
The Sensitivity of Tau Tracers for the Discrimination of Alzheimer’s Disease Patients and Healthy Controls by PET

Abstract
:1. Introduction
Molecular Description of Tau Aggregates
2. Development of Tau Tracers for PET
2.1. [18F]FDDNP, the Accidental First Tau PET Tracer
2.1.1. In Vitro Studies
2.1.2. In Vivo PET Studies of [18F]FDDNP in AD
2.2. Arylquinoline and Arylquinoxaline Derivatives
2.2.1. [18F]THK-523 (BF-242)
2.2.2. PET Studies with [18F]THK-523
2.2.3. [18F]THK-951
2.2.4. [18F]THK-5105 and [18F]THK-5117
Human PET Studies of [18F]THK-5105 and [18F]THK-5117
2.2.5. [18F]THK-5351
Human PET Studies of [18F]THK-5351
2.2.6. 2-Phenylquinoxaline Derivatives
2.3. Derivatives of Lanzoprasole and Astemizole
2.4. Benzimidazopyridine and Benzimidazopyrimidine Derivatives: Flortaucipir and T808
2.4.1. In Vitro Studies with [18F]Flortaucipir and [18F]T808
2.4.2. PET Studies with [18F]Flortaucipir and [18F]T808
2.5. Pyridinyl-Butadienyl-Benzothiazole (PBB) Derivatives
2.5.1. [11C]PBB3
2.5.2. [18F]PM-PBB3
PET Studies PM-PBB3
2.6. Pyrrolo-Pyridineisoquinolineamines
2.6.1. [18F]MK-6240
2.6.2. [18F]GTP1
2.6.3. [11C]RO6931643, [18F]RO6958948, and [11C]RO6924963
2.7. Napthyridine Derivatives or Janssen Series (JNJ)
2.7.1. [18F]JNJ-311
2.7.2. JNJ-067
2.8. [18F]PI-2620
2.9. Radioiodinated Benzoimidazopyridine (BIP) Derivatives
3. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Compound, Term, Acronym | Figure | Names, Synonyms |
| Astemizole | Figure 4b | hismanal; histaminos; paralergin; loaridal; retolen; astemison; 1-[(4-fluorophenyl)methyl]-N-[1-[2-(4-methoxyphenyl)ethyl]piperidin-4-yl] benzimidazol-2-amine; CAS-RN: 68844-77-9 |
| AV-1451 | T807; 7-(6-fluoropyridin-3-yl)-5H-pyrido[4,3-b]indole; CAS RN: 1415379-56-4 | |
| [18F]AV1451 | Figure 5a | [18F]flortaucipir; [18F]T807; [18F]LY 3191748; [18F]AV-1451; 7-(6-[18F]fluoro-pyridin-3-yl)-5H-pyrido[4,3-b]indole; CAS RN: 1522051-90-6 |
| BBB | blood brain barrier | |
| BF-158 | Figure 2a | N-methyl-4-(quinolin-2-yl)aniline; CAS RN: 682763-67-3 |
| [11C]BF-158 | Figure 2e | N-[11C]methyl-4-(quinolin-2-yl)aniline |
| BF-168 | Figure 2c | (E)-4-(2-(6-(2-fluoroethoxy)benzo[d]oxazol-2-yl)vinyl)-N-methylaniline; CAS RN: 634911-47-0 |
| BF-170 | Figure 2b | 2-(4-aminophenyl)quinoline |
| [11C]BF-227 | Figure 2g | (E)-5-(2-(6-(2-fluoroethoxy)benzo[d]oxazol-2-yl)vinyl)-N-methyl-N-[11C]methyl-thiazol-2-amine |
| BF-242 | Figure 2d | THK-523; 4-[6-(2-fluoroethoxy)-2-quinolinyl]aniline; CAS RN: 1573029-17-0 |
| [18F]BF-242 | Figure 2f | [18F]THK-523; 4-[6-(2-[18F]fluoroethoxy)-2-quinolinyl]aniline |
| BIP | benzimidazopyridine | |
| [125I]BIP-NMe2 | Figure 9a | 7-[125I]iodo-N,N-dimethylbenzo[[4,5]]imidazo[1,2-a]pyridin-3-amine |
| [18F]Br-BIPF | Figure 9d | 7-bromo-N-(2-([18F]fluoroethyl)-N-methylbenzo[[4,5]]imidazo[1,2-a]pyridin-3-amine |
| [18F]Cl-BIPF | Figure 9e | 7-chloro-N-(2-[18F]fluoroethyl)-N-methylbenzo[[4,5]]imidazo[1,2-a]pyridin-3-amine |
| [18F]IBIPF1 | Figure 9b | N-(2-[18F]fluoroethyl)-7-iodo-N-methylbenzo[[4,5]]imidazo[1,2-a]pyridin-3-amine |
| [18F]IBIPF2 | Figure 9c | N-(2-[18F]fluoroethyl)-7-iodobenzo[[4,5]]imidazo[1,2-a]pyridin-3-amine |
| [18F]Me-BIPF | Figure 9f | N-(2-[18F]fluoroethyl)-N,7-dimethylbenzo[[4,5]]imidazo[1,2-a]pyridin-3-amine |
| [18F]OMe-BIPF | Figure 9g | N-(2-([18F]fluoroethyl)-7-methoxy-N-methylbenzo[[4,5]]imidazo[1,2-a]pyridin-3-amine |
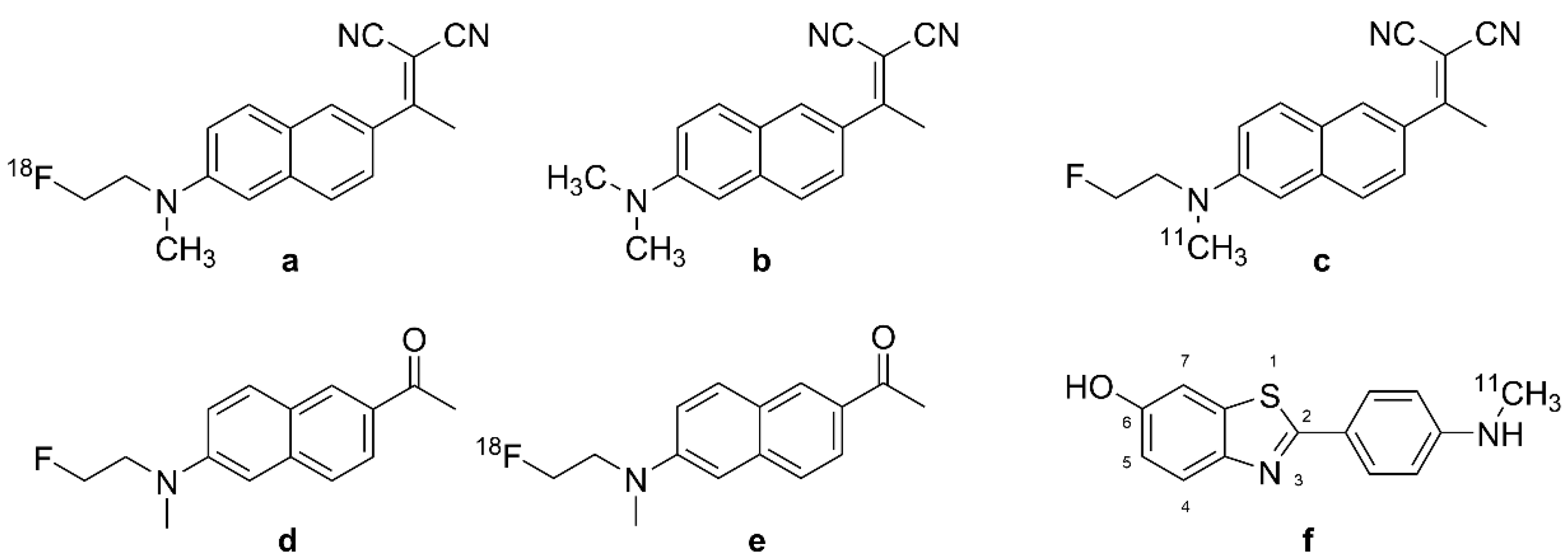
| DDNP | Figure 1b | 1,1-dicyano-2-[6-(dimethylamino)naphthalene-2-yl]propene; 2-[1-[1-(dimethylamino)naphthalen-2-yl]ethylidene]propanedinitrile; CAS RN: 178385-38-1 |
| FDDNP | FDDNP; DMFEAN 2-[1-[6-[2-fluoroethyl(methyl)amino]naphthalen-2-yl]ethylidene]propanedinitrile CAS RN: 590365-47-2 | |
| [18F]FDDNP | Figure 1a | Fddnp F-18; [18F]UNII-3J4JP3286H 2-[1-[6-[2-[18F]fluoroethyl(methyl)amino]naphthalen-2-yl]ethylidene] propanedinitrile; CAS RN: 259738-99-3 |
| FENE | Figure 1d | 1-(6-[(2-fluoroethyl)(methyl)amino]naphthalen-2-yl)ethanone CAS RN: 1260894-66-3 |
| [18F]FENE | Figure 1e | 1-(6-[(2-[18F]fluoroethyl)(methyl)amino]naphthalen-2-yl)ethanone |
| flortaucipir | T807; 7-(6-fluoropyridin-3-yl)-5H-pyrido[4,3-b]indole; CAS RN: 1415379-56-4 | |
| [18F]flortaucipir | Figure 5a | [18F]T807; [18F]LY 3191748; [18F]AV-1451; 7-(6-[18F]fluoro-pyridin-3-yl)-5H-pyrido[4,3-b]indole; CAS RN: 1522051-90-6 |
| [18F]GTP1 | Figure 7b | Genetech Tau Probe 1 |
| [18F]JNJ-64326067 | Figure 8d | [18F]JNJ067; N-(4-[18F]Fluoro-5-methylpyridin-2-yl)isoquinolin-6-amine N-[4-[18F]fluoro-5-methyl-2-pyridinyl]-6-isoquinolinamine, CAS RN: 2173357-42-9 |
| [18F]JNJ-64349311 | Figure 8c | [18F]JNJ311; 18F-JNJ 311; 6-[18F]fluoro-N-(2-methyl-4-pyridinyl)-1,5-naphthyridin-2-amine; CAS RN: 2121497-78-5 |
| [18F]JNJ (Co. No. 2) | Figure 8b | 6-[18F]fluoro-N-(3-methyl-4-pyridinyl)-1,5-naphthyridin-2-amineCAS RN: 2121497-79-6; Moechars, D.W.E. et al. AU Pat. 2017216212 |
| [18F]JNJ (Co. Nr. 3) | Figure 8a | 6-[18F]fluoro-N-4-pyridinyl-1,5-naphthyridin-2-amine CAS RN: 2121497-80-9, Moechars, D.W.E. et al. AU Pat. 2017216212 |
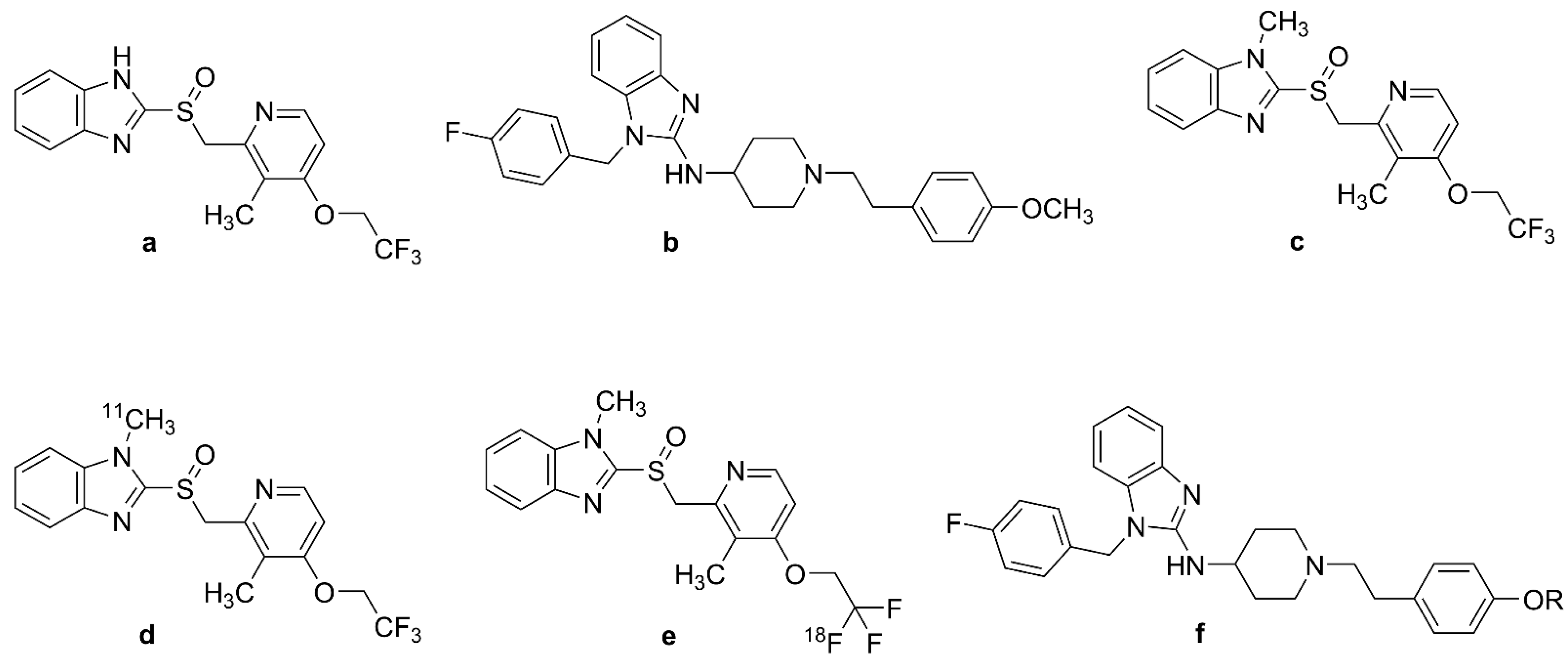
| Lansoprazole | Figure 4a | prevacid; bamalite; monolitum; ogastro; agopton; 2-[[3-methyl-4-(2,2,2-trifluoroethoxy)pyridin-2-yl]methylsulfinyl]-1H-benzimidazole CAS RN: 103577-45-3 |
| MK-6240 | 6-fluoro-3-(1H-pyrrolo[2,3-c]pyridin-1-yl)isoquinolin-5-amine CAS RN: 1841078-87-2 | |
| [18F]MK-6240 | Figure 7a | [18F]MNI-946; 6-[18F]fluoro-3-(1H-pyrrolo[2,3-c]pyridin-1-yl)isoquinolin-5-amine |
| NML | Figure 4c | N-Methyl-lansoprazole; 1-methyl-2-[[3-methyl-4-(2,2,2-trifluoroethoxy)pyridin-2-yl]methylsulfinyl]benzimidazole |
| [11C]NML | Figure 4d | [11C]-N-Methyl-lansoprazole; 1-[11C]methyl-2-[[3-methyl-4-(2,2,2-trifluoroethoxy)pyridin-2-yl]methylsulfinyl]benzimidazole |
| [18F]NML | Figure 4e | [18F]-N-Methyl-lansoprazole; 2-(((4-(2,2-difluoro-2-[18F]fluoro-ethoxy)-3-methylpyridin-2-yl)methyl)sulfinyl)-1-methyl-1H-benzo[d]imidazole |
| PI-2620 | 2-(2-[18F]fluoro-pyridin-4-yl)-9H-pyrrolo[2,3-b:4,5-c’]dipyridine | |
| [18F]PI-2620 | Figure 7f | [18F]MNI-960; 11-(2-[18F]fluoranylpyridin-4-yl)-4,8,10-triazatricyclo[7.4.0.02,7]trideca-1(9),2(7), 3,5,10,12-hexaene; 2-(2-([18F]fluoro-4-pyridinyl)-9H-pyrrolo(2,3-b:4,5-c’)dipyridine; CAS RN: 2173353-61-0 |
| PBB | pyridinyl-butadienyl-benzothiazole | |
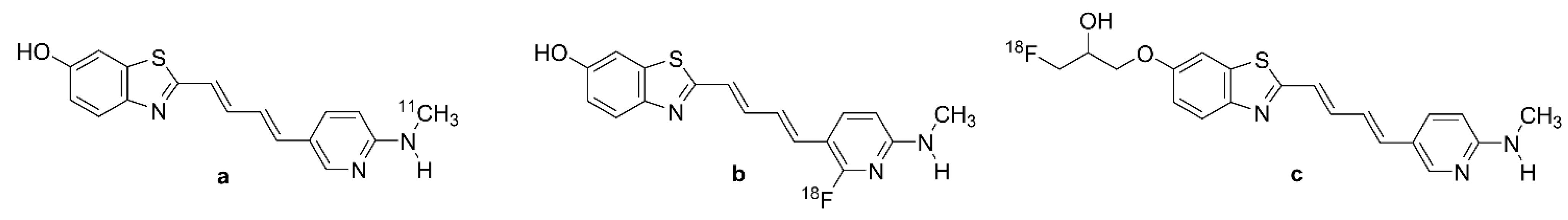
| PBB3 | 2-[(1E,3E)-4-[6-(methylamino)pyridin-3-yl]buta-1,3-dienyl]-1,3-benzothiazol-6-ol CAS RN: 1565796-97-5 | |
| [11C]PBB3 | Figure 6a | 2-[(1E,3E)-4-[6-[11C]methylamino)pyridin-3-yl]buta-1,3-dienyl]-1,3-benzothiazol-6-ol; CAS RN: 1565797-40-1 |
| [18F]PBB3 | Figure 6b | 2-((1E,3E)-4-(2-[18F]fluoro)-6-(methylamino)pyridin-3-yl)buta-1,3-dien-1-yl)benzo[d]thiazol-6-ol |
| PIB | 6-OH-BTA-1; Pittsburgh Compound B 2-[4-(methylamino)phenyl]-6-benzothiazolol CAS RN: 566169-93-5 | |
| [11C]PIB | Figure 1f | [N-methyl-11C]-6-OH-BTA-1, CAS RN: 566170-04-5 |
| PM-PBB3 | APN-1607; 1-fluoro-3-((2-((1E,3E)-4-(6-(methylamino)pyridin-3-yl)buta-1,3-dien-1-yl)benzo[d]thiazol-6-yl)oxy)propan-2-ol | |
| [18F]PM-PBB3 | Figure 6c | [18F]APN-1607; [18F]MNI-958; 1-[18F]fluoro-3-((2-((1E,3E)-4-(6-(methylamino)pyridine-3-yl)buta-1,3-dien-1-yl)benzo[d]thiazol-6-yl)oxy)propan-2-ol, CAS RN 1565797-57-0 |
| RO-6924963 | Ro963; 2-(4-methoxyphenyl)imidazo[1,2-a]pyridin-7-amine | |
| [11C]RO-6924963 | Figure 7e | [11C]R0963; 2-(4-([11C]methoxy)phenyl)imidazo[1,2-a]pyridin-7-amine |
| RO-6931643 | Ro643; N-methyl-2-(m-tolyl)imidazo[1,2-a]pyrimidin-7-amine | |
| [11C]RO-6931643 | Figure 7c | [11C]Ro643; N-[11C]methyl-2-(m-tolyl)imidazo[1,2-a]pyrimidin-7-amine |
| RO-6958948 | RO-948; 2-(6-fluoro-pyridin-3-yl)-9H-pyrrolo[2,3-b:4,5-c’]dipyridine | |
| [18F]RO-6958948 | Figure 7d | [18F]Ro948; 2-(6-([18F]fluoro-)pyridin-3-yl)-9H-pyrrolo[2,3-b:4,5-c’]dipyridine |
| [18F]S16 | Figure 3i | (S)-1-(4-(6-(dimethylamino)quinoxalin-2-yl)phenoxy)-3-fluoropropan-2-ol |
| T807 | flortaucipir; LY 3191748; AV-1451 | |
| [18F]T807 | Figure 5a | [18F]flortaucipir; [18F]LY 3191748; [18F]AV-1451 |
| T808 | AV-680; 2-[4-(2-Fluoroethyl)-1-piperidinyl]pyrimido[1,2-a]benzimidazole CAS RN; 1320211-61-7 | |
| [18F]T808 | Figure 5b | [18F]AV-680; 2-[4-(2-[18F]Fluoroethyl)-1-piperidinyl]pyrimido[1,2-a]benzimidazole |
| Tauvid | Figure 5a | 18F-AV-1451, 18F-T807, Flortaucipir F-18 |
| THK-523 | Figure 2d | 4-[6-(2-fluoroethoxy)-2-quinolinyl]aniline 2-(4-Aminophenyl)-6-(2-(fluoroethoxy))quinoline; CAS RN: 1573029-17-0 |
| [18F]THK-523 | Figure 2f | 4-[6-(2-[18F]fluoroethoxy)-2-quinolinyl]aniline 2-(4-Aminophenyl)-6-(2-([18F]fluoroethoxy))quinoline |
| THK-951 | 2-[(N-Methyl-4-amino)phenyl]quinolin-7-ol | |
| [11C]THK-951 | Figure 3a | 2-(4-([11C]methyl)-amino)phenyl)quinolin-7-ol |
| THK-5105 | 1-((2-(4-(dimethylamino)phenyl)quinolin-6-yl)oxy)-3-fluoropropan-2-ol | |
| [18F]THK-5105 | Figure 3b | 1-((2-(4-(dimethylamino)phenyl)quinolin-6-yl)oxy)-3-[18F]fluoropropan-2-ol |
| [18F]THK-5116 | Figure 3c | 1-((2-(4-aminophenyl)quinolin-6-yl)oxy)-3-[18F]fluoropropan-2-ol |
| THK-5117 | 1-fluoro-3-[2-[4-(methylamino)phenyl]quinolin-6-yl]oxypropan-2-ol CAS RN: 1374107-54-6 | |
| [18F]THK-5117 | Figure 3d | 1-[18F]fluoro-3-[2-[4-(methylamino)phenyl]quinolin-6-yl]oxypropan-2-ol |
| THK-5317 | 6-((3-fluoro-2-hydroxy)propoxy)-2-(4-methylaminophenyl)quinoline | |
| [18F]THK-5317 | Figure 3f | 6-((3-[18F]fluoro-2-hydroxy)propoxy)-2-(4-methylaminophenyl)quinoline |
| THK-5351 | GE-216; (2R)-1-fluoro-3-[2-[6-(methylamino)pyridin-3-yl]quinolin-6-yl]oxypropan-2-ol CAS RN: 2101218-44-2 | |
| [11C]THK-5351 | Figure 3h | (2R)-1-fluoro-3-[2-[6-([11C]methylamino)pyridin-3-yl]quinolin-6-yl]oxypropan-2-ol |
