
Association between Impaired Ketogenesis and Metabolic-Associated Fatty Liver Disease


Abstract
:1. Introduction
2. Lipid Metabolism in the Pathogenesis of MAFLD and the Ketogenic Pathway
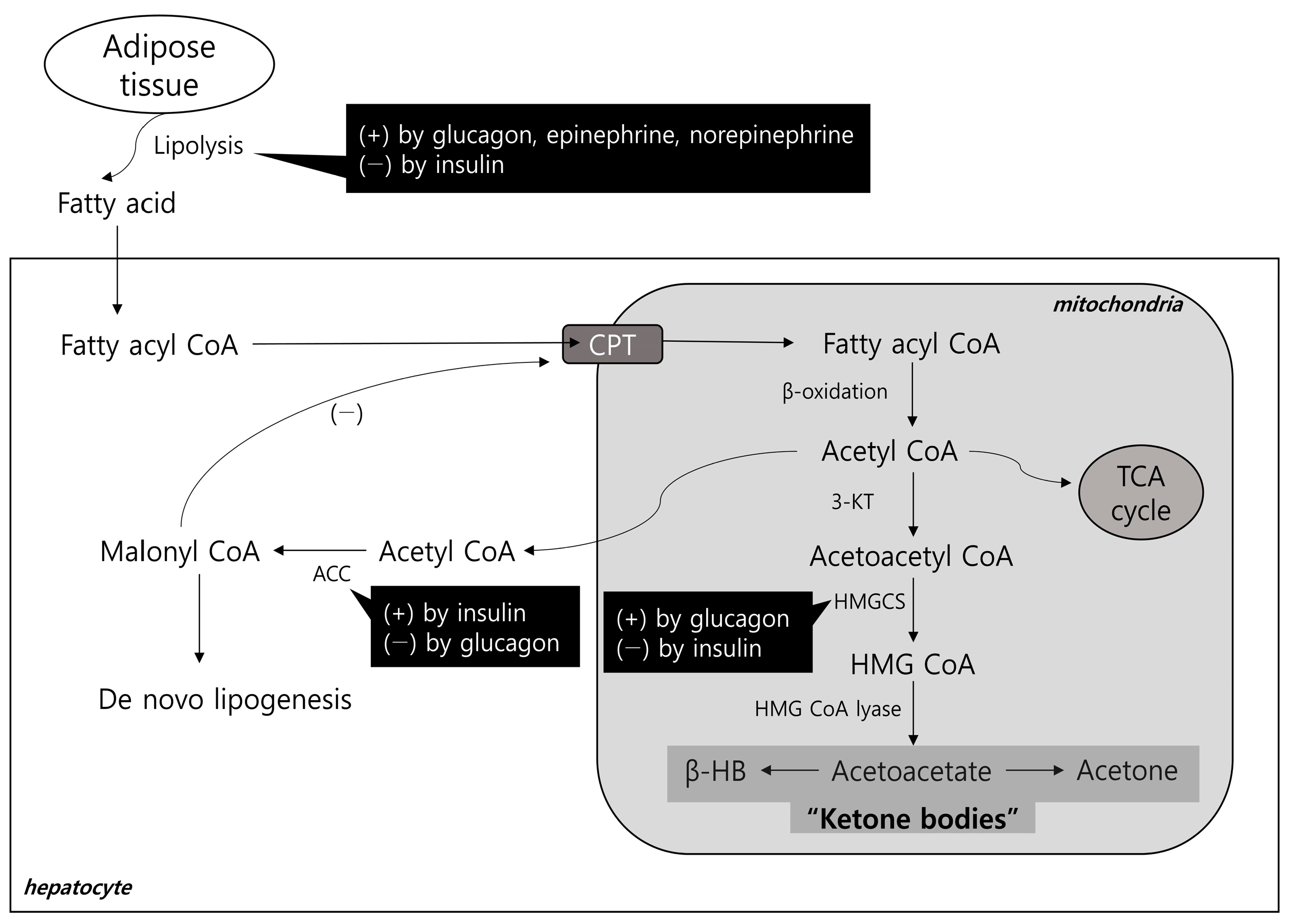
2.1. Hepatic Lipid Input
2.2. Hepatic Lipid Output and Ketogenesis
3. Ketogenesis and MAFLD
3.1. Previous Studies on the Association between Ketogenesis and MAFLD
3.2. How Ketogenesis Affects MAFLD
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef]
- Astrup, A.; Meinert Larsen, T.; Harper, A. Atkins and other low-carbohydrate diets: Hoax or an effective tool for weight loss? Lancet 2004, 364, 897–899. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; De Nigris, V.; Micheloni, S.; La Sala, L.; Ceriello, A. Increases in circulating levels of ketone bodies and cardiovascular protection with sglt2 inhibitors: Is low-grade inflammation the neglected component? Diabetes Obes. Metab. 2018, 20, 2515–2522. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. Cv protection in the empa-reg outcome trial: A “thrifty substrate” hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E. Sodium-glucose co-transporters and their inhibition: Clinical physiology. Cell Metab. 2017, 26, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Joo, N.S.; Lee, D.J.; Kim, K.M.; Kim, B.T.; Kim, C.W.; Kim, K.N.; Kim, S.M. Ketonuria after fasting may be related to the metabolic superiority. J. Korean Med. Sci. 2010, 25, 1771–1776. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Lee, S.G.; Lee, B.W.; Kang, E.S.; Cha, B.S.; Ferrannini, E.; Lee, Y.H.; Cho, N.H. Spontaneous ketonuria and risk of incident diabetes: A 12 year prospective study. Diabetologia 2019, 62, 779–788. [Google Scholar] [CrossRef]
- Lee, S.; Bae, J.; Jo, D.R.; Lee, M.; Lee, Y.H.; Kang, E.S.; Cha, B.S.; Lee, B.W. Impaired ketogenesis is associated with metabolic-associated fatty liver disease in subjects with type 2 diabetes. Front. Endocrinol. 2023, 14, 1124576. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the american association for the study of liver diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (nafld) and nonalcoholic steatohepatitis (nash): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. Mafld: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Bhagat, N.; Mehta, M.; Taneja, S.; Duseja, A. Metabolic dysfunction-associated steatotic liver disease (masld) definition is better than mafld criteria for lean patients with non-alcoholic fatty liver disease (nafld). J. Hepatol. 2023; in press. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. Nafld: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Han, S.K.; Baik, S.K.; Kim, M.Y. Non-alcoholic fatty liver disease: Definition and subtypes. Clin. Mol. Hepatol. 2023, 29, S5–S16. [Google Scholar] [CrossRef]
- Ter Horst, K.W.; Serlie, M.J. Fructose consumption, lipogenesis, and non-alcoholic fatty liver disease. Nutrients 2017, 9, 981. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of nafld development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef]
