

Intracranial Aneurysms and Lipid Metabolism Disorders: From Molecular Mechanisms to Clinical Implications

Abstract
:1. Introduction
2. Dysregulated Lipid Metabolism in Vascular Diseases
2.1. Fatty Acyls
2.2. Triglycerides
2.3. Glycerophospholipids and Sphingolipids
2.4. Apolipoprotein
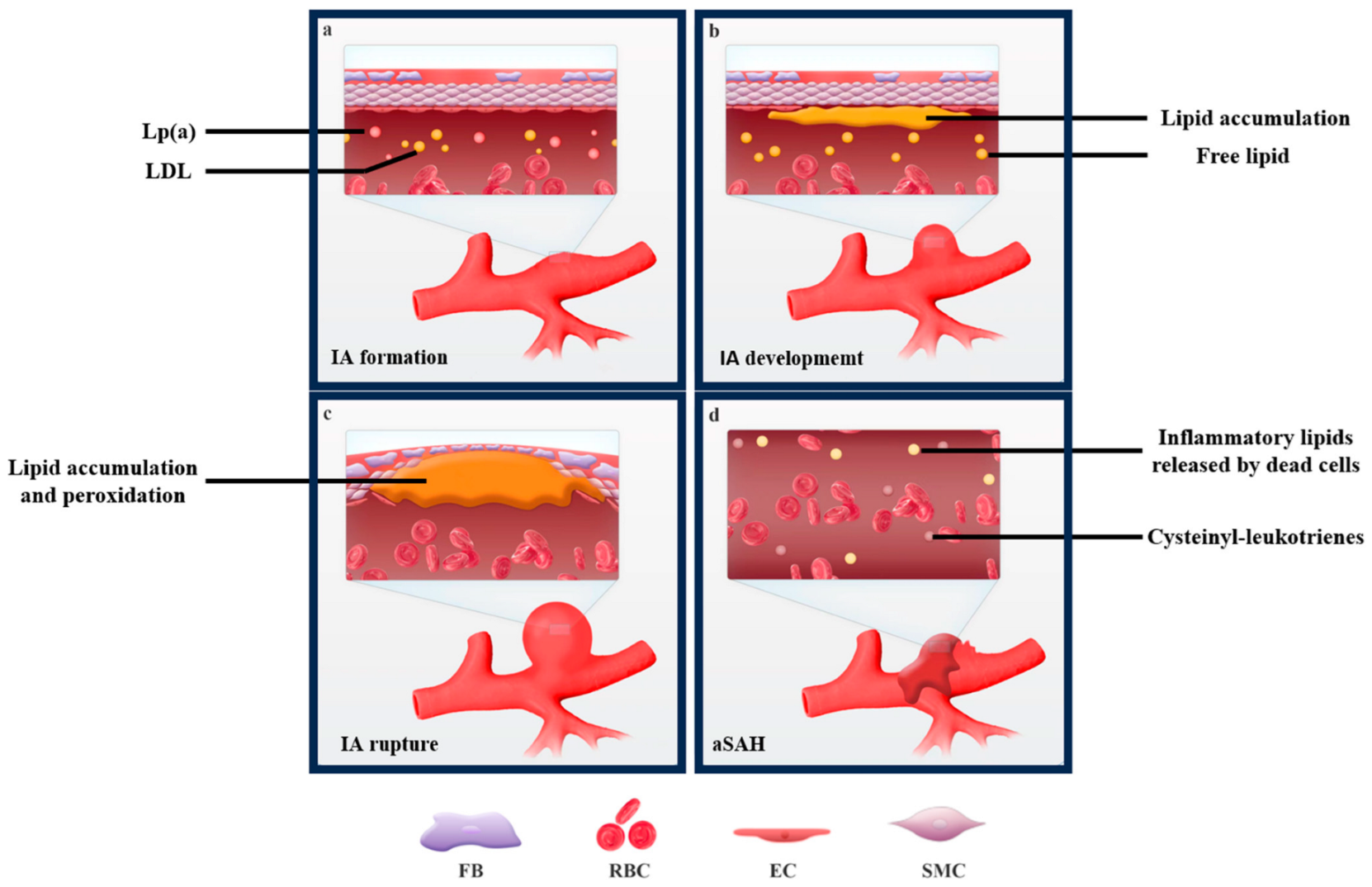
3. Lipid Metabolism and IA Formation
4. Lipid Metabolism and IA Rupture
5. Lipid Metabolism and aSAH
6. Lipid Metabolism as a Biomarker and Therapeutic Target for IA
6.1. Lipid Metabolites as Biomarkers for IA
6.2. Lipid Metabolism Disorder as a Therapeutic Target for IA
7. Conclusions
Funding
Conflicts of Interest
References
- Liu, J.; Zou, X.; Zhao, Y.; Jin, Z.; Tu, J.; Ning, X.; Li, J.; Yang, X.; Wang, J. Prevalence and Risk Factors for Unruptured Intracranial Aneurysms in the Population at High Risk for Aneurysm in the Rural Areas of Tianjin. Front. Neurol. 2022, 13, 853054. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ye, M.; Cimpoca, A.; Henkes, H.; Wang, H.; Xu, X.; Gu, Y.; Shi, H.; Ji, H.; Wang, F.; et al. Avenir® vs. AxiumTM Coils for the Treatment of Intracranial Aneurysms: Results of a Multicenter Randomized Controlled Trial With Short-Term Follow-Up. Front. Neurol. 2021, 12, 817989. [Google Scholar] [CrossRef]
- Jin, J.; Duan, J.; Du, L.; Xing, W.; Peng, X.; Zhao, Q. Inflammation and immune cell abnormalities in intracranial aneurysm subarachnoid hemorrhage (SAH): Relevant signaling pathways and therapeutic strategies. Front. Immunol. 2022, 13, 1027756. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Cho, K. Lipid metabolism, inflammation, and foam cell formation in health and metabolic disorders: Targeting mTORC1. J. Mol. Med. 2021, 99, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhu, N.; Yan, T.; Shi, Y.-N.; Chen, J.; Zhang, C.-J.; Xie, X.-J.; Liao, D.-F.; Qin, L. The crosstalk: Exosomes and lipid metabolism. Cell Commun. Signal 2020, 18, 119. [Google Scholar] [CrossRef] [PubMed]
- Løvik, K.; Laupsa-Borge, J.; Logallo, N.; Helland, C.A. Dyslipidemia and rupture risk of intracranial aneurysms—A systematic review. Neurosurg. Rev. 2021, 44, 3143–3150. [Google Scholar] [CrossRef]
- Ou, C.; Qian, Y.; Zhang, X.; Liu, J.; Liu, W.; Su, H.; Zhang, N.; Zhang, J.; He, X.; Duan, C.-Z. Elevated Lipid Infiltration Is Associated With Cerebral Aneurysm Rupture. Front. Neurol. 2020, 11, 154. [Google Scholar] [CrossRef]
- Frösen, J.; Tulamo, R.; Heikura, T.; Sammalkorpi, S.; Niemelä, M.; Hernesniemi, J.; Levonen, A.-L.; Hörkkö, S.; Ylä-Herttuala, S. Lipid accumulation, lipid oxidation, and low plasma levels of acquired antibodies against oxidized lipids associate with degeneration and rupture of the intracranial aneurysm wall. Acta Neuropathol. Commun. 2013, 1, 71. [Google Scholar] [CrossRef]
- van der Kamp, L.T.; Rinkel, G.J.E.; Verbaan, D.; van den Berg, R.; Vandertop, W.P.; Murayama, Y.; Ishibashi, T.; Lindgren, A.; Koivisto, T.; Teo, M.; et al. Risk of Rupture After Intracranial Aneurysm Growth. JAMA Neurol. 2021, 78, 1228–1235. [Google Scholar] [CrossRef]
- Renowden, S.; Nelson, R. Management of incidental unruptured intracranial aneurysms. Prac. Neurol. 2020, 20, 347–355. [Google Scholar] [CrossRef]
- Krischek, B.; Tatagiba, M. The influence of genetics on intracranial aneurysm formation and rupture: Current knowledge and its possible impact on future treatment. Adv. Tech. Stand. Neurosurg. 2008, 33, 131–147. [Google Scholar]
- Vanrossomme, A.E.; Eker, O.F.; Thiran, J.P.; Courbebaisse, G.P.; Zouaoui Boudjeltia, K. Intracranial Aneurysms: Wall Motion Analysis for Prediction of Rupture. AJNR. Am. J. Neuroradiol. 2015, 36, 1796–1802. [Google Scholar] [CrossRef]
- Tang, H.; Wang, Q.; Xu, F.; Zhang, X.; Zeng, Z.; Yan, Y.; Lu, Z.; Xue, G.; Zuo, Q.; Luo, Y.; et al. Underlying mechanism of hemodynamics and intracranial aneurysm. Chin. Neurosurg. J. 2021, 7, 44. [Google Scholar] [CrossRef]