| [18F]THK-5351 | Figure 3g | [18F]GE-216; (2R)-1-[18F]fluoro-3-[2-[6-(methylamino)pyridin-3-yl]quinolin-6-yl]oxypropan-2-ol |
References
- Kowall, N.W.; Kosik, K.S. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann. Neurol. 1987, 22, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Kouri, N.; Murray, M.E.; Josephs, K.A. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J. Mol. Neurosci. 2011, 45, 384–389. [Google Scholar]
- Villemagne, V.L.; Furumoto, S.; Fodero-Tavoletti, M.; Harada, R.; Mulligan, R.S.; Kudo, Y.; Masters, C.L.; Yanai, K.; Rowe, C.C.; Okamura, N. The challenges of tau imaging. Future Neurol. 2012, 7, 409–421. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Doré, V.; Burnham, S.C.; Masters, C.L.; Rowe, C.C. Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol. 2018, 14, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef]
- Mohorko, N.; Bresjanac, M. Tau protein and human tauopathies: An overwiev. Slov. Med. J. 2008, 77. [Google Scholar]
- Goedert, M.; Spillantini, M.; Cairns, N.; Crowther, R. Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992, 8, 159–168. [Google Scholar] [CrossRef]
- Crowther, R. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc. Natl. Acad. Sci. USA 1991, 88, 2288–2292. [Google Scholar] [CrossRef]
- Delacourte, A. Tauopathies: Recent insights into old diseases. Folia Neuropathol. 2005, 43, 244–257. [Google Scholar]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Bobinski, M.; Wegiel, J.; Wisniewski, H.M.; Tarnawski, M.; Bobinski, M.; Reisberg, B.; De Leon, M.J.; Miller, D.C. Neurofibrillary pathology—Correlation with hippocampal formation atrophy in Alzheimer disease. Neurobiol. Aging 1996, 17, 909–919. [Google Scholar] [PubMed]
- Harrison, T.M.; La Joie, R.; Maass, A.; Baker, S.L.; Bs, K.S.; Fenton, L.; Bs, T.J.M.; Edwards, L.; Pham, J.; Miller, B.L.; et al. Longitudinal tau accumulation and atrophy in aging and alzheimer disease. Ann. Neurol. 2019, 85, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef]
- Delaere, P.; Duyckaerts, C.; Brion, J.P.; Poulain, V.; Hauw, J.-J. Tau, paired helical filaments and amyloid in the neocortex: A morphometric study of 15 cases with graded intellectual status in aging and senile dementia of Alzheimer type. Acta Neuropathol. 1989, 77, 645–653. [Google Scholar] [CrossRef]
- Delaere, P.; Duyckaerts, C.; Masters, C.; Beyreuther, K.; Piette, F.; Hauw, J. Large amounts of neocortical βA4 deposits without neuritic plaques nor tangles in a psychometrically assessed, non-demented person. Neurosci. Lett. 1990, 116, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.C.; Ackerman, U.; Browne, W.; Mulligan, R.; Pike, K.L.; O’Keefe, G.; Tochon-Danguy, H.; Chan, G.; Berlangieri, S.U.; Jones, G.; et al. Imaging of amyloid β in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: Proof of mechanism. Lancet Neurol. 2008, 7, 129–135. [Google Scholar] [CrossRef]
- Katzman, R.; Terry, R.; DeTeresa, R.; Brown, T.; Davies, P.; Fuld, P.; Renbing, X.; Peck, A. Clinical, pathological, and neurochemical changes in dementia: A subgroup with preserved mental status and numerous neocortical plaques. Ann. Neurol. 1988, 23, 138–144. [Google Scholar] [CrossRef]
- Shoghi-Jadid, K.; Small, G.W.; Agdeppa, E.D.; Kepe, V.; Ercoli, L.M.; Siddarth, P.; Read, S.; Satyamurthy, N.; Petric, A.; Huang, S.C.; et al. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am. J. Geriatr. Psychiatry 2002, 10, 24–35. [Google Scholar] [CrossRef]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.-G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies—Still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; van der Kant, R.; Hansson, O. Tau biomarkers in Alzheimer’s disease: Towards implementation in clinical practice and trials. Lancet Neurol. 2022, 21, 726–734. [Google Scholar] [CrossRef]
- Maschio, C.; Ni, R. Amyloid and Tau Positron Emission Tomography Imaging in Alzheimer’s Disease and Other Tauopathies. Front. Aging Neurosci. 2022, 14, 838034. [Google Scholar] [CrossRef]
- Kallinen, A.; Kassiou, M. Tracer development for PET imaging of proteinopathies. Nucl. Med. Biol. 2022, 114–115, 115–127. [Google Scholar] [CrossRef]
- Jie, C.; Treyer, V.; Schibli, R.; Mu, L. Tauvid™: The First FDA-Approved PET Tracer for Imaging Tau Pathology in Alzheimer’s Disease. Pharmaceuticals 2021, 14, 110. [Google Scholar] [CrossRef] [PubMed]
- Cumming, P.; Burgher, B.; Patkar, O.; Breakspear, M.; Vasdev, N.; Thomas, P.; Liu, G.-J.; Banati, R. Sifting through the surfeit of neuroinflammation tracers. J. Cereb. Blood Flow Metab. 2018, 38, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- LoPresti, P.; Szuchet, S.; Papasozomenos, S.C.; Zinkowski, R.P.; Binder, L.I. Functional implications for the microtubule-associated protein tau: Localization in oligodendrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10369–10373. [Google Scholar] [CrossRef]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Mol. Brain Res. 1986, 1, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, A. Misregulation of tau alternative splicing in neurodegeneration and dementia. Altern. Splicing Dis. 2006, 44, 89–107. [Google Scholar]
- Lee, G.; Cowan, N.; Kirschner, M. The primary structure and heterogeneity of tau protein from mouse brain. Science 1988, 239, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22. [Google Scholar] [CrossRef]
- Schweers, O.; Schönbrunn-Hanebeck, E.; Marx, A.; Mandelkow, E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 1994, 269, 24290–24297. [Google Scholar] [CrossRef]
- Majounie, E.; Cross, W.; Newsway, V.; Dillman, A.; Vandrovcova, J.; Morris, C.M.; Nalls, M.A.; Ferrucci, L.; Owen, M.J.; O’Donovan, M.C.; et al. Variation in tau isoform expression in different brain regions and disease states. Neurobiol. Aging 2013, 34, 1922.e7–1922.e12. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef]
- Ackmann, M.; Wiech, H.; Mandelkow, E. Nonsaturable binding indicates clustering of tau on the microtubule surface in a paired helical filament-like conformation. J. Biol. Chem. 2000, 275, 30335–30343. [Google Scholar] [CrossRef]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.-J.; Mandelkow, E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef]
- Berriman, J.; Serpell, L.C.; Oberg, K.A.; Fink, A.L.; Goedert, M.; Crowther, R.A. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-β structure. Proc. Natl. Acad. Sci. USA 2003, 100, 9034–9038. [Google Scholar] [CrossRef]
- Lim, S.; Haque, M.M.; Kim, D.; Kim, D.J.; Kim, Y.K. Cell-based Models To Investigate Tau Aggregation. Comput. Struct. Biotechnol. J. 2014, 12, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.; Strijkstra, A.M.; Hut, R.A.; Rüdiger, J.; Van Der Zee, E.A.; Harkany, T.; Holzer, M.; Härtig, W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J. Neurosci. 2003, 23, 6972–6981. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.-X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Catafau, A.M. Molecular imaging insights into neurodegeneration: Focus on tau PET radiotracers. J. Nucl. Med. 2014, 55, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Furumoto, S.; Fodero-Tavoletti, M.T.; Mulligan, R.S.; Hodges, J.; Harada, R.; Yates, P.; Piguet, O.; Pejoska, S.; Doré, V.; et al. In vivo evaluation of a novel tau imaging tracer for Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 816–826. [Google Scholar] [CrossRef]
- Barrio, J.R.; Huang, S.C.; Cole, G.M.; Satyamurthy, N.M.; Petric, A.; Phelps, M.E.; Small, G.W. PET imaging of tangles and plaques in Alzheimer disease with a highly hydrophobic probe. J. Label. Compd. Radiopharm. 1999, 42, S194–S195. [Google Scholar]
- Noda, A.; Murakami, Y.; Nishiyama, S.; Fukumoto, D.; Miyoshi, S.; Tsukada, H.; Nishimura, S. Amyloid imaging in aged and young macaques with [11C]PIB and [18F]FDDNP. Synapse 2008, 62, 472–475. [Google Scholar] [CrossRef]
- Agdeppa, E.D.; Kepe, V.; Satyamurthy, N.; Liu, J.; Huang, S.-C.; Small, G.W.; Cole, G.M.; Barrio, J.R. In vitro detection of (S)-naproxen and ibuprofen binding to plaques in the Alzheimer’s brain using the positron emission tomography molecular imaging probe 2-(1-{6-[(2-[18F]fluoroethyl)(methyl) amino]-2-naphthyl} ethylidene) malononitrile. Neuroscience 2003, 117, 723–730. [Google Scholar] [PubMed]
- Landau, M.; Sawaya, M.; Faull, K.F.; Laganowsky, A.; Jiang, L.; Sievers, S.A.; Liu, J.; Barrio, J.R.; Eisenberg, D. Towards a pharmacophore for amyloid. PLoS Biol. 2011, 9, e1001080. [Google Scholar] [CrossRef]
- Agdeppa, E.D.; Kepe, V.; Liu, J.; Flores-Torres, S.; Satyamurthy, N.; Petric, A.; Cole, G.M.; Small, G.W.; Huang, S.C.; Barrio, J.R. Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for β-amyloid plaques in Alzheimer’s disease. J. Neurosci. 2001, 21, RC189. [Google Scholar] [CrossRef] [PubMed]
- Harada, R.; Okamura, N.; Furumoto, S.; Tago, T.; Maruyama, M.; Higuchi, M.; Yoshikawa, T.; Arai, H.; Iwata, R.; Kudo, Y.; et al. Comparison of the binding characteristics of [18F]THK-523 and other amyloid imaging tracers to Alzheimer’s disease pathology. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Smid, L.M.; Vovko, T.D.; Popovic, M.; Petrič, A.; Kepe, V.; Barrio, J.R.; Vidmar, G.; Bresjanac, M. The 2,6-disubstituted naphthalene derivative FDDNP labeling reliably predicts Congo red birefringence of protein deposits in brain sections of selected human neurodegenerative diseases. Brain Pathol. 2006, 16, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Suemoto, T.; Okamura, N.; Shiomitsu, T.; Suzuki, M.; Shimadzu, H.; Akatsu, H.; Yamamoto, T.; Kudo, Y.; Sawada, T. In vivo labeling of amyloid with BF-108. Neurosci. Res. 2004, 48, 65–74. [Google Scholar] [CrossRef]