- Byrne, C.D. Dorothy hodgkin lecture 2012: Non-alcoholic fatty liver disease, insulin resistance and ectopic fat: A new problem in diabetes management. Diabet. Med. A J. Br. Diabet. Assoc. 2012, 29, 1098–1107. [Google Scholar] [CrossRef]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Liu, W.; Tao, M.H.; Patel, R.; Nowak, N.J.; Baker, R.D. Lipid in the livers of adolescents with nonalcoholic steatohepatitis: Combined effects of pathways on steatosis. Metab. Clin. Exp. 2011, 60, 1001–1011. [Google Scholar] [CrossRef]
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Sirén, J.; Hamsten, A.; Fisher, R.M.; Yki-Järvinen, H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007, 56, 2759–2765. [Google Scholar] [CrossRef]
- Greco, D.; Kotronen, A.; Westerbacka, J.; Puig, O.; Arkkila, P.; Kiviluoto, T.; Laitinen, S.; Kolak, M.; Fisher, R.M.; Hamsten, A.; et al. Gene expression in human nafld. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1281–G1287. [Google Scholar] [CrossRef]
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sánchez-Campos, S.; García-Mediavilla, M.V.; Fernández-Bermejo, M.; Lozano-Rodríguez, T.; Vargas-Castrillón, J.; Buqué, X.; Ochoa, B.; Aspichueta, P.; et al. Hepatic fatty acid translocase cd36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis c. Gut 2011, 60, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Sanders, F.W.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef]
- Geidl-Flueck, B.; Gerber, P.A. Insights into the hexose liver metabolism-glucose versus fructose. Nutrients 2017, 9, 1026. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. CMLS 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. Srebp transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Jensen-Urstad, A.P.; Semenkovich, C.F. Fatty acid synthase and liver triglyceride metabolism: Housekeeper or messenger? Biochim. Et Biophys. Acta 2012, 1821, 747–753. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef]
- Horton, J.D.; Shimano, H.; Hamilton, R.L.; Brown, M.S.; Goldstein, J.L. Disruption of ldl receptor gene in transgenic srebp-1a mice unmasks hyperlipidemia resulting from production of lipid-rich vldl. J. Clin. Investig. 1999, 103, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes/Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Mooli, R.G.R.; Ramakrishnan, S.K. Emerging role of hepatic ketogenesis in fatty liver disease. Front. Physiol. 2022, 13, 946474. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.A.; Kassovska-Bratinova, S.; Boukaftane, Y.; Robert, M.F.; Wang, S.P.; Ashmarina, L.; Lambert, M.; Lapierre, P.; Potier, E. Medical aspects of ketone body metabolism. Clin. Investig. Med. Med. Clin. Et Exp. 1995, 18, 193–216. [Google Scholar]
- Abdul Kadir, A.; Clarke, K.; Evans, R.D. Cardiac ketone body metabolism. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165739. [Google Scholar] [CrossRef]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Satapati, S.; Fu, X.; He, T.; Mehdibeigi, R.; Spring-Robinson, C.; Duarte, J.; Potthoff, M.J.; Browning, J.D.; Burgess, S.C. Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1226–E1235. [Google Scholar] [CrossRef] [PubMed]