- Budohoski, K.P.; Guilfoyle, M.; Helmy, A.; Huuskonen, T.; Czosnyka, M.; Kirollos, R.; Menon, D.K.; Pickard, J.D.; Kirkpatrick, P.J. The pathophysiology and treatment of delayed cerebral ischaemia following subarachnoid haemorrhage. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1343–1353. [Google Scholar] [CrossRef]
- Yu, W. Vasospasm and Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage: Recent Advances and Future Directions in Translational Research. Transl. Stroke Res. 2023, 14, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.A. Management of delayed cerebral ischemia after subarachnoid hemorrhage. CONTINUUM Lifelong Learn. Neurol. 2012, 18, 579–597. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xiong, X.; Wu, X.; Ye, Y.; Jian, Z.; Zhi, Z.; Gu, L. Targeting Oxidative Stress and Inflammation to Prevent Ischemia-Reperfusion Injury. Front. Mol. Neurosci. 2020, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K. Fatty acids act on vascular endothelial cells and influence the development of cardiovascular disease. Prostaglandins Other Lipid Mediat 2023, 165, 106704. [Google Scholar] [CrossRef]
- Vafeiadou, K.; Weech, M.; Sharma, V.; Yaqoob, P.; Todd, S.; Williams, C.M.; Jackson, K.G.; Lovegrove, J.A. A review of the evidence for the effects of total dietary fat, saturated, monounsaturated and n-6 polyunsaturated fatty acids on vascular function, endothelial progenitor cells and microparticles. Br. J. Nutr. 2012, 107, 303–324. [Google Scholar] [CrossRef]
- Du, Y.; Taylor, C.G.; Zahradka, P. Modulation of endothelial cell responses and vascular function by dietary fatty acids. Nutr. Rev. 2019, 77, 614–629. [Google Scholar] [CrossRef]
- Jackson, K.G.; Newens, K.J.; Fry, M.J.; Thompson, A.K.; Williams, C.M. Differential effects of single fatty acids and fatty acid mixtures on the phosphoinositide 3-kinase/Akt/eNOS pathway in endothelial cells. Eur. J. Nutr. 2022, 61, 2463–2473. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gonzalez, O.; Shen, X.; Barnhart, S.; Kramer, F.; Kanter, J.E.; Vivekanandan-Giri, A.; Tsuchiya, K.; Handa, P.; Pennathur, S.; et al. Endothelial acyl-CoA synthetase 1 is not required for inflammatory and apoptotic effects of a saturated fatty acid-rich environment. Arter. Thromb. Vasc. Biol. 2013, 33, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Mantzaris, M.D.; Tsianos, E.V.; Galaris, D. Interruption of triacylglycerol synthesis in the endoplasmic reticulum is the initiating event for saturated fatty acid-induced lipotoxicity in liver cells. FEBS J. 2011, 278, 519–530. [Google Scholar] [CrossRef]
- Anderson, E.K.; Hill, A.A.; Hasty, A.H. Stearic acid accumulation in macrophages induces toll-like receptor 4/2-independent inflammation leading to endoplasmic reticulum stress-mediated apoptosis. Arter. Thromb. Vasc. Biol. 2012, 32, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.H.; Kim, J.-A.; Lee, J.Y. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur. J. Pharmacol. 2016, 785, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef]
- Dallak, M.A. Acylated ghrelin induces but deacylated ghrelin prevents hepatic steatosis and insulin resistance in lean rats: Effects on DAG/ PKC/JNK pathway. Biomed. Pharmacother. 2018, 105, 299–311. [Google Scholar] [CrossRef]
- Gupta, P.; Sharma, G.; Lahiri, A.; Barthwal, M.K. FOXO3a acetylation regulates PINK1, mitophagy, inflammasome activation in murine palmitate-conditioned and diabetic macrophages. J. Leukoc. Biol. 2022, 111, 611–627. [Google Scholar] [CrossRef]
- Sardar, A.; Dewangan, N.; Panda, B.; Bhowmick, D.; Tarafdar, P.K. Lipid and Lipidation in Membrane Fusion. J. Membr. Biol. 2022, 255, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef]
- Mazzocchi, A.; De Cosmi, V.; Risé, P.; Milani, G.P.; Turolo, S.; Syrén, M.-L.; Sala, A.; Agostoni, C. Bioactive Compounds in Edible Oils and Their Role in Oxidative Stress and Inflammation. Front. Physiol. 2021, 12, 659551. [Google Scholar] [CrossRef]
- Chipurupalli, S.; Samavedam, U.; Robinson, N. Crosstalk Between ER Stress, Autophagy and Inflammation. Front. Med. 2021, 8, 758311. [Google Scholar] [CrossRef]