- Thompson, P.W.; Ye, L.; Morgenstern, J.L.; Sue, L.; Beach, T.G.; Judd, D.J.; Shipley, N.J.; Libri, V.; Lockhart, A. Interaction of the amyloid imaging tracer FDDNP with hallmark Alzheimer’s disease pathologies. J. Neurochem. 2009, 109, 623–630. [Google Scholar] [CrossRef]
- Cole, G.B.; Satyamurthy, N.; Liu, J.; Wong, K.-P.; Small, G.W.; Huang, S.-C.; Košmrlj, J.; Barrio, J.R.; Petrič, A. The Value of In Vitro Binding as Predictor of In Vivo Results: A Case for [18F]FDDNP PET. Mol. Imaging Biol. 2019, 21, 25–34. [Google Scholar] [CrossRef]
- Murugan, N.A.; Nordberg, A.; Ågren, H. Different Positron Emission Tomography Tau Tracers Bind to Multiple Binding Sites on the Tau Fibril: Insight from Computational Modeling. ACS Chem. Neurosci. 2018, 9, 1757–1767. [Google Scholar]
- Small, G.W.; Kepe, V.; Ercoli, L.M.; Siddarth, P.; Bookheimer, S.Y.; Miller, K.J.; Lavretsky, H.; Burggren, A.C.; Cole, G.M.; Vinters, H.V.; et al. PET of brain amyloid and tau in mild cognitive impairment. N. Engl. J. Med. 2006, 355, 2652–2663. [Google Scholar] [CrossRef]
- Tauber, C.; Beaufils, E.; Hommet, C.; Ribeiro, M.J.; Vercouillie, J.; Vierron, E.; Mondon, K.; Cottier, J.P.; Gissot, V.; Guilloteau, D.; et al. Brain [18F]FDDNP binding and glucose metabolism in advanced elderly healthy subjects and Alzheimer’s disease patients. J. Alzheimer’s Dis. 2013, 36, 311–320. [Google Scholar]
- Tolboom, N.; van der Flier, W.M.; Yaqub, M.; Boellaard, R.; Verwey, N.A.; Blankenstein, M.A.; Windhorst, A.D.; Scheltens, P.; Lammertsma, A.A.; van Berckel, B.N. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J. Nucl. Med. 2009, 50, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Lee, S.-Y.; Kim, S.-H.; Kim, Y.-B.; Cho, S.-J. Multitracer PET imaging of amyloid plaques and neurofibrillary tangles in Alzheimer’s disease. Neuroimage 2008, 43, 236–244. [Google Scholar] [CrossRef]
- Okamura, N.; Furumoto, S.; Fodero-Tavoletti, M.T.; Mulligan, R.S.; Harada, R.; Yates, P.; Pejoska, S.; Kudo, Y.; Masters, C.L.; Yanai, K.; et al. Non-invasive assessment of Alzheimer’s disease neurofibrillary pathology using 18F-THK5105 PET. Brain 2014, 137, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Harada, R.; Okamura, N.; Furumoto, S.; Furukawa, K.; Ishiki, A.; Tomita, N.; Hiraoka, K.; Watanuki, S.; Shidahara, M.; Miyake, M.; et al. [18F]THK-5117 PET for assessing neurofibrillary pathology in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zhou, K.; Zhang, X.; Zhang, J.; Cui, M.; Xu, B.; Tian, J. In vivo imaging of neurofibrillary tau PET radioligand 18F-S16 in comparison with 18F-THK5317 in Alzheimer’s disease. J. Nucl. Med. 2020, 61, 1545. [Google Scholar]
- Chanisa, C.; Monchaya, N.; Anchisa, K.; Chetsadaporn, P.; Attapon, J. Analysis of amyloid and tau deposition in Alzheimer’s disease using 11C-Pittsburgh compound B and 18F-THK 5351 positron emission tomography imaging. World J. Nucl. Med. 2020, 20, 61–72. [Google Scholar] [CrossRef]
- Ezura, M.; Kikuchi, A.; Okamura, N.; Ishiki, A.; Hasegawa, T.; Harada, R.; Watanuki, S.; Funaki, Y.; Hiraoka, K.; Baba, T.; et al. 18F-THK5351 Positron Emission Tomography Imaging in Neurodegenerative Tauopathies. Front. Aging Neurosci. 2021, 13, 761010. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Pirraglia, E.; Okamura, N.; Rusinek, H.; De Leon, M.J. Quantitative evaluation of tau PET tracers 18F-THK5351 and 18F-AV-1451 in Alzheimer’s disease with standardized uptake value peak-alignment (SUVP) normalization. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1596–1604. [Google Scholar] [CrossRef]
- Kang, J.M.; Lee, S.Y.; Seo, S.; Jeong, H.J.; Woo, S.H.; Lee, H.; Lee, Y.B.; Yeon, B.K.; Shin, D.H.; Park, K.H.; et al. Tau positron emission tomography using [18F]THK5351 and cerebral glucose hypometabolism in Alzheimer’s disease. Neurobiol. Aging 2017, 59, 210–219. [Google Scholar] [CrossRef]
- Leuzy, A.; Pascoal, T.A.; Strandberg, O.; Insel, P.; Smith, R.; Mattsson-Carlgren, N.; Benedet, A.L.; Cho, H.; Lyoo, C.H.; La Joie, R.; et al. A multicenter comparison of [18F]flortaucipir, [18F]RO948, and [18F]MK6240 tau PET tracers to detect a common target ROI for differential diagnosis. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2295–2305. [Google Scholar] [CrossRef]
- Li, C.-H.; Chen, T.-F.; Chiu, M.-J.; Yen, R.-F.; Shih, M.-C.; Lin, C.-H. Integrated 18F-T807 Tau PET, Structural MRI, and Plasma Tau in Tauopathy Neurodegenerative Disorders. Front. Aging Neurosci. 2021, 13, 133. [Google Scholar] [CrossRef] [PubMed]
- Wolters, E.E.; Ossenkoppele, R.; Verfaillie, S.C.J.; Coomans, E.M.; Timmers, T.; Visser, D.; Tuncel, H.; Golla, S.S.V.; Windhorst, A.D.; Boellaard, R.; et al. Regional [18F]flortaucipir PET is more closely associated with disease severity than CSF p-tau in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2866–2878. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Rabinovici, G.D.; Smith, R.; Cho, H.; Schöll, M.; Strandberg, O.; Palmqvist, S.; Mattsson, N.; Janelidze, S.; Santillo, A.; et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA 2018, 320, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Pontecorvo, M.J.; Devous Sr, M.D.; Navitsky, M.; Lu, M.; Salloway, S.; Schaerf, F.W.; Jennings, D.; Arora, A.K.; McGeehan, A.; Lim, N.C.; et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 2017, 140, 748–763. [Google Scholar] [CrossRef]
- Cho, H.; Choi, J.Y.; Hwang, M.S.; Lee, J.H.; Kim, Y.J.; Lee, H.M.; Lyoo, C.H.; Ryu, Y.H.; Lee, M.S. Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 2016, 87, 375–383. [Google Scholar] [CrossRef]
- Kitamura, S.; Shimada, H.; Niwa, F.; Endo, H.; Shinotoh, H.; Takahata, K.; Higuchi, M. Tau-induced focal neurotoxicity and network disruption related to apathy in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1208–1214. [Google Scholar] [CrossRef]
- Shimada, H.; Kitamura, S.; Shinotoh, H.; Endo, H.; Niwa, F.; Hirano, S.; Kimura, Y.; Zhang, M.R.; Kuwabara, S.; Suhara, T.; et al. Association between Aβ and tau accumulations and their influence on clinical features in aging and Alzheimer’s disease spectrum brains: A [11C]PBB3-PET study. Alzheimer’s Dement. 2017, 6, 11–20. [Google Scholar] [CrossRef]
- Hsu, J.-L.; Lin, K.-J.; Hsiao, I.-T.; Huang, K.-L.; Liu, C.-H.; Wu, H.-C.; Weng, Y.-C.; Huang, C.-Y.; Chang, C.-C.; Yen, T.-C.; et al. The Imaging Features and Clinical Associations of a Novel Tau PET Tracer—18F-APN1607 in Alzheimer Disease. Clin. Nucl. Med. 2020, 45, 747–756. [Google Scholar] [CrossRef]
- Lu, J.; Bao, W.; Li, M.; Li, L.; Zhang, Z.; Alberts, I.; Brendel, M.; Cumming, P.; Lu, H.; Xiao, Z.; et al. Associations of [18F]-APN-1607 Tau PET Binding in the Brain of Alzheimer’s Disease Patients with Cognition and Glucose Metabolism. Front. Neurosci. 2020, 14, 604. [Google Scholar] [CrossRef]
- Therriault, J.; Pascoal, T.A.; Lussier, F.Z.; Tissot, C.; Chamoun, M.; Bezgin, G.; Servaes, S.; Benedet, A.L.; Ashton, N.J.; Karikari, T.K.; et al. Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nat. Aging 2022, 2, 526–535. [Google Scholar] [CrossRef]
- Ashton, N.J.; Pascoal, T.A.; Karikari, T.K.; Benedet, A.L.; Lantero-Rodriguez, J.; Brinkmalm, G.; Snellman, A.; Schöll, M.; Troakes, C.; Hye, A.; et al. Plasma p-tau231: A new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol. 2021, 141, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Therriault, J.; Pascoal, T.A.; Savard, M.; Benedet, A.L.; Chamoun, M.; Tissot, C.; Lussier, F.; Kang, M.S.; Thomas, E.; Terada, T.; et al. Topographic distribution of Amyloid-β, Tau, and Atrophy in patients with behavioral/dysexecutive Alzheimer disease. Neurology 2021, 96, e81–e92. [Google Scholar] [CrossRef] [PubMed]
- Therriault, J.; Pascoal, T.A.; Benedet, A.L.; Tissot, C.; Savard, M.; Chamoun, M.; Lussier, F.; Kang, M.S.; Berzgin, G.; Wang, T.; et al. Frequency of biologically defined Alzheimer disease in relation to age, sex, APOE ε4, and cognitive impairment. Neurology 2021, 96, e975–e985. [Google Scholar] [CrossRef] [PubMed]
- Tissot, C.; Therriault, J.; Pascoal, T.A.; Chamoun, M.; Lussier, F.Z.; Savard, M.; Mathotaarachchi, S.S.; Benedet, A.L.; Thomas, E.M.; Parsons, M.; et al. Association between regional tau pathology and neuropsychiatric symptoms in aging and dementia due to Alzheimer’s disease. Alzheimer’s Dement. 2021, 7, e12154. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Therriault, J.; Benedet, A.L.; Savard, M.; Lussier, F.Z.; Chamoun, M.; Tissot, C.; Qureshi, M.N.I.; Kang, M.S.; Mathotaarachchi, S.; et al. 18F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain 2020, 143, 2818–2830. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Toth, B.; Manser, P.T.; Sanabria-Bohórquez, S.; Teng, E.; Keeley, M.; Bateman, R.J.; Weimer, R.M.; Wildsmith, K.R. Site-Specific Cerebrospinal Fluid Tau Hyperphosphorylation in Response to Alzheimer’s Disease Brain Pathology: Not All Tau Phospho-Sites are Hyperphosphorylated. J. Alzheimer’s Dis. 2022, 85, 415–429. [Google Scholar] [CrossRef]
- Leuzy, A.; Smith, R.; Cullen, N.C.; Strandberg, O.; Vogel, J.W.; Binette, A.P.; Borroni, E.; Janelidze, S.; Ohlsson, T.; Jögi, J.; et al. Biomarker-based prediction of longitudinal tau positron emission tomography in Alzheimer disease. JAMA Neurol. 2022, 79, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Smith, R.; Ossenkoppele, R.; Santillo, A.; Borroni, E.; Klein, G.; Ohlsson, T.; Jögi, J.; Palmqvist, S.; Mattsson-Carlgren, N.; et al. Diagnostic performance of RO948 F 18 tau positron emission tomography in the differentiation of Alzheimer disease from other neurodegenerative disorders. JAMA Neurol. 2020, 77, 955–965. [Google Scholar] [CrossRef]