- Fougerat, A.; Schoiswohl, G.; Polizzi, A.; Régnier, M.; Wagner, C.; Smati, S.; Fougeray, T.; Lippi, Y.; Lasserre, F.; Raho, I.; et al. Atgl-dependent white adipose tissue lipolysis controls hepatocyte pparα activity. Cell Rep. 2022, 39, 110910. [Google Scholar] [CrossRef] [PubMed]
- Koeslag, J.H.; Noakes, T.D.; Sloan, A.W. Post-exercise ketosis. J. Physiol. 1980, 301, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef] [PubMed]
- Keller, U.; Lustenberger, M.; Müller-Brand, J.; Gerber, P.P.; Stauffacher, W. Human ketone body production and utilization studied using tracer techniques: Regulation by free fatty acids, insulin, catecholamines, and thyroid hormones. Diabetes/Metab. Rev. 1989, 5, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Liljenquist, J.E.; Bomboy, J.D.; Lewis, S.B.; Sinclair-Smith, B.C.; Felts, P.W.; Lacy, W.W.; Crofford, O.B.; Liddle, G.W. Effects of glucagon on lipolysis and ketogenesis in normal and diabetic men. J. Clin. Investig. 1974, 53, 190–197. [Google Scholar] [CrossRef]
- Alberti, K.G.; Johnston, D.G.; Gill, A.; Barnes, A.J.; Orskov, H. Hormonal regulation of ketone-body metabolism in man. Biochem. Soc. Symp. 1978, 43, 163–182. [Google Scholar]
- Chakrabarti, P.; Kim, J.Y.; Singh, M.; Shin, Y.K.; Kim, J.; Kumbrink, J.; Wu, Y.; Lee, M.J.; Kirsch, K.H.; Fried, S.K.; et al. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mtorc1-egr1-atgl-mediated pathway. Mol. Cell. Biol. 2013, 33, 3659–3666. [Google Scholar] [CrossRef]
- McGarry, J.D.; Woeltje, K.F.; Kuwajima, M.; Foster, D.W. Regulation of ketogenesis and the renaissance of carnitine palmitoyltransferase. Diabetes/Metab. Rev. 1989, 5, 271–284. [Google Scholar] [CrossRef]
- Inokuchi, T.; Orita, M.; Imamura, K.; Takao, T.; Isogai, S. Resistance to ketosis in moderately obese patients: Influence of fatty liver. Intern. Med. 1992, 31, 978–983. [Google Scholar] [CrossRef]
- Croci, I.; Byrne, N.M.; Choquette, S.; Hills, A.P.; Chachay, V.S.; Clouston, A.D.; O’Moore-Sullivan, T.M.; Macdonald, G.A.; Prins, J.B.; Hickman, I.J. Whole-body substrate metabolism is associated with disease severity in patients with non-alcoholic fatty liver disease. Gut 2013, 62, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Mey, J.T.; Erickson, M.L.; Axelrod, C.L.; King, W.T.; Flask, C.A.; McCullough, A.J.; Kirwan, J.P. Β-hydroxybutyrate is reduced in humans with obesity-related nafld and displays a dose-dependent effect on skeletal muscle mitochondrial respiration in vitro. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E187–E195. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-coa oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 5, e127737. [Google Scholar] [CrossRef]
- Thoma, C.; Day, C.P.; Trenell, M.I. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: A systematic review. J. Hepatol. 2012, 56, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.; Reeds, D.N.; Finck, B.N.; Mayurranjan, S.M.; Patterson, B.W.; Klein, S. Dietary fat and carbohydrates differentially alter insulin sensitivity during caloric restriction. Gastroenterology 2009, 136, 1552–1560. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.M.; Hakkarainen, A.; Lehtimäki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Järvinen, H. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef]
