosis. J. Cyst. Fibros. 2020, 19, 245–254. [Google Scholar] [CrossRef]
- Putman, M.S.; Greenblatt, L.B.; Bruce, M.; Joseph, T.; Lee, H.; Sawicki, G.; Uluer, A.; Sicilian, L.; Neuringer, I.; Gordon, C.M.; et al. The Effects of Ivacaftor on Bone Density and Microarchitecture in Children and Adults with Cystic Fibrosis. J. Clin. Endocrinol. Metab. 2021, 106, e1248–e1261. [Google Scholar] [CrossRef]
- Sermet-Gaudelus, I.; Delion, M.; Durieu, I.; Jacquot, J.; Hubert, D. Bone demineralization is improved by ivacaftor in patients with cystic fibrosis carrying the p.Gly551Asp mutation. J. Cyst. Fibros. 2016, 15, e67–e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velard, F.; Delion, M.; Lemaire, F.; Tabary, O.; Guillaume, C.; Le Pimpec Barthès, F.; Touqui, L.; Gangloff, S.; Sermet-Gaudelus, I.; Jacquot, J. Cystic fibrosis bone disease: Is the CFTR corrector C18 an option for therapy? Eur. Respir. J. 2015, 45, 845–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sermet-Gaudelus, I.; Bianchi, M.L.; Garabédian, M.; Aris, R.M.; Morton, A.; Hardin, D.S.; Elkin, S.L.; Compston, J.E.; Conway, S.P.; Castanet, M.; et al. European cystic fibrosis bone mineralisation guidelines. J. Cyst. Fibros. 2011, 10 (Suppl. 2), S16–S23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, M.; Holfelder, C.; Kneppo, C.; Kieser, M.; Kasperk, C.; Schoenau, E.; Sommerburg, O.; Tönshoff, B. Multiple prevalent fractures in relation to macroscopic bone architecture in patients with cystic fibrosis. J. Cyst. Fibros. 2018, 17, 114–120. [Google Scholar] [CrossRef] [Green Version]
- Ratti, G.A.; Fernandez, G.S.; Schechter, M.S.; Stalvey, M.S.; Ostrenga, J.; Fink, A.K.; Jain, R. Bone mineral density screening by DXA for people with cystic fibrosis: A registry analysis of patient and program factors influencing rates of screening. J. Cyst. Fibros. 2022, 21, 784–791. [Google Scholar] [CrossRef]
- Silva Júnior, C.C.D.; Marques Queiroz, D.J.; de Paiva, M.P.; Lopes, M.T.; da Cunha Costa, M.; de Matos Bezerra, P.G.; de Carvalho Costa, M.J.; Silva, A.S.; Filho, J.M.; Braga Cartaxo, C.G.; et al. Evaluation of anthropometry as an alternative to DXA as predictor of low bone mineral density in children and adolescents with cystic fibrosis. Clin. Nutr. ESPEN 2021, 45, 229–235. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca, Ó.; Gomes, M.S.; Amorim, M.A.; Gomes, A.C. Cystic Fibrosis Bone Disease: The Interplay between CFTR Dysfunction and Chronic Inflammation. Biomolecules 2023, 13, 425. https://doi.org/10.3390/biom13030425
Fonseca Ó, Gomes MS, Amorim MA, Gomes AC. Cystic Fibrosis Bone Disease: The Interplay between CFTR Dysfunction and Chronic Inflammation. Biomolecules. 2023; 13(3):425. https://doi.org/10.3390/biom13030425
Chicago/Turabian StyleFonseca, Óscar, Maria Salomé Gomes, Maria Adelina Amorim, and Ana Cordeiro Gomes. 2023. "Cystic Fibrosis Bone Disease: The Interplay between CFTR Dysfunction and Chronic Inflammation" Biomolecules 13, no. 3: 425. https://doi.org/10.3390/biom13030425