- Wong, D.F.; Comley, R.A.; Kuwabara, H.; Rosenberg, P.B.; Resnick, S.M.; Ostrowitzki, S.; Vozzi, C.; Boess, F.; Oh, E.; Lyketsos, C.G.; et al. Characterization of 3 novel tau radiopharmaceuticals, 11C-RO-963, 11C-RO-643, and 18F-RO-948, in healthy controls and in Alzheimer subjects. J. Nucl. Med. 2018, 59, 1869–1876. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.E.; Janssens, L.; Moechars, D.; Rombouts, F.J.R.; Timmers, M.; Barret, O.; Constantinescu, C.C.; Madonia, J.; Russell, D.S.; Sandiego, C.M.; et al. Clinical evaluation of [18F]JNJ-64326067, a novel candidate PET tracer for the detection of tau pathology in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 3176–3185. [Google Scholar] [CrossRef]
- Bun, S.; Moriguchi, S.; Tezuka, T.; Sato, Y.; Takahata, K.; Seki, M.; Nakajima, S.; Yamamoto, Y.; Sano, Y.; Suzuki, N.; et al. Findings of 18F-PI-2620 tau PET imaging in patients with Alzheimer’s disease and healthy controls in relation to the plasma P-tau181 levels in a Japanese sample. Neuropsychopharmacol. Rep. 2022, 42, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Jantarato, A.; Vachatimanont, S.; Boonkawin, N.; Yaset, S.; Kunawudhi, A.; Promteangtrong, C.; Assanasen, J.; Mahanonda, N.; Chotipanich, C. The Evaluation of Tau Deposition with [18F]PI-2620 by Using a Semiquantitative Method in Cognitively Normal Subjects and Patients with Mild Cognitive Impairment and Alzheimer’s Disease. Mol. Imaging 2021, 2021, 6640054. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.; Bullich, S.; Barret, O.; Madonia, J.; Berndt, M.; Papin, C.; Perrotin, A.; Koglin, N.; Kroth, H.; Pfeifer, A.; et al. Tau PET imaging with 18F-PI-2620 in patients with Alzheimer disease and healthy controls: A first-in-humans study. J. Nucl. Med. 2020, 61, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zhang, J.; Zhou, K.; Zhang, X.; Xie, H.; Zhu, M.; Cui, M.; Wang, R. In vivo imaging of tau deposition in Alzheimer’s disease using both [18F]-THK5317 and [18F]-S16: A pilot human study. Front. Aging Neurosci. 2022, 14, 994750. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cai, L.; Zhou, K.; Cui, M.; Yao, S. Biodistribution and dosimetry evaluation for a novel tau tracer [18F]-S16 in healthy volunteers and its application in assessment of tau pathology in Alzheimer’s disease. Front. Bioeng. Biotechnol. 2022, 9, 1515. [Google Scholar] [CrossRef] [PubMed]
- Tolboom, N.; Yaqub, M.; van der Flier, W.M.; Boellaard, R.; Luurtsema, G.; Windhorst, A.D.; Barkhof, F.; Scheltens, P.; Lammertsma, A.A.; van Berckel, B.N. Detection of Alzheimer pathology in vivo using both 11C-PIB and 18F-FDDNP PET. J. Nucl. Med. 2009, 50, 191–197. [Google Scholar] [CrossRef]
- Small, G.W.; Siddarth, P.; Burggren, A.C.; Kepe, V.; Ercoli, L.M.; Miller, K.J.; Lavretsky, H.; Thompson, P.; Cole, G.M.; Huang, S.C.; et al. Influence of cognitive status, age, and APOE-4 genetic risk on brain FDDNP positron-emission tomography imaging in persons without dementia. Arch. Gen. Psychiatry 2009, 66, 81–87. [Google Scholar] [CrossRef]
- Tolboom, N.; Koedam, E.L.; Schott, J.M.; Yaqub, M.; Blankenstein, M.A.; Barkhof, F.; Pijnenburg, Y.A.; Lammertsma, A.A.; Scheltens, P.; van Berckel, B.N. Dementia mimicking Alzheimer’s disease Owing to a tau mutation: CSF and PET findings. Alzheimer Dis. Assoc. Disord. 2010, 24, 303–307. [Google Scholar] [CrossRef]
- Okamura, N.; Suemoto, T.; Furumoto, S.; Suzuki, M.; Shimadzu, H.; Akatsu, H.; Yamamoto, T.; Fujiwara, H.; Nemoto, M.; Maruyama, M.; et al. Quinoline and benzimidazole derivatives: Candidate probes for in vivo imaging of tau pathology in Alzheimer’s disease. J. Neurosci. 2005, 25, 10857–10862. [Google Scholar] [CrossRef] [PubMed]
- Fodero-Tavoletti, M.T.; Okamura, N.; Furumoto, S.; Mulligan, R.S.; Connor, A.R.; McLean, C.A.; Cao, D.; Rigopoulos, A.; Cartwright, G.A.; O’Keefe, G.; et al. 18F-THK523: A novel in vivo tau imaging ligand for Alzheimer’s disease. Brain 2011, 134, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Bouras, C.; Hof, P.R.; Giannakopoulos, P.; Michel, J.-P.; Morrison, J.H. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: A quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb. Cortex 1994, 4, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Tago, T.; Furumoto, S.; Okamura, N.; Harada, R.; Ishikawa, Y.; Arai, H.; Yanai, K.; Iwata, R.; Kudo, Y. Synthesis and preliminary evaluation of 2-arylhydroxyquinoline derivatives for tau imaging. J. Label. Compd. Radiopharm. 2014, 57, 18–24. [Google Scholar] [CrossRef]
- Cai, L.; Qu, B.; Hurtle, B.T.; Dadiboyena, S.; Diaz-Arrastia, R.; Pike, V.W. Candidate PET radioligand development for neurofibrillary tangles: Two distinct radioligand binding sites identified in postmortem Alzheimer’s disease brain. ACS Chem. Neurosci. 2016, 7, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Murugan, N.A.; Chiotis, K.; Rodriguez-Vieitez, E.; Lemoine, L.; Ågren, H.; Nordberg, A. Cross-interaction of tau PET tracers with monoamine oxidase B: Evidence from in silico modelling and in vivo imaging. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Okamura, N.; Furumoto, S.; Harada, R.; Tago, T.; Yoshikawa, T.; Fodero-Tavoletti, M.; Mulligan, R.S.; Villemagne, V.L.; Akatsu, H.; Yamamoto, T.; et al. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J. Nucl. Med. 2013, 54, 1420–1427. [Google Scholar] [CrossRef]
- Fodero-Tavoletti, M.T.; Furumoto, S.; Taylor, L.; McLean, C.A.; Mulligan, R.S.; Birchall, I.; Harada, R.; Masters, C.L.; Yanai, K.; Kudo, Y.; et al. Assessing THK523 selectivity for tau deposits in Alzheimer’s disease and non–Alzheimer’s disease tauopathies. Alzheimer’s Res. Ther. 2014, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Mukaetova-Ladinska, E.; Harrington, C.; Roth, M.; Wischik, C. Biochemical and anatomical redistribution of tau protein in Alzheimer’s disease. Am. J. Pathol. 1993, 143, 565. [Google Scholar]
- Tago, T.; Furumoto, S.; Okamura, N.; Harada, R.; Adachi, H.; Ishikawa, Y.; Yanai, K.; Iwata, R.; Kudo, Y. Preclinical evaluation of [18F]THK-5105 enantiomers: Effects of chirality on its effectiveness as a tau imaging radiotracer. Mol. Imaging Biol. 2016, 18, 258–266. [Google Scholar] [CrossRef]
- Lemoine, L.; Saint-Aubert, L.; Marutle, A.; Antoni, G.; Eriksson, J.P.; Ghetti, B.; Okamura, N.; Nennesmo, I.; Gillberg, P.-G.; Nordberg, A. Visualization of regional tau deposits using 3H-THK5117 in Alzheimer brain tissue. Acta Neuropathol. Commun. 2015, 3, 40. [Google Scholar] [CrossRef]
- Lemoine, L.; Saint-Aubert, L.; Nennesmo, I.; Gillberg, P.-G.; Nordberg, A. Cortical laminar tau deposits and activated astrocytes in Alzheimer’s disease visualised by 3H-THK5117 and 3H-deprenyl autoradiography. Sci. Rep. 2017, 7, srep45496. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.C.; Andrés, J.I. Tau Positron Emission Tomography Imaging. Cold Spring Harb. Perspect. Biol. 2017, 9, a023721. [Google Scholar] [CrossRef] [PubMed]
- Ishiki, A.; Harada, R.; Kai, H.; Sato, N.; Totsune, T.; Tomita, N.; Watanuki, S.; Hiraoka, K.; Ishikawa, Y.; Funaki, Y.; et al. Neuroimaging-pathological correlations of [18F]THK5351 PET in progressive supranuclear palsy. Acta Neuropathol. Commun. 2018, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Ishiki, A.; Okamura, N.; Furukawa, K.; Furumoto, S.; Harada, R.; Tomita, N.; Hiraoka, K.; Watanuki, S.; Ishikawa, Y.; Tago, T.; et al. Longitudinal assessment of tau pathology in patients with Alzheimer’s disease using [18F] THK-5117 positron emission tomography. PLoS ONE 2015, 10, e0140311. [Google Scholar] [CrossRef]
- Jonasson, M.; Wall, A.; Chiotis, K.; Saint-Aubert, L.; Wilking, H.; Sprycha, M.; Borg, B.; Thibblin, A.; Eriksson, J.; Eriksson, J.; et al. Tracer kinetic analysis of (S)-18F-THK5117 as a PET tracer for assessing tau pathology. J. Nucl. Med. 2016, 57, 574–581. [Google Scholar] [CrossRef]
- Chiotis, K.; Saint-Aubert, L.; Savitcheva, I.; Jelic, V.; Andersen, P.; Jonasson, M.; Eriksson, J.; Lubberink, M.; Almkvist, O.; Wall, A.; et al. Imaging in-vivo tau pathology in Alzheimer’s disease with THK5317 PET in a multimodal paradigm. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1686–1699. [Google Scholar] [CrossRef]
- Harada, R.; Okamura, N.; Furumoto, S.; Furukawa, K.; Ishiki, A.; Tomita, N.; Tago, T.; Hiraoka, K.; Watanuki, S.; Shidahara, M.; et al. 18F-THK5351: A novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer disease. J. Nucl. Med. 2016, 57, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Tago, T.; Toyohara, J.; Harada, R.; Furumoto, S.; Okamura, N.; Kudo, Y.; Takahashi-Fujigasaki, J.; Murayama, S.; Ishii, K. Characterization of the binding of tau imaging ligands to melanin-containing cells: Putative off-target-binding site. Ann. Nucl. Med. 2019, 33, 375–382. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Fodero-Tavoletti, M.T.; Masters, C.L.; Rowe, C.C. Tau imaging: Early progress and future directions. Lancet Neurol. 2015, 14, 114–124. [Google Scholar] [CrossRef]
- Lockhart, S.N.; Baker, S.L.; Okamura, N.; Furukawa, K.; Ishiki, A.; Furumoto, S.; Tashiro, M.; Yanai, K.; Arai, H.; Kudo, Y.; et al. Dynamic PET measures of tau accumulation in cognitively normal older adults and Alzheimer’s disease patients measured using [18F] THK-5351. PLoS ONE 2016, 11, e0158460. [Google Scholar] [CrossRef]
- Sone, D.; Imabayashi, E.; Maikusa, N.; Okamura, N.; Furumoto, S.; Kudo, Y.; Ogawa, M.; Takano, H.; Yokoi, Y.; Sakata, M.; et al. Regional tau deposition and subregion atrophy of medial temporal structures in early Alzheimer’s disease: A combined positron emission tomography/magnetic resonance imaging study. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2017, 9, 35–40. [Google Scholar] [CrossRef]