- Hart, C.M.; Tolson, J.K.; Block, E.R. Fatty acid supplementation protects pulmonary artery endothelial cells from oxidant injury. Am. J. Respir. Cell Mol. Biol. 1990, 3, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Sekharam, K.M.; Patel, J.M.; Block, E.R. Effect of polyunsaturated fatty acids and phospholipids on [3H]-vitamin E incorporation into pulmonary artery endothelial cell membranes. J. Cell. Physiol. 1990, 145, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Block, E.R.; Patel, J.M.; Sheridan, N.P. Endotoxin protects against hyperoxic decrease in membrane fluidity in endothelial cells but not in fibroblasts. Lab. Invest. 1986, 54, 146–153. [Google Scholar] [PubMed]
- Duran, E.K.; Aday, A.W.; Cook, N.R.; Buring, J.E.; Ridker, P.M.; Pradhan, A.D. Triglyceride-Rich Lipoprotein Cholesterol, Small Dense LDL Cholesterol, and Incident Cardiovascular Disease. J. Am. Coll. Cardiol. 2020, 75, 2122–2135. [Google Scholar] [CrossRef] [PubMed]
- Esan, O.; Wierzbicki, A.S. Triglycerides and cardiovascular disease. Curr. Opin. Cardiol. 2021, 36, 469–477. [Google Scholar] [CrossRef]
- Carrasquilla, G.D.; Christiansen, M.R.; Kilpeläinen, T.O. The Genetic Basis of Hypertriglyceridemia. Curr. Atheroscler. Rep. 2021, 23, 39. [Google Scholar] [CrossRef]
- Hussain, A.; Ballantyne, C.M.; Saeed, A.; Virani, S.S. Triglycerides and ASCVD Risk Reduction: Recent Insights and Future Directions. Curr. Atheroscler. Rep. 2020, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Sillars, A.; Sattar, N. Management of Lipid Abnormalities in Patients with Diabetes. Curr. Cardiol. Rep. 2019, 21, 147. [Google Scholar] [CrossRef]
- Iqbal, J.; Walsh, M.T.; Hammad, S.M.; Hussain, M.M. Sphingolipids and Lipoproteins in Health and Metabolic Disorders. Trends Endocrinol. Metab. 2017, 28, 506–518. [Google Scholar] [CrossRef]
- Jiang, W.; Chen, J.; Gong, L.; Liu, F.; Zhao, H.; Mu, J. Alteration of Glycerophospholipid Metabolism in Hippocampus of Post-stroke Depression Rats. Neurochem. Res. 2022, 47, 2052–2063. [Google Scholar] [CrossRef] [PubMed]
- Shaik, N.F.; Regan, R.F.; Naik, U.P. Platelets as drivers of ischemia/reperfusion injury after stroke. Blood Adv. 2021, 5, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Zong, N.; Hu, Y.; Chen, Y.; Xu, Y. Neuronal Death Mechanisms and Therapeutic Strategy in Ischemic Stroke. Neurosci. Bull. 2022, 38, 1229–1247. [Google Scholar] [CrossRef]
- Magaye, R.R.; Savira, F.; Hua, Y.; Kelly, D.J.; Reid, C.; Flynn, B.; Liew, D.; Wang, B.H. The role of dihydrosphingolipids in disease. Cell Mol. Life Sci. 2019, 76, 1107–1134. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Nolde, J.M.; Chan, J.; Carnagarin, R.; Watts, G.F.; Schlaich, M.P. Lipoprotein (a) and Hypertension. Curr. Hypertens Rep. 2021, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Rhainds, D.; Brodeur, M.R.; Tardif, J.-C. Lipoprotein (a): When to Measure and How to Treat? Curr. Atheroscler Rep. 2021, 23, 51. [Google Scholar] [CrossRef]
- Saeed, A.; Kinoush, S.; Virani, S.S. Lipoprotein (a): Recent Updates on a Unique Lipoprotein. Curr. Atheroscler Rep. 2021, 23, 41. [Google Scholar] [CrossRef]
- Coassin, S.; Kronenberg, F. Lipoprotein(a) beyond the kringle IV repeat polymorphism: The complexity of genetic variation in the LPA gene. Atherosclerosis 2022, 349, 17–35. [Google Scholar] [CrossRef]
- Grüneis, R.; Weissensteiner, H.; Lamina, C.; Schönherr, S.; Forer, L.; Di Maio, S.; Streiter, G.; Peters, A.; Gieger, C.; Kronenberg, F.; et al. The kringle IV type 2 domain variant 4925G>A causes the elusive association signal of the LPA pentanucleotide repeat. J. Lipid Res. 2022, 63, 100306. [Google Scholar] [CrossRef]