- Chalasani, N.; Gorski, J.C.; Asghar, M.S.; Asghar, A.; Foresman, B.; Hall, S.D.; Crabb, D.W. Hepatic cytochrome p450 2e1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology 2003, 37, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Seppälä-Lindroos, A.; Vehkavaara, S.; Bergholm, R.; Frayn, K.N.; Fielding, B.A.; Yki-Järvinen, H. Liver fat and lipid oxidation in humans. Liver Int. Off. J. Int. Assoc. Study Liver 2009, 29, 1439–1446. [Google Scholar] [CrossRef]
- Post, A.; Garcia, E.; van den Berg, E.H.; Flores-Guerrero, J.L.; Gruppen, E.G.; Groothof, D.; Westenbrink, B.D.; Connelly, M.A.; Bakker, S.J.L.; Dullaart, R.P.F. Nonalcoholic fatty liver disease, circulating ketone bodies and all-cause mortality in a general population-based cohort. Eur. J. Clin. Investig. 2021, 51, e13627. [Google Scholar] [CrossRef]
- Kim, Y.; Chang, Y.; Kwon, M.J.; Hong, Y.S.; Kim, M.K.; Sohn, W.; Cho, Y.K.; Shin, H.; Wild, S.H.; Byrne, C.D.; et al. Fasting ketonuria and the risk of incident nonalcoholic fatty liver disease with and without liver fibrosis in nondiabetic adults. Am. J. Gastroenterol. 2021, 116, 2270–2278. [Google Scholar] [CrossRef]
- Lim, K.; Kang, M.; Park, J. Association between fasting ketonuria and advanced liver fibrosis in non-alcoholic fatty liver disease patients without prediabetes and diabetes mellitus. Nutrients 2021, 13, 3400. [Google Scholar] [CrossRef] [PubMed]
- Männistö, V.T.; Simonen, M.; Hyysalo, J.; Soininen, P.; Kangas, A.J.; Kaminska, D.; Matte, A.K.; Venesmaa, S.; Käkelä, P.; Kärjä, V.; et al. Ketone body production is differentially altered in steatosis and non-alcoholic steatohepatitis in obese humans. Liver Int. Off. J. Int. Assoc. Study Liver 2015, 35, 1853–1861. [Google Scholar] [CrossRef]
- McGarry, J.D.; Foster, D.W. Regulation of ketogenesis and clinical aspects of the ketotic state. Metab. Clin. Exp. 1972, 21, 471–489. [Google Scholar] [CrossRef] [PubMed]
- Balasse, E.O. Kinetics of ketone body metabolism in fasting humans. Metab. Clin. Exp. 1979, 28, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; d’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef] [PubMed]
- d’Avignon, D.A.; Puchalska, P.; Ercal, B.; Chang, Y.; Martin, S.E.; Graham, M.J.; Patti, G.J.; Han, X.; Crawford, P.A. Hepatic ketogenic insufficiency reprograms hepatic glycogen metabolism and the lipidome. JCI Insight 2018, 3, e99762. [Google Scholar] [CrossRef] [PubMed]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: Implications for prevention and therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by pparalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, M.; Kim, S.H.; Kim, S.R.; Lee, B.W.; Kang, E.S.; Cha, B.S.; Cho, J.W.; Lee, Y.H. Sodium-glucose cotransporter 2 inhibitors regulate ketone body metabolism via inter-organ crosstalk. Diabetes Obes. Metab. 2019, 21, 801–811. [Google Scholar] [CrossRef]
- Watanabe, M.; Singhal, G.; Fisher, F.M.; Beck, T.C.; Morgan, D.A.; Socciarelli, F.; Mather, M.L.; Risi, R.; Bourke, J.; Rahmouni, K.; et al. Liver-derived fgf21 is essential for full adaptation to ketogenic diet but does not regulate glucose homeostasis. Endocrine 2020, 67, 95–108. [Google Scholar] [CrossRef]