- Betthauser, T.J.; Lao, P.J.; Murali, D.; Barnhart, T.E.; Furumoto, S.; Okamura, N.; Stone, C.K.; Johnson, S.C.; Christian, B.T. In vivo comparison of tau radioligands 18F-THK-5351 and 18F-THK-5317. J. Nucl. Med. 2017, 58, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Chiotis, K.; Stenkrona, P.; Almkvist, O.; Stepanov, V.; Ferreira, D.; Arakawa, R.; Takano, A.; Westman, E.; Varrone, A.; Okamura, N.; et al. Dual tracer tau PET imaging reveals different molecular targets for 11C-THK5351 and 11C-PBB3 in the Alzheimer brain. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1605–1617. [Google Scholar] [CrossRef]
- Oh, M.; Oh, S.J.; Lee, S.J.; Oh, J.S.; Roh, J.H.; Chung, S.J.; Lee, J.-H.; Lee, C.S.; Kim, J.S. Clinical Evaluation of 18F-PI-2620 as a Potent PET Radiotracer Imaging Tau Protein in Alzheimer Disease and Other Neurodegenerative Diseases Compared with 18F-THK-5351. Clin. Nucl. Med. 2020, 45, 841–847. [Google Scholar] [CrossRef]
- Jang, Y.K.; Lyoo, C.H.; Park, S.; Oh, S.J.; Cho, H.; Oh, M.; Ryu, Y.H.; Choi, J.Y.; Rabinovici, G.D.; Kim, H.J.; et al. Head to head comparison of [18F]AV-1451 and [18F] THK5351 for tau imaging in Alzheimer’s disease and frontotemporal dementia. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 432–442. [Google Scholar] [CrossRef]
- Lemoine, L.; Gillberg, P.-G.; Svedberg, M.; Stepanov, V.; Jia, Z.; Huang, J.; Nag, S.; Tian, H.; Ghetti, B.; Okamura, N.; et al. Comparative binding properties of the tau PET tracers THK5117, THK5351, PBB3, and T807 in postmortem Alzheimer brains. Alzheimer’s Res. Ther. 2017, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Therriault, J.; Kang, M.S.; Struyfs, H.; Pascoal, T.A.; Mathotaarachchi, S.; Shin, M.; Benedet, A.L.; Massarweh, G.; Soucy, J.-P.; et al. Rasagiline, a monoamine oxidase B inhibitor, reduces in vivo [18F]THK5351 uptake in progressive supranuclear palsy. NeuroImage Clin. 2019, 24, 102091. [Google Scholar] [CrossRef]
- Lemoine, L.; Leuzy, A.; Chiotis, K.; Rodriguez-Vieitez, E.; Nordberg, A. Tau positron emission tomography imaging in tauopathies: The added hurdle of off-target binding. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2018, 10, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Harada, R.; Ishiki, A.; Kai, H.; Sato, N.; Furukawa, K.; Furumoto, S.; Tago, T.; Tomita, N.; Watanuki, S.; Hiraoka, K.; et al. Correlations of 18F-THK5351 PET with postmortem burden of tau and astrogliosis in Alzheimer disease. J. Nucl. Med. 2018, 59, 671–674. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Shin, M.; Kang, M.S.; Chamoun, M.; Chartrand, D.; Mathotaarachchi, S.; Bennacef, I.; Therriault, J.; Ng, K.P.; Hopewell, R.; et al. In vivo quantification of neurofibrillary tangles with [18F]MK-6240. Alzheimer’s Res. Ther. 2018, 10, 74. [Google Scholar] [CrossRef]
- Zhou, K.; Yang, F.; Li, Y.; Chen, Y.; Zhang, X.; Zhang, J.; Wang, J.; Dai, J.; Cai, L.; Cui, M. Synthesis and evaluation of fluorine-18 labeled 2-phenylquinoxaline derivatives as potential tau imaging agents. Mol. Pharm. 2021, 18, 1176–1195. [Google Scholar] [CrossRef]
- Rojo, L.E.; Alzate-Morales, J.; Saavedra, I.N.; Davies, P.; Maccioni, R.B. Selective interaction of lansoprazole and astemizole with tau polymers: Potential new clinical use in diagnosis of Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 19, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Fawaz, M.V.; Brooks, A.F.; Rodnick, M.E.; Carpenter, G.M.; Shao, X.; Desmond, T.J.; Sherman, P.; Quesada, C.A.; Hockley, B.G.; Kilbourn, M.R.; et al. High affinity radiopharmaceuticals based upon lansoprazole for PET imaging of aggregated tau in Alzheimer’s disease and progressive supranuclear palsy: Synthesis, preclinical evaluation, and lead selection. ACS Chem. Neurosci. 2014, 5, 718–730. [Google Scholar] [CrossRef]
- Riss, P.J.; Brichard, L.; Ferrari, V.; Williamson, D.J.; Fryer, T.D.; Hong, Y.T.; Baron, J.-C.; Aigbirhio, F.I. Radiosynthesis and characterization of astemizole derivatives as lead compounds toward PET imaging of τ-pathology. MedChemComm 2013, 4, 852–855. [Google Scholar] [CrossRef]
- Kramer, V.; Brooks, A.F.; Haeger, A.; Kuljis, R.O.; Rafique, W.; Koeppe, R.A.; Raffel, D.M.; Frey, K.A.; Amaral, H.; Scott, P.J.H.; et al. Evaluation of [18F]-N-Methyl lansoprazole as a Tau PET Imaging Agent in First-in-Human Studies. ACS Chem. Neurosci. 2020, 11, 427–435. [Google Scholar] [CrossRef]
- Shao, X.; Carpenter, G.M.; Desmond, T.J.; Sherman, P.; Quesada, C.A.; Fawaz, M.; Brooks, A.F.; Kilbourn, M.R.; Albin, R.L.; Frey, K.A.; et al. Evaluation of [11C]N-methyl lansoprazole as a radiopharmaceutical for PET imaging of tau neurofibrillary tangles. ACS Med. Chem. Lett. 2012, 3, 936–941. [Google Scholar] [CrossRef]
- Rafique, W.; Kramer, V.; Pardo, T.; Smits, R.; Spilhaug, M.M.; Hoepping, A.; Savio, E.; Engler, H.; Kuljs, R.; Amaral, H.; et al. Image-guided development of heterocyclic sulfoxides as ligands for tau neurofibrillary tangles: From first-in-man to second-generation ligands. ACS Omega 2018, 3, 7567–7579. [Google Scholar] [CrossRef] [PubMed]
- Betthauser, T.J. 1cii—AD molecular: Imaging tau aggregates with positron emissions tomography. In Progress in Molecular Biology and Translational Science; Becker, J.T., Cohen, A.D., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 165, pp. 107–138. [Google Scholar]
- Xia, C.-F.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; Mocharla, V.P.; et al. [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s Dement. 2013, 9, 666–676. [Google Scholar] [CrossRef]
- Zhang, W.; Arteaga, J.; Cashion, D.K.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; et al. A highly selective and specific PET tracer for imaging of tau pathologies. J. Alzheimer’s Dis. 2012, 31, 601–612. [Google Scholar] [CrossRef]
- Chien, D.T.; Bahri, S.; Szardenings, A.K.; Walsh, J.C.; Mu, F.; Su, M.-Y.; Shankle, W.R.; Elizarov, A.; Kolb, H.C. Early Clinical PET Imaging Results with the Novel PHF-Tau Radioligand [F-18]-T807. J. Alzheimer’s Dis. 2013, 34, 457–468. [Google Scholar] [CrossRef]
- Marquié, M.; Normandin, M.D.; Vanderburg, C.R.; Costantino, I.M.; Bien, E.A.; Rycyna, L.G.; Klunk, W.E.; Mathis, C.A.; Ikonomovic, M.D.; Debnath, M.L.; et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann. Neurol. 2015, 78, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Chhatwal, J.P.; Schultz, A.P.; Marshall, G.A.; Boot, B.; Gomez-Isla, T.; Dumurgier, J.; LaPoint, M.; Scherzer, C.; Roe, A.D.; Hyman, B.T.; et al. Temporal T807 binding correlates with CSF tau and phospho-tau in normal elderly. Neurology 2016, 87, 920–926. [Google Scholar] [CrossRef]
- Lowe, V.J.; Curran, G.; Fang, P.; Liesinger, A.M.; Josephs, K.A.; Parisi, J.E.; Kantarci, K.; Boeve, B.F.; Pandey, M.K.; Bruinsma, T.; et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol. Commun. 2016, 4, 58. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Puschmann, A.; Schöll, M.; Ohlsson, T.; van Swieten, J.; Honer, M.; Englund, E.; Hansson, O. 18F-AV-1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain 2016, 139, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.A.; Friedrichsen, K.; Brier, M.; Blazey, T.; Su, Y.; Christensen, J.; Aldea, P.; McConathy, J.; Holtzman, D.M.; Cairns, N.J.; et al. The relationship between cerebrospinal fluid markers of Alzheimer pathology and positron emission tomography tau imaging. Brain 2016, 139, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Schöll, M.; Strandberg, O.; Smith, R.; Palmqvist, S.; Insel, P.S.; Hägerström, D.; Ohlsson, T.; Zetterberg, H.; Jögi, J.; et al. 18F-AV-1451 and CSF T-tau and P-tau as biomarkers in Alzheimer’s disease. EMBO Mol. Med. 2017, 9, 1212–1223. [Google Scholar] [CrossRef]
- Vermeiren, C.; Motte, P.; Viot, D.; Mairet-Coello, G.; Courade, J.P.; Citron, M.; Mercier, J.; Hannestad, J.; Gillard, M. The tau positron-emission tomography tracer AV-1451 binds with similar affinities to tau fibrils and monoamine oxidases. Mov. Disord. 2018, 33, 273–281. [Google Scholar] [CrossRef]
- Hansen, A.K.; Brooks, D.J.; Borghammer, P. MAO-B inhibitors do not block in vivo flortaucipir ([18F]-AV-1451) binding. Mol. Imaging Biol. 2018, 20, 356–360. [Google Scholar] [CrossRef]
- Chien, D.T.; Szardenings, A.K.; Bahri, S.; Walsh, J.C.; Mu, F.; Xia, C.; Shankle, W.R.; Lerner, A.J.; Su, M.-Y.; Elizarov, A.; et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F18]-T808. J. Alzheimer’s Dis. 2014, 38, 171–184. [Google Scholar] [CrossRef]
- Mattsson, N.; Insel, P.S.; Donohue, M.; Jögi, J.; Ossenkoppele, R.; Olsson, T.; Schöll, M.; Smith, R.; Hansson, O. Predicting diagnosis and cognition with 18F-AV-1451 tau PET and structural MRI in Alzheimer’s disease. Alzheimer’s Dement. 2019, 15, 570–580. [Google Scholar] [CrossRef]
- Brier, M.R.; Gordon, B.; Friedrichsen, K.; McCarthy, J.; Stern, A.; Christensen, J.; Owen, C.; Aldea, P.; Su, Y.; Hassenstab, J.; et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 338ra66. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K.; et al. Tau positron emission tomographic imaging in aging and early A lzheimer disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef]
- Schöll, M.; Lockhart, S.N.; Schonhaut, D.R.; O’Neil, J.P.; Janabi, M.; Ossenkoppele, R.; Baker, S.L.; Vogel, J.W.; Faria, J.; Schwimmer, H.D.; et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron 2016, 89, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.J.; Yu, P.; Miller, B.B.; Shcherbinin, S.; Dickson, J.; Navitsky, M.; Joshi, A.D.; Devous Sr, M.D.; Mintun, M.S. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016, 139, 1539–1550. [Google Scholar] [CrossRef]