- Tomova, V.D.; Alexandrova, M.L.; Atanasova, M.A.; Tzekova, M.L.; Rashev, T.R.; Ahmad, S. Plasma lipoprotein(a) concentration as an independent predictor of hemodynamic progression of aortic valve stenosis. Mol. Cell. Biochem. 2020, 472, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.M.; Kennedy, D.J.; Morton, R.E.; Febbraio, M. CD36/SR-B2-TLR2 Dependent Pathways Enhance Porphyromonas gingivalis Mediated Atherosclerosis in the Ldlr KO Mouse Model. PLoS ONE 2015, 10, e0125126. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, H.S.; Wilkinson, M.J. Lipoprotein(a): Evidence for Role as a Causal Risk Factor in Cardiovascular Disease and Emerging Therapies. J. Clin. Med. 2022, 11, 6040. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.B. Inflammatory and atherogenic effects of oxidized phospholipids. Nat. Rev. Cardiol. 2018, 15, 441. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Sá, M.P.B.; Cavalcanti, L.R.P.; Perazzo, Á.M.; Gomes, R.A.F.; Clavel, M.-A.; Pibarot, P.; Biondi-Zoccai, G.; Zhigalov, K.; Weymann, A.; Ruhparwar, A.; et al. Calcific Aortic Valve Stenosis and Atherosclerotic Calcification. Curr. Atheroscler Rep. 2020, 22, 2. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Vasquez, N.; Ayers, C.R.; Patel, J.; Hooda, A.; Khera, A.; Blumenthal, R.S.; Shapiro, M.D.; Rodriguez, C.J.; Tsai, M.Y.; et al. Independent Association of Lipoprotein(a) and Coronary Artery Calcification With Atherosclerotic Cardiovascular Risk. J. Am. Coll. Cardiol. 2022, 79, 757–768. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, S.; Miao, Y.; Song, G.; Yuan, F.; Liu, L.; Xia, S.; Qin, Y.; Huo, X.; Wu, Z.; et al. Effects of blood lipids and lipid-modifying drugs on intracranial aneurysms. Eur. J. Neurol. 2022, 29, 2967–2975. [Google Scholar] [CrossRef]
- Karhunen, V.; Bakker, M.K.; Ruigrok, Y.M.; Gill, D.; Larsson, S.C. Modifiable Risk Factors for Intracranial Aneurysm and Aneurysmal Subarachnoid Hemorrhage: A Mendelian Randomization Study. J. Am. Heart Assoc. 2021, 10, e022277. [Google Scholar] [CrossRef]
- Liu, H.; Mao, P.; Xie, C.; Xie, W.; Wang, M.; Jiang, H. Apolipoprotein E polymorphism and the risk of intracranial aneurysms in a Chinese population. BMC Neurol. 2016, 16, 14. [Google Scholar] [CrossRef]
- Caird, J.; Burke, M.; Roberts, G.; Brett, F.; Phillips, J.; Usher, D.; Bouchier-Hayes, D.; Farrell, M. Apolipoprotein(A) expression in intracranial aneurysms. Neurosurgery 2003, 52, 854–859. [Google Scholar] [CrossRef]
- Phillips, J.; Roberts, G.; Bolger, C.; el Baghdady, A.; Bouchier-Hayes, D.; Farrell, M.; Collins, P. Lipoprotein (a): A potential biological marker for unruptured intracranial aneurysms. Neurosurgery 1997, 40, 1112–1117. [Google Scholar] [CrossRef]
- Bolger, C.; Phillips, J.; Gilligan, S.; Zourob, T.; Farrell, M.; Croake, D.; Collins, P.; Bouchier-Hayes, D. Elevated levels of lipoprotein (a) in association with cerebrovascular saccular aneurysmal disease. Neurosurgery 1995, 37, 241–245. [Google Scholar] [CrossRef]
- Roberts, G.A.; Corcoran, B.T.; Pfouts, L.L.; Phillips, J.P.; Farrell, M.A.; Bouchier-Hayes, D.J.; Collins, P.B. Genetic evaluation of lipoprotein(a) in intracranial aneurysm disease. Neurosurgery 2001, 49, 133–142. [Google Scholar]
- Synowiec, E.; Wojcik, K.A.; Wójcik, R.; Wiśniewski, K.; Fila, M.; Tokarz, P.; Bieńkowski, M.; Jaskolski, D.; Blasiak, J. Expression and variability of lipid metabolism genes in intracranial aneurysm. Cell Mol. Biol. 2016, 62, 73–82. [Google Scholar] [PubMed]
- van ‘t Hof, F.N.G.; Ruigrok, Y.M.; Baas, A.F.; Kiemeney, L.A.L.M.; Vermeulen, S.H.; Uitterlinden, A.G.; Hofman, A.; Rivadeneira, F.; Rinkel, G.J.E.; de Bakker, P.I.W. Impact of inherited genetic variants associated with lipid profile, hypertension, and coronary artery disease on the risk of intracranial and abdominal aortic aneurysms. Circ. Cardiovasc. Genet. 2013, 6, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Takeda, R.; Islam, A.; Sato, T.; Kurita, H.; Kahyo, T.; Urano, T.; Setou, M. The stability of the metabolic turnover of arachidonic acid in human unruptured intracranial aneurysmal walls is sustained. Clin. Neurol. Neurosurg. 2021, 208, 106881. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, E.; Tulamo, R.; Lehti, S.; Lee-Rueckert, M.; Hernesniemi, J.; Niemelä, M.; Ylä-Herttuala, S.; Kovanen, P.T.; Frösen, J. Smooth Muscle Cell Foam Cell Formation, Apolipoproteins, and ABCA1 in Intracranial Aneurysms: Implications for Lipid Accumulation as a Promoter of Aneurysm Wall Rupture. J. Neuropathol. Exp. Neurol. 2016, 75, 689–699. [Google Scholar] [CrossRef]
- Ishii, D.; Matsushige, T.; Sakamoto, S.; Shimonaga, K.; Akiyama, Y.; Okazaki, T.; Oshita, J.; Kurisu, K. Decreased Antiatherogenic Protein Levels are Associated with Aneurysm Structure Alterations in MR Vessel Wall Imaging. J. Stroke Cerebrovasc. Dis. 2019, 28, 2221–2227. [Google Scholar] [CrossRef]
- Quan, K.; Song, J.; Yang, Z.; Wang, D.; An, Q.; Huang, L.; Liu, P.; Li, P.; Tian, Y.; Zhou, L.; et al. Validation of Wall Enhancement as a New Imaging Biomarker of Unruptured Cerebral Aneurysm. Stroke 2019, 50, 1570–1573. [Google Scholar] [CrossRef]
- Ishii, D.; Zanaty, M.; Roa, J.A.; Li, L.; Lu, Y.; Sabotin, R.; Allan, L.; Samaniego, E.A.; Hasan, D.M. Concentration of Lp(a) (Lipoprotein[a]) in Aneurysm Sac Is Associated With Wall Enhancement of Unruptured Intracranial Aneurysm. Stroke 2021, 52, 1465–1468. [Google Scholar] [CrossRef]
- Shimizu, K.; Miyata, H.; Abekura, Y.; Oka, M.; Kushamae, M.; Kawamata, T.; Mizutani, T.; Kataoka, H.; Nozaki, K.; Miyamoto, S.; et al. High-Fat Diet Intake Promotes the Enlargement and Degenerative Changes in the Media of Intracranial Aneurysms in Rats. J. Neuropathol. Exp. Neurol. 2019, 78, 798–807. [Google Scholar] [CrossRef]
- Jin, T.; Wang, L.; Li, D.; Yang, T.; Zhou, Y. Testosterone aggravates cerebral vascular injury by reducing plasma HDL levels. Open Life Sci. 2020, 15, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Seifert, V.; Stolke, D.; Kaever, V.; Dietz, H. Arachidonic acid metabolism following aneurysm rupture. Evaluation of cerebrospinal fluid and serum concentration of 6-keto-prostaglandin F1 alpha and thromboxane B2 in patients with subarachnoid hemorrhage. Surg. Neurol. 1987, 27, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, N.; Matsukado, Y.; Oribe, Y.; Sano, Y.; Ito, Y.; Kuratsu, J.; Seto, H.; Miura, G. Lipids metabolism of the patients with subarachnoid hemorrhage due to ruptured intracranial aneurysm, in comparison with other cerebrovascular diseases. No Shinkei 1984, 36, 389–395. [Google Scholar]
- Seifert, V.; Stolke, D.; Kaever, V.; Dietz, H. Arachidonic acid metabolism following aneurysm rupture. Eur. Arch. Psychiatry Neurol. Sci. 1986, 236, 94–101. [Google Scholar] [CrossRef]
- Gaetani, P.; Marzatico, F.; Rodriguez y Baena, R. Ex vivo release of eicosanoids after aneurysmal subarachnoid hemorrhage: A preliminary experience in humans. Acta Neurol. Scand. 1992, 86, 184–189. [Google Scholar] [CrossRef]
- Jarocka-Karpowicz, I.; Syta-Krzyżanowska, A.; Kochanowicz, J.; Mariak, Z.D. Clinical Prognosis for SAH Consistent with Redox Imbalance and Lipid Peroxidation. Molecules 2020, 25, 1921. [Google Scholar] [CrossRef]
- Paoletti, P.; Gaetani, P.; Grignani, G.; Pacchiarini, L.; Silvani, V.; Rodriguez y Baena, R. CSF leukotriene C4 following subarachnoid hemorrhage. J. Neurosurg. 1988, 69, 488–493. [Google Scholar] [CrossRef]
- Asaeda, M.; Sakamoto, M.; Kurosaki, M.; Tabuchi, S.; Kamitani, H.; Yokota, M.; Watanabe, T. A non-enzymatic derived arachidonyl peroxide, 8-iso-prostaglandin F2 alpha, in cerebrospinal fluid of patients with aneurysmal subarachnoid hemorrhage participates in the pathogenesis of delayed cerebral vasospasm. Neurosci. Lett. 2005, 373, 222–225. [Google Scholar] [CrossRef]
- Nam, D.H.; Kim, J.S.; Hong, S.C.; Lee, W.H.; Lee, J.I.; Shin, H.J.; Park, K.; Eoh, W.; Han, D.H.; Kim, J.H. Expression of interleukin-1 beta in lipopolysaccharide stimulated monocytes derived from patients with aneurysmal subarachnoid hemorrhage is correlated with cerebral vasospasm. Neurosci. Lett. 2001, 312, 41–44. [Google Scholar] [CrossRef]
- Kamezaki, T.; Yanaka, K.; Nagase, S.; Fujita, K.; Kato, N.; Nose, T. Increased levels of lipid peroxides as predictive of symptomatic vasospasm and poor outcome after aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2002, 97, 1302–1305. [Google Scholar] [CrossRef]
- Sano, K.; Asano, T.; Tanishima, T.; Sasaki, T. Lipid peroxidation as a cause of cerebral vasospasm. Neurol. Res. 1980, 2, 253–272. [Google Scholar] [CrossRef]