- Devous Sr, M.D.; Joshi, A.D.; Navitsky, M.; Southekal, S.; Pontecorvo, M.J.; Shen, H.; Lu, M.; Shankle, W.R.; Seibyl, J.P.; Marek, K.; et al. Test–retest reproducibility for the tau PET imaging agent Flortaucipir F18. J. Nucl. Med. 2018, 59, 937–943. [Google Scholar] [CrossRef]
- Soleimani-Meigooni, D.N.; Iaccarino, L.; La Joie, R.; Baker, S.; Bourakova, V.; Boxer, A.L.; Edwards, L.; Eser, R.; Gorno-Tempini, M.-L.; Jagust, W.J.; et al. 18F-flortaucipir PET to autopsy comparisons in Alzheimer’s disease and other neurodegenerative diseases. Brain 2020, 143, 3477–3494. [Google Scholar] [CrossRef] [PubMed]
- Schöll, M.; Ossenkoppele, R.; Strandberg, O.; Palmqvist, S.; Jögi, J.; Ohlsson, T.; Smith, R.; Hansson, O. The Swedish BioFINDER Study. Distinct 18F-AV-1451 tau PET retention patterns in early-and late-onset Alzheimer’s disease. Brain 2017, 140, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. Alzheimer disease: [11C]PBB3—A new PET ligand that identifies tau pathology in the brains of patients with AD. Nat. Rev. Neurol. 2013, 9, 599. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Shimada, H.; Suhara, T.; Shinotoh, H.; Ji, B.; Maeda, J.; Zhang, M.R.; Trojanowski, J.Q.; Lee, V.M.; Ono, M.; et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 2013, 79, 1094–1108. [Google Scholar] [CrossRef]
- Ni, R.; Ji, B.; Ono, M.; Sahara, N.; Zhang, M.-R.; Aoki, I.; Nordberg, A.; Suhara, T.; Higuchi, M. Comparative In Vitro and In Vivo Quantifications of Pathologic Tau Deposits and Their Association with Neurodegeneration in Tauopathy Mouse Models. J. Nucl. Med. 2018, 59, 960–966. [Google Scholar] [CrossRef]
- Okamura, N.; Harada, R.; Furumoto, S.; Arai, H.; Yanai, K.; Kudo, Y. Tau PET imaging in Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 2014, 14, 500. [Google Scholar] [CrossRef]
- Ono, M.; Sahara, N.; Kumata, K.; Ji, B.; Ni, R.; Koga, S.; Dickson, D.W.; Trojanowski, J.Q.; Lee, V.M.-Y.; Yoshida, M.; et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 2017, 140, 764–780. [Google Scholar] [CrossRef] [PubMed]
- Declercq, L.; Celen, S.; Lecina, J.; Ahamed, M.; Tousseyn, T.; Moechars, D.; Alcazar, J.; Ariza, M.; Fierens, K.; Bottelbergs, A.; et al. Comparison of New Tau PET-Tracer Candidates with [18F]T808 and [18F]T807. Mol. Imaging 2016, 15, 1536012115624920. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Ono, M.; Sahara, N.; Higuchi, M.; Dickson, D.W. Fluorescence and autoradiographic evaluation of tau PET ligand PBB3 to α-synuclein pathology. Mov. Disord. 2017, 32, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Ichise, M.; Ito, H.; Shimada, H.; Ikoma, Y.; Seki, C.; Takano, H.; Kitamura, S.; Shinotoh, H.; Kawamura, K.; et al. PET quantification of tau pathology in human brain with 11C-PBB3. J. Nucl. Med. 2015, 56, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-T.; Li, X.-Y.; Lu, J.-Y.; Wu, P.; Li, L.; Liang, X.-N.; Ju, Z.-Z.; Jiao, F.-Y.; Chen, M.-J.; Ge, J.-J.; et al. 18F-Florzolotau Tau Positron Emission Tomography Imaging in Patients with Multiple System Atrophy–Parkinsonian Subtype. Mov. Disord. 2022, 37, 1915–1923. [Google Scholar] [CrossRef]
- Hashimoto, H.; Kawamura, K.; Igarashi, N.; Takei, M.; Fujishiro, T.; Aihara, Y.; Shiomi, S.; Muto, M.; Ito, T.; Furutsuka, K.; et al. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C-PBB3 as a clinically useful PET probe for imaging of tau pathology. J. Nucl. Med. 2014, 55, 1532–1538. [Google Scholar] [CrossRef]
- Weng, C.-C.; Hsiao, I.-T.; Yang, Q.-F.; Yao, C.-H.; Tai, C.-Y.; Wu, M.-F.; Yen, T.-C.; Jang, M.-K.; Lin, K.-J. Characterization of 18F-PM-PBB3 (18F-APN-1607) Uptake in the rTg4510 Mouse Model of Tauopathy. Molecules 2020, 25, 1750. [Google Scholar] [CrossRef]
- Kawamura, K.; Hashimoto, H.; Furutsuka, K.; Ohkubo, T.; Fujishiro, T.; Togashi, T.; Arashi, D.; Sakai, T.; Muto, M.; Ogawa, M.; et al. Radiosynthesis and quality control testing of the tau imaging positron emission tomography tracer [18F]PM-PBB3 for clinical applications. J. Label. Compd. Radiopharm. 2021, 64, 109–119. [Google Scholar] [CrossRef]
- Tagai, K.; Ono, M.; Kubota, M.; Kitamura, S.; Takahata, K.; Seki, C.; Takado, Y.; Shinotoh, H.; Sano, Y.; Yamamoto, Y.; et al. High-contrast in vivo imaging of tau pathologies in Alzheimer’s and non-Alzheimer’s disease tauopathies. Neuron 2021, 109, 42–58. [Google Scholar] [CrossRef]
- Shimada, H.; Kitamura, S.; Ono, M.; Kimura, Y.; Ichise, M.; Takahata, K.; Moriguchi, S.; Kubota, M.; Ishii, T.; Takado, Y.; et al. [IC-P-198]: First-in-Human Pet Study with 18F-AM-PBB3 and 18F-PM-PBB3. Alzheimer’s Dement. 2017, 13, P146. [Google Scholar]
- Walji, A.M.; Hostetler, E.D.; Selnick, H.; Zeng, Z.; Miller, P.; Bennacef, I.; Salinas, C.; Connolly, B.; Gantert, L.; Holahan, M.; et al. Discovery of 6-(Fluoro-18F)-3-(1H-pyrrolo[2,3-c]pyridin-1-yl)isoquinolin-5-amine ([18F]-MK-6240): A Positron Emission Tomography (PET) Imaging Agent for Quantification of Neurofibrillary Tangles (NFTs). J. Med. Chem. 2016, 59, 4778–4789. [Google Scholar] [CrossRef] [PubMed]
- Hostetler, E.D.; Walji, A.M.; Zeng, Z.; Miller, P.; Bennacef, I.; Salinas, C.; Connolly, B.; Gantert, L.; Haley, H.; Holahan, M.; et al. Preclinical Characterization of 18F-MK-6240, a Promising PET Tracer for In Vivo Quantification of Human Neurofibrillary Tangles. J. Nucl. Med. 2016, 57, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Malarte, M.L.; Nordberg, A.; Lemoine, L. Characterization of MK6240, a tau PET tracer, in autopsy brain tissue from Alzheimer’s disease cases. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Aguero, C.; Dhaynaut, M.; Normandin, M.D.; Amaral, A.C.; Guehl, N.J.; Neelamegam, R.; Marquie, M.; Johnson, K.A.; El Fakhri, G.; Frosch, M.P.; et al. Autoradiography validation of novel tau PET tracer [F-18]-MK-6240 on human postmortem brain tissue. Acta Neuropathol. Commun. 2019, 7, 37. [Google Scholar] [CrossRef]
- Koole, M.; Lohith, T.G.; Valentine, J.L.; Bennacef, I.; Declercq, R.; Reynders, T.; Riffel, K.; Celen, S.; Serdons, K.; Bormans, G.; et al. Preclinical Safety Evaluation and Human Dosimetry of [18F]MK-6240, a Novel PET Tracer for Imaging Neurofibrillary Tangles. Mol. Imaging Biol. 2020, 22, 173–180. [Google Scholar] [CrossRef]
- Betthauser, T.J.; Cody, K.A.; Zammit, M.D.; Murali, D.; Converse, A.K.; Barnhart, T.E.; Stone, C.K.; Rowley, H.A.; Johnson, S.C.; Christian, B.T. In Vivo Characterization and Quantification of Neurofibrillary Tau PET Radioligand 18F-MK-6240 in Humans from Alzheimer Disease Dementia to Young Controls. J. Nucl. Med. 2019, 60, 93–99. [Google Scholar] [CrossRef]
- Lohith, T.G.; Bennacef, I.; Vandenberghe, R.; Vandenbulcke, M.; Salinas, C.A.; Declercq, R.; Reynders, T.; Telan-Choing, N.F.; Riffel, K.; Celen, S.; et al. Brain Imaging of Alzheimer Dementia Patients and Elderly Controls with 18F-MK-6240, a PET Tracer Targeting Neurofibrillary Tangles. J. Nucl. Med. 2019, 60, 107–114. [Google Scholar] [CrossRef]
- Salinas, C.; Lohith, T.G.; Purohit, A.; Struyk, A.; Sur, C.; Bennacef, I.; Beaver, J.; Martarello, L. Test-retest characteristic of [18F]MK-6240 quantitative outcomes in cognitively normal adults and subjects with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2020, 40, 2179–2187. [Google Scholar] [CrossRef]
- Gogola, A.; Minhas, D.S.; Villemagne, V.L.; Cohen, A.D.; Mountz, J.M.; Pascoal, T.A.; Laymon, C.M.; Mason, N.S.; Ikonomovic, M.D.; Mathis, C.A.; et al. Direct comparison of the tau PET tracers [18F]flortaucipir and [18F]MK-6240 in human subjects. J. Nucl. Med. 2021, 63, 108–116. [Google Scholar] [CrossRef]
- Sanabria-Bohórquez, S.; Marik, J.; Ogasawara, A.; Tinianow, J.N.; Gill, H.S.; Barret, O.; Tamagnan, G.; Alagille, D.; Ayalon, G.; Manser, P.; et al. [18F]GTP1 (Genentech Tau Probe 1), a radioligand for detecting neurofibrillary tangle tau pathology in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2077–2089. [Google Scholar] [CrossRef]
- Marik, J.T.J.; Ogasawara, A. [18F]GTP1—A tau specific tracer for imaging tau-pathology in AD. In Proceedings of the Human Amyloid Imaging Conference, Miami Beach, FL, USA, 13–15 January 2016. [Google Scholar]
- Teng, E.; Ward, M.; Manser, P.T.; Sanabria-Bohorquez, S.; Ray, R.D.; Wildsmith, K.R.; Baker, S.; Kerchner, G.A.; Weimer, R.M. Cross-sectional associations between [18F]GTP1 tau PET and cognition in Alzheimer’s disease. Neurobiol. Aging 2019, 81, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohorquez, S.S.; Barret, O.; Tamagnan, G.; Alagille, D.; Marik, J.; Ayalon, G.; Bengtsson, T.; de Crespigny, A.; Jennings, D.; Seibyl, J.P.; et al. P4-351: Evaluation of TAU Burden in a Cross-Sectional Cohort of Alzheimer’s Disease Subjects Using [18F] GTP1 (Genetech tau probe 1). Alzheimer’s Dement. 2016, 12, P1172. [Google Scholar] [CrossRef]
- Honer, M.; Gobbi, L.; Knust, H.; Kuwabara, H.; Muri, D.; Koerner, M.; Valentine, H.; Dannals, R.F.; Wong, D.F.; Borroni, E. Preclinical evaluation of 18F-RO6958948, 11C-RO6931643, and 11C-RO6924963 as novel PET radiotracers for imaging tau aggregates in Alzheimer disease. J. Nucl. Med. 2018, 59, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, H.; Comley, R.A.; Borroni, E.; Honer, M.; Kitzmiller, K.; Roberts, J.; Gapasin, L.; Mathur, A.; Klein, G.; Wong, D.F. Evaluation of 18F-RO-948 PET for quantitative assessment of tau accumulation in the human brain. J. Nucl. Med. 2018, 59, 1877–1884. [Google Scholar] [CrossRef]