- Promyslov, M.S.; Levchenko, L.I.; Demchuk, M.L.; Miakota, A.E. Lipid peroxidation of the cerebrospinal fluid in patients with arterial cerebral aneurysms. Zhurnal Vopr. Neirokhirurgii Im. NN Burdenko 1991, 3, 27–29. [Google Scholar]
- Rodriguez y Baena, R.; Gaetani, P.; Folco, G.; Viganó, T.; Paoletti, P. Arachidonate metabolites and vasospasm after subarachnoid haemorrhage. Neurol. Res. 1986, 8, 25–32. [Google Scholar] [CrossRef]
- Guo, Z.-D.; Sun, X.-C.; Zhang, J.H. The role of apolipoprotein e in the pathological events following subarachnoid hemorrhage: A review. Acta Neurochirurgica. Suppl. 2011, 110 Pt 2, 5–7. [Google Scholar] [CrossRef]
- Tang, J.; Zhao, J.; Zhao, Y.; Wang, S.; Chen, B.; Zeng, W. Apolipoprotein E epsilon4 and the risk of unfavorable outcome after aneurysmal subarachnoid hemorrhage. Surg. Neurol. 2003, 60, 391–396. [Google Scholar] [CrossRef]
- Leung, C.H.S.; Poon, W.S.; Yu, L.M.; Wong, G.K.C.; Ng, H.K. Apolipoprotein e genotype and outcome in aneurysmal subarachnoid hemorrhage. Stroke 2002, 33, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Juvela, S.; Siironen, J.; Lappalainen, J. Apolipoprotein E genotype and outcome after aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2009, 110, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.T.; Stewart, E.; Murray, G.D.; Nicoll, J.A.; Teasdale, G.M. The influence of apolipoprotein E genotype on outcome after spontaneous subarachnoid hemorrhage: A preliminary study. Neurosurgery 2001, 48, 1006–1011. [Google Scholar] [PubMed]
- Huang, Q.; Shang-Guan, H.-C.; Wu, S.-Y.; Yao, P.-S.; Sun, Y.; Zeng, Y.-L.; Zheng, S.-F.; Chen, G.-R.; Lin, Y.-X.; Kang, D.-Z. High-Density Lipoprotein Is Associated with Progression of Intracranial Aneurysms. World Neurosurg. 2018, 120, e234–e240. [Google Scholar] [CrossRef]
- Syta-Krzyżanowska, A.; Jarocka-Karpowicz, I.; Kochanowicz, J.; Turek, G.; Rutkowski, R.; Gorbacz, K.; Mariak, Z.; Skrzydlewska, E. F2-isoprostanes and F4-neuroprostanes as markers of intracranial aneurysm development. Adv. Clin. Exp. Med. 2018, 27, 673–680. [Google Scholar] [CrossRef]
- Wiśniewski, K.; Jóźwik-Pruska, J.; Bieńkowski, M.; Bobeff, E.J.; Bryl, M.; Kałużna-Czaplińska, J.; Jaskólski, D.J. Isoprostanes as potential cerebral vasospasm biomarkers. Neurol. I Neurochir. Pol. 2018, 52, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; McIntyre, M.; Gandhi, C.; Halabi, M.; Long, A.; Van Hoof, A.; Afridi, A.; Schmidt, M.; Cole, C.; Santarelli, J.; et al. Low total cholesterol and high density lipoprotein are independent predictors of poor outcomes following aneurysmal subarachnoid hemorrhage: A preliminary report. Clin. Neurol. Neurosurg. 2020, 197, 106062. [Google Scholar] [CrossRef] [PubMed]
- Winking, M.; Müller, H.W.; Deinsberger, W.; Joedicke, A.; Boeker, D.K. Levels of immunoreactive cysteinyl-leukotrienes in CSF after subarachnoid haemorrhage correlate with blood flow-velocity in TCD. Acta Neurochir. 1997, 139, 764–769. [Google Scholar] [CrossRef]
- Aoki, T.; Kataoka, H.; Ishibashi, R.; Nozaki, K.; Hashimoto, N. Simvastatin suppresses the progression of experimentally induced cerebral aneurysms in rats. Stroke 2008, 39, 1276–1285. [Google Scholar] [CrossRef]
- Liu, P.; An, Q.; Chen, X.; Huang, J.; Yang, G.-Y.; Zhu, W. Rosuvastatin for enhancement of aneurysm neck endothelialization after coil embolization: Promotion of endothelial progenitor cells in a rodent model. J. Neurosurg. 2016, 124, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Yang, M.; Yu, K.; Dong, W.; Liang, W.; Wang, Z.; Jiang, R.; Zhang, J. Atorvastatin Protects Against Cerebral Aneurysmal Degenerative Pathology by Promoting Endothelial Progenitor Cells (EPC) Mobilization and Attenuating Vascular Deterioration in a Rat Model. Med. Sci. Monit. 2019, 25, 928–936. [Google Scholar] [CrossRef]
- Tseng, M.-Y.; Czosnyka, M.; Richards, H.; Pickard, J.D.; Kirkpatrick, P.J. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: A phase II randomized placebo-controlled trial. Stroke 2005, 36, 1627–1632. [Google Scholar] [CrossRef]
- Chen, J.; Li, M.; Zhu, X.; Chen, L.; Yang, S.; Zhang, C.; Wu, T.; Feng, X.; Wang, Y.; Chen, Q. Atorvastatin reduces cerebral vasospasm and infarction after aneurysmal subarachnoid hemorrhage in elderly Chinese adults. Aging 2020, 12, 2939–2951. [Google Scholar] [CrossRef]
- Akhigbe, T.; Zolnourian, A.; Bulters, D. Cholesterol-Reducing Agents for Treatment of Aneurysmal Subarachnoid Hemorrhage: Systematic Review and Meta-Analysis of Randomized Controlled Trials. World Neurosurg. 2017, 101, 476–485. [Google Scholar] [CrossRef]
- Tseng, M.-Y.; Hutchinson, P.J.; Turner, C.L.; Czosnyka, M.; Richards, H.; Pickard, J.D.; Kirkpatrick, P.J. Biological effects of acute pravastatin treatment in patients after aneurysmal subarachnoid hemorrhage: A double-blind, placebo-controlled trial. J. Neurosurg. 2007, 107, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.H.; Gurka, M.J.; Nathan, B.; Dumont, A.S.; Kassell, N.F.; Bleck, T.P. Statin use was not associated with less vasospasm or improved outcome after subarachnoid hemorrhage. Neurosurgery 2008, 62, 422–430. [Google Scholar] [CrossRef]
- Vergouwen, M.D.I.; Meijers, J.C.M.; Geskus, R.B.; Coert, B.A.; Horn, J.; Stroes, E.S.G.; van der Poll, T.; Vermeulen, M.; Roos, Y.B.W.E.M. Biologic effects of simvastatin in patients with aneurysmal subarachnoid hemorrhage: A double-blind, placebo-controlled randomized trial. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2009, 29, 1444–1453. [Google Scholar] [CrossRef]
- McGirt, M.J.; Garces Ambrossi, G.L.; Huang, J.; Tamargo, R.J. Simvastatin for the prevention of symptomatic cerebral vasospasm following aneurysmal subarachnoid hemorrhage: A single-institution prospective cohort study. J. Neurosurg. 2009, 110, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Peña, P.; Nouet, A.; Clarençon, F.; Colonne, C.; Jean, B.; Le Jean, L.; Fonfrede, M.; Aout, M.; Vicaut, E.; Puybasset, L. Atorvastatin decreases computed tomography and S100-assessed brain ischemia after subarachnoid aneurysmal hemorrhage: A comparative study. Crit. Care Med. 2012, 40, 594–602. [Google Scholar] [CrossRef]
- Naraoka, M.; Matsuda, N.; Shimamura, N.; Asano, K.; Akasaka, K.; Takemura, A.; Hasegawa, S.; Ohkuma, H. Long-acting statin for aneurysmal subarachnoid hemorrhage: A randomized, double-blind, placebo-controlled trial. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2018, 38, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, Y.; Tian, Z.; Zhu, W.; Liu, J.; Zhang, Y.; Yang, X.; Tian, D.-C. Statin treatment for unruptured intracranial aneurysms study: A study protocol for a double-blind, placebo-controlled trial. Stroke Vasc. Neurol. 2020, 5, 410–415. [Google Scholar] [CrossRef]
- Wang, J.; Weng, J.; Li, H.; Jiao, Y.; Fu, W.; Huo, R.; Yan, Z.; Xu, H.; Zhan, J.; Wang, S.; et al. Atorvastatin and growth, rupture of small unruptured intracranial aneurysm: Results of a prospective cohort study. Ther. Adv. Neurol. Disord. 2021, 14, 1756286420987939. [Google Scholar] [CrossRef]
- Turhon, M.; Kang, H.; Huang, J.; Li, M.; Liu, J.; Zhang, Y.; Wang, K.; Yang, X.; Zhang, Y. Atorvastatin for unruptured intracranial vertebrobasilar dissecting aneurysm (ATREAT-VBD): Protocol for a randomised, double-blind, blank-controlled trial. BMJ Open 2022, 12, e059616. [Google Scholar] [CrossRef]
- Kang, H.; Tian, D.-C.; Yang, X.; Zhang, Y.; Li, W.; Sui, B.; Duan, Y.; Zhang, Y.; Liu, J.; Wang, K.; et al. A Randomized Controlled Trial of Statins to Reduce Inflammation in Unruptured Cerebral Aneurysms. JACC. Cardiovasc. Imaging 2022, 15, 1668–1670. [Google Scholar] [CrossRef]
- Yoneda, H.; Shirao, S.; Kurokawa, T.; Fujisawa, H.; Kato, S.; Suzuki, M. Does eicosapentaenoic acid (EPA) inhibit cerebral vasospasm in patients after aneurysmal subarachnoid hemorrhage? Acta Neurol. Scand. 2008, 118, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, H.; Shirao, S.; Nakagawara, J.; Ogasawara, K.; Tominaga, T.; Suzuki, M. A prospective, multicenter, randomized study of the efficacy of eicosapentaenoic acid for cerebral vasospasm: The EVAS study. World Neurosurg. 2014, 81, 309–315. [Google Scholar] [CrossRef]