- Rombouts, F.J.; Andrés, J.I.; Ariza, M.; Alonso, J.M.; Austin, N.; Bottelbergs, A.; Chen, L.; Chupakhin, V.; Cleiren, E.; Fierens, K.; et al. Discovery of N-(Pyridin-4-yl)-1,5-naphthyridin-2-amines as Potential Tau Pathology PET Tracers for Alzheimer’s Disease. J. Med. Chem. 2017, 60, 1272–1291. [Google Scholar] [CrossRef]
- Declercq, L.; Rombouts, F.; Koole, M.; Fierens, K.; Mariën, J.; Langlois, X.; Andrés, J.I.; Schmidt, M.; Macdonald, G.; Moechars, D.; et al. Preclinical Evaluation of 18F-JNJ64349311, a Novel PET Tracer for Tau Imaging. J. Nucl. Med. 2017, 58, 975–981. [Google Scholar] [CrossRef]
- Rombouts, F.J.R.; Declercq, L.; Andrés, J.I.; Bottelbergs, A.; Chen, L.; Iturrino, L.; Leenaerts, J.E.; Mariën, J.; Song, F.; Wintmolders, C.; et al. Discovery of N-(4-[18F]Fluoro-5-methylpyridin-2-yl)isoquinolin-6-amine (JNJ-64326067), a New Promising Tau Positron Emission Tomography Imaging Tracer. J. Med. Chem. 2019, 62, 2974–2987. [Google Scholar] [CrossRef]
- Baker, S.L.; Provost, K.; Thomas, W.P.; Whitman, A.J.; Janabi, M.; Schmidt, M.E.; Timmers, M.; Kolb, H.C.; Rabinovici, G.D.; Jagust, W.J. Evaluation of 18F-JNJ-067 as a tau tracer. Alzheimer’s Dement. 2020, 16, e040651. [Google Scholar] [CrossRef]
- Kroth, H.; Oden, F.; Molette, J.; Schieferstein, H.; Capotosti, F.; Mueller, A.; Berndt, M.; Schmitt-Willich, H.; Darmency, V.; Gabellieri, E.; et al. Discovery and preclinical characterization of [18F]PI-2620, a next-generation tau PET tracer for the assessment of tau pathology in Alzheimer’s disease and other tauopathies. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2178–2189. [Google Scholar] [CrossRef]
- Mueller, A.; Kroth, H.; Berndt, M.; Capotosti, F.; Molette, J.; Schieferstein, H.; Oden, F.; Juergens, T.; Darmency, V.; Schmitt-Willich, H.; et al. Characterization of the novel PET Tracer PI-2620 for the assessment of Tau pathology in Alzheimer’s disease and other tauopathies. J. Nucl. Med. 2017, 58, 847. [Google Scholar]
- Bullich, S.; Barret, O.; Constantinescu, C.; Sandiego, C.; Mueller, A.; Berndt, M.; Papin, C.; Perrotin, A.; Koglin, N.; Kroth, H.; et al. Evaluation of Dosimetry, Quantitative Methods, and Test–Retest Variability of 18F-PI-2620 PET for the Assessment of Tau Deposits in the Human Brain. J. Nucl. Med. 2020, 61, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Brendel, M.; Barthel, H.; van Eimeren, T.; Marek, K.; Beyer, L.; Song, M.; Sauerbeck, J.; Barbe, M.; Schroeter, M.; Russell, D.; et al. 18F-PI2620 Tau-PET in Progressive Supranuclear Palsy-A multi-center evaluation. J. Nucl. Med. 2019, 60 (Suppl. S1), 54. [Google Scholar]
- Stephens, A.; Seibyl, J.; Mueller, A.; Barret, O.; Berndt, M.; Madonia, J.; Kroth, H.; Bullich, S.; Pfeifer, A.; Muhs, A.; et al. IC-P-220: Clinical Update: 18F-PI-2620, a Next Generation Tau Pet Agent Evaluated in Subjects with Alzheimer’s Disease and Progressive Supranuclear Palsy. Alzheimer’s Dement. 2018, 14, P179. [Google Scholar] [CrossRef]
- Villemagne, V.; Dore, V.; Mulligan, R.; Lamb, F.; Bourgeat, P.; Salvado, O.; Masters, C.; Rowe, C. Evaluation of 18F-PI-2620, a second-generation selective tau tracer for the assessment of Alzheimer’s and non-Alzheimer’s tauopathies. J. Nucl. Med. 2018, 59 (Suppl. S1), 410. [Google Scholar]
- Barret, O.; Seibyl, J.; Stephens, A.; Madonia, J.; Alagille, D.; Mueller, A.; Bendt, M.; Kroth, H.; Muhs, A.; Pfeifer, A.; et al. Initial clinical PET studies with the novel tau agent 18F PI-2620 in Alzheimer’s disease and controls. J. Nucl. Med. 2017, 58 (Suppl. S1), 630. [Google Scholar]
- Brendel, M.; Palleis, C.; Prix, C.; Finze, A.; Boetzel, K.; Danek, A.; Höllerhage, M.; Beyer, L.; Sauerbeck, J.; Rauchmann, B.; et al. 18F-PI-2620 tau-PET in corticobasal syndrome (ActiGliA cohort). Alzheimer’s Dement. 2020, 16, e041469. [Google Scholar] [CrossRef]
- Chotipanich, C.; Nivorn, M.; Kunawudhi, A.; Promteangtrong, C.; Boonkawin, N.; Jantarato, A. Evaluation of Imaging Windows for Tau PET Imaging Using 18F-PI2620 in Cognitively Normal Individuals, Mild Cognitive Impairment, and Alzheimer’s Disease Patients. Mol. Imaging 2020, 19, 1536012120947582. [Google Scholar] [CrossRef]
- Mormino, E.C.; Toueg, T.N.; Azevedo, C.; Castillo, J.B.; Guo, W.; Nadiadwala, A.; Corso, N.K.; Hall, J.N.; Fan, A.; Trelle, A.N.; et al. Tau PET imaging with 18F-PI-2620 in aging and neurodegenerative diseases. Eur. J. Nucl. Med. Mol. Imaging 2020, 48, 2233–2244. [Google Scholar] [CrossRef] [PubMed]
- Kaide, S.; Ono, M.; Watanabe, H.; Kitada, A.; Yoshimura, M.; Shimizu, Y.; Saji, H. Structure–Activity Relationships of Radioiodinated Benzoimidazopyridine Derivatives for Detection of Tau Pathology. ACS Med. Chem. Lett. 2018, 9, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Tatsumi, H.; Kaide, S.; Shimizu, Y.; Iikuni, S.; Ono, M. Structure-Activity Relationships of Radioiodinated 6,5,6-Tricyclic Compounds for the Development of Tau Imaging Probes. ACS Med. Chem. Lett. 2020, 11, 120–126. [Google Scholar] [CrossRef]
- Watanabe, H.; Tarumizu, Y.; Kaide, S.; Shimizu, Y.; Iikuni, S.; Nakamoto, Y.; Ono, M. Structure–Activity and Brain Kinetics Relationships of 18F-Labeled Benzimidazopyridine Derivatives as Tau PET Tracers. ACS Med. Chem. Lett. 2021, 12, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.F.; Schöll, M.; Almkvist, O.; Wall, A.; Engler, H.; Långström, B.; Nordberg, A. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: A multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 2012, 53, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Watanabe, H.; Kitada, A.; Matsumura, K.; Ihara, M.; Saji, H. Highly selective tau-SPECT imaging probes for detection of neurofibrillary tangles in Alzheimer’s disease. Sci. Rep. 2016, 6, srep34197. [Google Scholar] [CrossRef] [PubMed]
- Kaide, S.; Watanabe, H.; Shimizu, Y.; Tatsumi, H.; Iikuni, S.; Nakamoto, Y.; Togashi, K.; Ihara, M.; Saji, H.; Ono, M. 18F-labeled benzimidazopyridine derivatives for PET imaging of tau pathology in Alzheimer’s disease. Bioorganic Med. Chem. 2019, 27, 3587–3594. [Google Scholar] [CrossRef]
- Krause-Sorio, B.; Siddarth, P.; Laird, K.T.; Ercoli, L.; Narr, K.; Barrio, J.R.; Small, G.; Lavretsky, H. [18F]FDDNP PET binding predicts change in executive function in a pilot clinical trial of geriatric depression. Int. Psychogeriatr. 2021, 33, 149–156. [Google Scholar] [CrossRef]
- Wright, J.P.; Goodman, J.R.; Lin, Y.G.; Lieberman, B.P.; Clemens, J.; Gomez, L.F.; Liang, Q.; Hoye, A.T.; Pontecorvo, M.J.; Conway, K.A. Monoamine oxidase binding not expected to significantly affect [18F]flortaucipir PET interpretation. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 3797–3808. [Google Scholar] [CrossRef]
- Nakamura, S.; Kawamata, T.; Akiguchi, I.; Kameyama, M.; Nakamura, N.; Kimura, H. Expression of monoamine oxidase B activity in astrocytes of senile plaques. Acta Neuropathol. 1990, 80, 419–425. [Google Scholar] [CrossRef]
- Ng, K.P.; Pascoal, T.A.; Mathotaarachchi, S.; Therriault, J.; Kang, M.S.; Shin, M.; Guiot, M.C.; Guo, Q.; Harada, R.; Comley, R.A.; et al. Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain. Alzheimer’s Res. Ther. 2017, 9, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tracers | Author (Reference) | Reference Region | n | MMSE ± SD | SUVr of HC | SUVr of AD | Cohen’s d | SUVr of HC | SUVr of AD | Cohen’s d | SUVr of HC | SUVr of AD | Cohen’s d | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HC | AD | ||||||||||||||
| [18F]FDDNP | Tauber et al., 2013 [60] | Cerebellum | HC = 8 AD = 7 | 29.0 ± 1.2 | 22.1 ± 2.5 | Frontal cortex | Occipital cortex | Medial temporal cortex | |||||||
| 1.06 ± 0.07 | 1.20 ± 0.05 | 2.30 | 0.91 ± 0.07 | 0.93 ± 0.05 | 0.32 | 1.30 ± 0.10 | 1.32 ± 0.09 | 0.21 | |||||||
| Tolboom et al., 2009 [61] | Cerebellar gray matter | HC = 13 AD = 14 | 29 ± 1 | 23 ± 3 | Frontal cortex | Temporal cortex | Parietal cortex | ||||||||
| 0.05 ± 0.04 | 0.10 ± 0.02 | 1.58 | 0.07 ± 0.03 | 0.10 ± 0.03 | 1.0 | 0.03 ± 0.04 | 0.06 ± 0.03 | 0.84 | |||||||
| Shin et al., 2008 [62] | Cerebellum | HC = 10 AD = 10 | 28.4 ± 0.9 | 13 ± 5 | Lateral temporal cortex | Neocortex | Posterior cingulate | ||||||||
| 1.08 ± 0.04 | 1.24 ± 0.05 | 3.53 | 1.06 ± 0.04 | 1.19 ± 0.05 | 2.87 | 1.05 ± 0.05 | 1.20 ± 0.06 | 2.71 | |||||||
| [18F]THK523 | Villemagne et al., 2014 [47] | Cerebellar cortex | HC = 10 AD = 10 | 29.3 ± 1.1 | 16.7 ± 6.6 | Neocortex | Inferior temporal cortex | Insula | |||||||
| 0.82 ± 0.10 | 1.13 ± 0.07 | 3.42 | 0.96 ± 0.16 | 1.81 ± 0.58 | 2.00 | 0.85 ± 0.16 | 1.09 ± 0.22 | 1.30 | |||||||
| [18F]THK5105 | Okamura et al., 2014 [63] | Cerebellar cortex | HC = 8 AD = 8 | 28.8 ± 1.5 | 17.3 ± 6.6 | Inferior temporal cortex | Superior temporal cortex | Neocortex | |||||||
| 1.09 ± 0.04 | 1.32 ± 0.08 | 3.58 | 1.04 ± 0.06 | 1.22 ± 0.07 | 2.75 | 1.05 ± 0.05 | 1.23 ± 0.08 | 2.68 | |||||||
| [18F]THK5117 | Harada et al., 2015 [64] | Cerebellar cortex | HC = 5 AD = 5 | 28.7 ± 1.6 | 18.5 ± 4.6 | Neocortex | Posterior cingulate | Inferior temporal cortex | |||||||
| 1.13 ± 0.05 | 1.42 ± 0.13 | 3.05 | 1.11 ± 0.07 | 1.43 ± 0.14 | 2.77 | 1.15 ± 0.02 | 1.61 ± 0.23 | 2.74 | |||||||
| [18F]THK5317 | Fu et al., 2020 [65] | Cerebellum | HC = 6 AD = 5 | 28.8 ± 0.7 | 17.2 ± 6.0 | Occipital cortex | Lateral temporal cortex | Parietal cortex | |||||||
| 1.12 ± 0.04 | 1.36 ± 0.10 | 3.15 | 1.15 ± 0.02 | 1.45 ± 0.14 | 3 | 1.07 ± 0.07 | 1.27 ± 0.09 | 2.48 | |||||||
| [18F]THK5351 | Chanisa et al., 2021 [66] | Cerebellum | Age ≤ 60 HC = 13 AD = 6 | N/A | N/A | Inferior temporal cortex | Occipital cortex | Posterior cingulate | |||||||
| 1.37 ± 0.04 | 1.60 ± 0.21 | 1.52 | 1.16 ± 0.05 | 1.35 ± 0.17 | 1.43 | 1.45 ± 0.06 | 1.68 ± 0.24 | 1.31 | |||||||