- Nakagawa, I.; Yokoyama, S.; Omoto, K.; Takeshima, Y.; Matsuda, R.; Nishimura, F.; Yamada, S.; Yokota, H.; Motoyama, Y.; Park, Y.-S.; et al. ω-3 Fatty Acids Ethyl Esters Suppress Cerebral Vasospasm and Improve Clinical Outcome Following Aneurysmal Subarachnoid Hemorrhage. World Neurosurg. 2017, 99, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Abekura, Y.; Ono, I.; Kawashima, A.; Takizawa, K.; Koseki, H.; Miyata, H.; Shimizu, K.; Oka, M.; Kushamae, M.; Miyamoto, S.; et al. Eicosapentaenoic acid prevents the progression of intracranial aneurysms in rats. J. Neuroinflammation 2020, 17, 129. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| References | Sample Size (E/C) | Diagnosis Standard | Experimental Group | Control Group | Treatment Duration | Outcome Measures |
|---|---|---|---|---|---|---|
| Tseng MY et al., 2005 [98] | 40/40 aSAH | CT | Pravastatin 40 mg/d | Placebo | 14 d | Postoperative complications (cerebral vasospasm, cerebral autoregulation, vasospasm-related DID) |
| Tseng MY et al., 2007 [101] | 38/42 aSAH | CT/DSA | Pravastatin 40 mg/d | Placebo | 14 d | Laboratory parameters, postoperative complications (DIND), clinical symptom scores (6-month mRS) |
| Kramer AH et al., 2008 [102] | 71/79 aSAH | CT/CTA/DSA | Simvastatin 80 mg/d | Nonstatin | 14 d | Postoperative complications (cerebral vasospasm, delayed infarction), clinical symptom scores (GOS of 1 to 3) |
| Vergouwen MD et al., 2009 [103] | 16/16 aSAH | CT/CTA | Simvastatin 80 mg/d | Placebo | 15 d | Laboratory parameters, postoperative complications, and clinical symptom scores (3- and 6-month GOS after SAH) |
| McGirt MJ et al., 2009 [104] | 170/170 aSAH | CT/CTA/DSA | Simvastatin 80 mg/d | Nonstatin | 14 d | Postoperative complications (cerebral vasospasm) and clinical symptom scores (perioperative death, length of hospital stay, discharged GOS) |
| Sanchez-Peña P et al., 2012 [105] | 142/136 aSAH | CT/DSA | Atorvastatin 40 mg/d | Nonstatin | 21 d | Clinical symptom scores (discharged and 1-yr GOS and modified Rankin Scale) |
| Naraoka M et al., 2018 [106] | 54/54 aSAH | CT | Pitavastatin 4 mg/d | Placebo | 14 d | Postoperative complications (DIND, cerebral vasospasm, vasospasm-related new cerebral infarctions) |
| Chen J et al., 2020 [99] | 150/150 aSAH | CT/CTA/DSA | Atorvastatin 20 mg/d | Placebo | 14 d | Postoperative complications (CVS, infarction, DIND) and clinical symptom scores (6-month GOS after SAH, 30-day mortality) |
| Li W et al., 2020 [107] | 30/30 UIA | CTA/MRA/DSA/HR-MRI | Atorvastatin 20 mg/d | Placebo | 12 M | AWE, aneurysm morphology, inflammatory factors (CRP, TNF-α, IL-1β, and IL-6) |
| Wang J et al., 2021 [108] | 489/598 UIA | CTA/MRA/DSA | Atorvastatin 20 mg/d | Nonstatin | 3 yrs | Aneurysm rupture (confirmed with CT or MRI) |
| Turhon M et al., 2022 [109] | 20/20 UIA | CTA/MRA/DSA/HR-MRI | Atorvastatin 20 mg/d | Nonstatin | 6 M | AWE, aneurysm morphology, inflammatory factors (CRP, TNF-α, IL-1β, and IL-6) |
| Kang H et al., 2022 [110] | 30/30 UIA | VW-MRI | Atorvastatin 20 mg/d | Placebo | 6 M | WEI, 3D-WEVR, aneurysm morphology, inflammatory factors |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, T.; Shi, Y.; Yu, G.; Mamtimin, A.; Zhu, W. Intracranial Aneurysms and Lipid Metabolism Disorders: From Molecular Mechanisms to Clinical Implications. Biomolecules 2023, 13, 1652. https://doi.org/10.3390/biom13111652
Pan T, Shi Y, Yu G, Mamtimin A, Zhu W. Intracranial Aneurysms and Lipid Metabolism Disorders: From Molecular Mechanisms to Clinical Implications. Biomolecules. 2023; 13(11):1652. https://doi.org/10.3390/biom13111652
Chicago/Turabian StylePan, Tonglin, Yuan Shi, Guo Yu, Abdureshid Mamtimin, and Wei Zhu. 2023. "Intracranial Aneurysms and Lipid Metabolism Disorders: From Molecular Mechanisms to Clinical Implications" Biomolecules 13, no. 11: 1652. https://doi.org/10.3390/biom13111652