| Age > 60 HC = 11 AD = 9 | N/A | N/A | Occipital cortex | Precuneus | Inferior temporal cortex | ||||||||||
| 1.18 ± 0.12 | 1.42 ± 0.15 | 1.76 | 1.26 ± 0.11 | 1.47 ± 0.18 | 1.40 | 1.48 ± 0.21 | 1.86 ± 0.32 | 1.37 | |||||||
| Ezura et al., 2021 [67] | Cerebellar cortex | HC = 9 AD = 10 | 28.8 ± 1.5 | 18.9 ± 4.6 | Inferior temporal gyrus | Fusiform gyrus | Parahippocampus | ||||||||
| 1.53 ± 0.12 | 2.11 ± 0.25 | 3.00 | 1.60 ± 0.11 | 2.06 ± 0.19 | 2.96 | 2.01 ± 0.12 | 2.43 ± 0.20 | 2.54 | |||||||
| Chen et al., 2018 [68] | SUVP | HC = 9 AD = 9 | 29 | 20 | Temporal cortex | Occipital cortex | Parietal cortex | ||||||||
| 1.57 ± 0.21 | 1.88 ± 0.22 | 1.44 | 1.22 ± 0.13 | 1.42 ± 0.16 | 1.37 | 1.30 ± 0.24 | 1.46 ± 0.12 | 0.84 | |||||||
| Kang et al., 2017 [69] | Cerebellar gray matter | HC = 43 AD = 51 | 28.5 ± 1.6 | 13.8 ± 6.0 | Frontal cortex | Medial temporal cortex | Hippocampus | ||||||||
| 1.35 ± 0.22 | 2.05 ± 0.34 | 2.44 | 2.42 ± 0.27 | 3.52 ± 0.59 | 2.39 | 2.44 ± 0.27 | 3.40 ± 0.55 | 2.21 | |||||||
| [18F]Flortaucipir | Leuzy et al., 2021 [70] | Inferior cerebellar cortex | HC = 638 AD = 159 | 28.9 ± 1.2 | 20.4 ± 5.0 | Entorhinal cortex | Early tau ROIs | Temporal meta-ROI | |||||||
| 1.14 ± 0.12 | 1.74 ± 0.32 | 2.48 | 1.19 ± 0.11 | 1.88 ± 0.46 | 2.06 | 1.18 ± 0.11 | 1.88 ± 0.47 | 2.05 | |||||||
| Li et al., 2021 [71] | Cerebellar gray matter | HC = 10 AD = 4 | 29.2 ± 1.3 | 8.5 ± 6.6 | Frontal regions | Lateral temporal region | Parietal region | ||||||||
| 1.32 ± 0.13 | 2.26 ± 0.12 | 7.51 | 1.35 ± 0.14 | 2.22 ± 0.29 | 3.82 | 1.38 ± 0.17 | 2.59 ± 0.43 | 3.70 | |||||||
| Wolters et al., 2020 [72] | Cerebellar gray matter | HC = 25 AD/MCI = 53 | 28 ± 1 | 23 ± 4 | Entorhinal cortex | Limbic region | Neocortex | ||||||||
| 1.1 ± 0.2 | 1.5 ± 0.2 | 2.00 | 1.2 ± 0.1 | 1.5 ± 0.2 | 1.89 | 1.0 ± 0.1 | 1.4 ± 0.3 | 1.87 | |||||||
| Chen et al., 2018 [68] | SUVP | HC = 20 AD = 12 | 29 | 21 | Temporal cortex | Occipital cortex | Frontal cortex | ||||||||
| 1.18 ± 0.16 | 1.41 ± 0.35 | 0.84 | 1.09 ± 0.09 | 1.17 ± 0.13 | 0.71 | 1.06 ± 0.07 | 1.18 ± 0.23 | 0.70 | |||||||
| Ossenkoppele et al., 2018 [73] | Cerebellar gray matter | HC = 254 AD = 179 | 23.6 ± 6.0 | 20.2 ± 5.5 | Entorhinal cortex | Inferior temporal cortex | Temporoparietal cortex | ||||||||
| 1.73 ± 0.31 | 1.18 ± 0.2 | 2.10 | 2.09 ± 0.56 | 1.23 ± 0.21 | 2.03 | 1.89 ± 0.53 | 1.15 ± 0.18 | 1.86 | |||||||
| Pontecorvo et al., 2017 [74] | Cerebellar gray matter | Aβ + OC = 58 Aβ + AD = 48 | 29.5 ± 0.5 | 22.1 ± 3.7 | Fusiform gyrus | Anterior parahippocampal gyrus | Temporal cortex | ||||||||
| 1.15 ± 0.10 | 1.66 ± 0.30 | 2.28 | 1.07 ± 0.13 | 1.49 ± 0.24 | 2.17 | 1.11 ± 0.09 | 1.64 ± 0.40 | 1.82 | |||||||
| Cho et al., 2016 [75] | Cerebellar cortex | HC = 20 AD = 20 | 27.5 ± 2.1 | 16.9 ± 6.6 | Entorhinal cortex | Parahippocampal cortex | Inferior temporal cortex | ||||||||
| 1.22 ± 0.16 | 1.80 ± 0.33 | 2.26 | 1.20 ± 0.17 | 1.49 ± 0.25 | 1.99 | 1.71 ± 0.34 | 1.22 ± 0.19 | 1.79 | |||||||
| [11C]PBB3 | Kitamura et al., 2018 [76] | Cerebellar gray matter | HC = 9 AD = 17 | 29.4 ± 0.7 | 21.4 ± 6.5 | Occipital cortex | Parietal cortex | Lateral temporal cortex | |||||||
| 0.90 ± 0.03 | 1.10 ± 0.1 | 2.70 | 0.83 ± 0.05 | 1.03 ± 0.1 | 2.52 | 0.95 ± 0.04 | 1.12 ± 0.11 | 2.05 | |||||||
| Shimada et al., 2017 [77] | Cerebellar cortex | HC = 18 AD = 17 | 28.9 ± 1.2 | 16.1 ± 5.1 | Mean cortical | ||||||||||
| 0.91 ± 0.06 | 1.10 ± 0.07 | 2.91 | |||||||||||||
| [18F]PM-PBB3 | Hsu et al., 2020 [78] | Cerebellar gray matter | HC = 12 AD = 10 | 29.3 ± 0.9 | 12.5 ± 8.9 | Parahippocampus | Temporal region | Posterior cingulate gyrus | |||||||
| 0.97 ± 0.11 | 2.06 ± 0.69 | 2.20 | 0.99 ± 0.10 | 2.46 ± 0.95 | 2.17 | 1.02 ± 0.09 | 2.63 ± 1.09 | 2.08 | |||||||
| Lu et al., 2020 [79] | Cerebellar cortex | HC = 11 AD = 19 | N/A | 17.0 ± 7.6 | Temporal lobe | Frontal lobe | Occipital lobe | ||||||||
| 0.91 ± 0.06 | 1.64 ± 0.50 | 2.06 | 0.85 ± 0.06 | 1.43 ± 0.42 | 1.92 | 0.95 ± 0.06 | 1.58 ± 0.47 | 1.91 | |||||||
| [18F]MK6240 | Therriault et al., 2022 [80] | Inferior cerebellar cortex | HC = 179 AD = 65 | 29.1 ± 1.4 | 19.7 ± 6.2 | Temporal meta-ROIs | |||||||||
| 1.06 ± 0.15 | 2.82 ± 1.03 | 2.39 | |||||||||||||
| Ashton et al., 2021 [81] | Inferior cerebellar cortex | HC = 159 AD = 42 | 29.1 ± 1.1 | 18.5 ± 5.7 | Braak I–II | Braak III–IV | Braak V-VI | ||||||||
| 0.97 ± 0.2 | 1.98 ± 0.6 | 2.25 | 0.95 ± 0.1 | 2.61 ± 1.1 | 2.12 | 0.97 ± 0.1 | 2.25 ± 2.1 | 0.86 | |||||||
| Leuzy et al., 2021 [70] | Inferior cerebellar cortex | HC = 218 AD = 50 | 29.2 ± 1.0 | 18.4 ± 5.9 | Early tau ROIs | Entorhinal cortex | Temporal meta-ROIs | ||||||||
| 0.87 ± 0.12 | 2.80 ± 0.64 | 4.19 | 0.93 ± 0.23 | 2.4 ± 0.56 | 3.48 | 0.86 ± 0.11 | 2.84 ± 0.66 | 3.42 | |||||||
| Therriault et al., 2021 [82] | Inferior cerebellar cortex | HC = 131 AD = 25 | 29.1 ± 1.2 | 20.1 ± 5.7 | Braak III–IV | Braak V–VI | Braak I–II | ||||||||
| 1.04 ± 0.17 | 2.89 ± 0.87 | 2.95 | 1.09 ± 0.13 | 2.47 ± 0.96 | 2.01 | 0.99 ± 0.29 | 2.02 ± 0.86 | 1.60 | |||||||
| Therriault et al., 2021 [83] | Inferior cerebellar cortex | HC = 166 AD = 62 | 29.1 ± 1.0 | 19.2 ± 6.2 | AD signature meta-ROI | Braak I–II | |||||||||
| 1.08 ± 0.24 | 3.3 ± 1.4 | 2.21 | 0.98 ± 0.22 | 2.04 ± 0.75 | 1.91 | ||||||||||
| Tissot et al., 2021 [84] | Inferior cerebellar cortex | HC = 143 AD = 26 | 29.0 ± 1.2 | 19.3 ± 7.1 | Braak III–IV | Braak I–II | Braak V-VI | ||||||||
| 1.07 ± 0.12 | 2.51 ± 0.91 | 2.21 | 0.97 ± 0.18 | 1.63 ± 0.40 | 2.12 | 1.10 ± 0.14 | 2.24 ± 0.87 | 1.82 | |||||||
| Pascoal et al., 2020 [85] | Inferior cerebellum | HC = 101 LOAD = 21 | 29.1 ± 1.1 | 21.2 ± 5.3 | Braak II | Braak I | Braak III | ||||||||
| 1.14 ± 0.15 | 2.6 ± 0.97 | 2.10 | 1.13 ± 0.17 | 3.05 ± 1.32 | 2.04 | 1.17 ± 0.1 | 2.6 ± 1.18 | 1.93 | |||||||
| [18F]GTP1 | Barthelemy et al., 2022 [86] | Cerebellar gray matter | Aβ+ HC = 5 MAD = 10 | 29.0 ± 0.7 | 17.6 ± 2.7 | Temporal meta-ROIs | Braak III–IV | Braak I–II | |||||||
| 1.26 ± 0.05 | 1.67 ± 0.32 | 1.79 | 1.2 ± 0.05 | 1.55 ± 0.35 | 1.4 | 1.27 ± 0.04 | 1.48 ± 0.26 | 1.12 | |||||||
| [18F]RO948 | Leuzy et al., 2022 [87] | Inferior cerebellar cortex | Aβ− HC = 137 AD = 63 | 28.9 ± 1.2 | 19.7 ± 4.2 | EBM stage I | EBM stage II | EBM stage III | |||||||
| 0.97 ± 0.13 | 1.71 ± 0.36 | 2.80 | 1.27 ± 0.11 | 2.62 ± 0.94 | 2.01 | 1.26 ± 0.12 | 2.37 ± 0.91 | 1.71 | |||||||
| Leuzy et al., 2021 [70] | Inferior cerebellar cortex | HC = 208 AD = 142 | 28.7 ± 1.2 | 20.3 ± 4.1 | Entorhinal cortex | Early tau ROIs | Temporal meta-ROIs | ||||||||
| 1.23 ± 0.22 | 2.00 ± 0.40 | 3.78 | 1.23 ± 0.18 | 2.15 ± 0.66 | 1.90 | 1.22 ± 0.18 | 2.13 ± 0.66 | 1.88 | |||||||
| Leuzy et al., 2020 [88] | Inferior cerebellar cortex | HC = 257 AD = 100 | 29 ± 1.1 | 20 ± 4.3 | Braak stage V–VI | Entorhinal cortex | Braak stage I-IV | ||||||||
| 1.06 ± 0.10 | 1.53 ± 0.21 | 2.85 | 1.16 ± 0.22 | l2.02 ± 0.40 | 2.66 | 1.17 ± 0.15 | 2.19 ± 0.58 | 2.40 | |||||||
| Wong et al., 2018 [89] | Cerebellar cortex | HC = 5 AD = 11 | N/A | 20.8 ± 2.8 | Parahippocampus | Entorhinal area | Anterior temporal lobe | ||||||||
| 0.93 ± 0.2 | 1.95 ± 0.7 | 1.98 | 0.93 ± 0.2 | 1.91 ± 0.7 | 1.89 | 1.01 ± 0.1 | 1.71 ± 0.6 | 1.62 | |||||||
| [18F]JNJ067 | Schmidt et al., 2020 [90] | Ventral cerebellar cortex | HC = 5 AD = 5 | 28.4 ± 0.5 | 19.2 ± 5.9 | Medial temporal cortex | Amygdala | Occipital cortex | |||||||
| 1.18 ± 0.09 | 1.76 ± 0.47 | 1.71 | 0.96 ± 0.12 | 1.51 ± 0.46 | 1.63 | 1.14 ± 0.10 | 1.54 ± 0.40 | 1.37 | |||||||
| [18F]PI2620 | Bun et al., 2022 [91] | Cerebellum | HC = 7 AD = 7 | 28.4 ± 1.5 | 18.7 ± 5.1 | Right amygdala | Left amygdala | Left hippocampus | |||||||
| 1.14 ± 0.08 | 1.98 ± 0.31 | 3.63 | 1.19 ± 0.14 | 2.07 ± 0.44 | 2.66 | 1.42 ± 0.14 | 1.81 ± 0.21 | 2.08 | |||||||
| Jantarato et al., 2021 [92] | Cerebellum | HC = 26 AD = 7 | 27.3 | 18.4 | Occipital cortex | Precuneus | Caudate | ||||||||
| 1.1 ± 0.12 | 1.14 ± 0.21 | 0.93 | 1.02 ± 0.12 | 1.27 ± 0.45 | 0.75 | 0.80 ± 0.13 | 0.7 ± 0.14 | 0.74 | |||||||
| Mueller et al., 2020 [93] | Cerebellum | HC = 10 AD = 12 | 29 | 20.5 | Inferior temporal | Fusiform gyrus | Occipital | ||||||||
| 1.08 ± 0.08 | 1.70 ± 0.32 | 2.59 | 1.08 ± 0.09 | 1.57 ± 0.24 | 2.55 | 1.06 ± 0.07 | 1.39 ± 0.19 | 2.13 | |||||||
| [18F]S16 | Fu et al., 2022 [65,94] | Cerebellar gray matter | HC = 6 AD = 5 | 28.8 ± 0.7 | 17.2 ± 6.0 | Occipital cortex | Lateral temporal cortex | Medial temporal cortex | |||||||
| 1.10 ± 0.07 | 1.21 ± 0.08 | 1.46 | 1.22 ± 0.09 | 1.30 ± 0.10 | 0.84 | 1.11 ± 0.10 | 1.18 ± 0.12 | 0.63 | |||||||
| Wang et al., 2021 [95] | Cerebellum | HC = 6 AD = 15 | 29.8 ± 0.3 | 18.6 ± 5.3 | Parietal cortex | Posterior cingulate and precuneus | Occipital cortex | ||||||||
| 1.10 ± 0.05 | 1.35 ± 0.19 | 1.78 | 1.17 ± 0.04 | 1.35 ± 0.14 | 1.70 | 1.05 ± 0.03 | 1.19 ± 0.12 | 1.49 | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammadi, Z.; Alizadeh, H.; Marton, J.; Cumming, P. The Sensitivity of Tau Tracers for the Discrimination of Alzheimer’s Disease Patients and Healthy Controls by PET. Biomolecules 2023, 13, 290. https://doi.org/10.3390/biom13020290
Mohammadi Z, Alizadeh H, Marton J, Cumming P. The Sensitivity of Tau Tracers for the Discrimination of Alzheimer’s Disease Patients and Healthy Controls by PET. Biomolecules. 2023; 13(2):290. https://doi.org/10.3390/biom13020290
Chicago/Turabian StyleMohammadi, Zohreh, Hadi Alizadeh, János Marton, and Paul Cumming. 2023. "The Sensitivity of Tau Tracers for the Discrimination of Alzheimer’s Disease Patients and Healthy Controls by PET" Biomolecules 13, no. 2: 290. https://doi.org/10.3390/biom13020290