- Itoh, N. Fgf21 as a hepatokine, adipokine, and myokine in metabolism and diseases. Front. Endocrinol. 2014, 5, 107. [Google Scholar] [CrossRef]
- Itoh, N.; Nakayama, Y.; Konishi, M. Roles of fgfs as paracrine or endocrine signals in liver development, health, and disease. Front. Cell Dev. Biol. 2016, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for nafld/nash. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef]
- Berglund, E.D.; Li, C.Y.; Bina, H.A.; Lynes, S.E.; Michael, M.D.; Shanafelt, A.B.; Kharitonenkov, A.; Wasserman, D.H. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology 2009, 150, 4084–4093. [Google Scholar] [CrossRef]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The effects of ly2405319, an fgf21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. TEM 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Taggart, A.K.; Kero, J.; Gan, X.; Cai, T.Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.J.; et al. (d)-beta-hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor puma-g. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Liu, X.; Chen, X.; Zhang, S.; Chen, Y.; Chen, J.; Chen, J.; Wu, F.; Chen, G.Q. 3-hydroxybutyrate ameliorates insulin resistance by inhibiting pparγ ser273 phosphorylation in type 2 diabetic mice. Signal Transduct. Target. Ther. 2023, 8, 190. [Google Scholar] [CrossRef]
- Bates, M.W.; Linn, L.C. Blood d-3-hydroxybutyrate and the regulation of plasma concentrations of free fatty acids in the fasted rat. Metab. Clin. Exp. 1976, 25, 685–695. [Google Scholar] [CrossRef]
- Senior, B.; Loridan, L. Direct regulatory effect of ketones on lipolysis and on glucose concentrations in man. Nature 1968, 219, 83–84. [Google Scholar] [CrossRef]
- Van Hove, J.L.; Grünewald, S.; Jaeken, J.; Demaerel, P.; Declercq, P.E.; Bourdoux, P.; Niezen-Koning, K.; Deanfeld, J.E.; Leonard, J.V. D,l-3-hydroxybutyrate treatment of multiple acyl-coa dehydrogenase deficiency (madd). Lancet 2003, 361, 1433–1435. [Google Scholar] [CrossRef]
- Soto-Mota, A.; Norwitz, N.G.; Evans, R.; Clarke, K.; Barber, T.M. Exogenous ketosis in patients with type 2 diabetes: Safety, tolerability and effect on glycaemic control. Endocrinol. Diabetes Metab. 2021, 4, e00264. [Google Scholar] [CrossRef]
- Lee, A.K.; Kim, D.H.; Bang, E.; Choi, Y.J.; Chung, H.Y. Β-hydroxybutyrate suppresses lipid accumulation in aged liver through gpr109a-mediated signaling. Aging Dis. 2020, 11, 777–790. [Google Scholar] [CrossRef]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via g protein-coupled receptor 41 (gpr41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef]
- Xu, B.T.; Teng, F.Y.; Wu, Q.; Wan, S.R.; Li, X.Y.; Tan, X.Z.; Xu, Y.; Jiang, Z.Z. Bdh1 overexpression ameliorates hepatic injury by activation of nrf2 in a mafld mouse model. Cell Death Discov. 2022, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks nlrp3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. Β-hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via ampk activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef] [PubMed]
- Carnagarin, R.; Tan, K.; Adams, L.; Matthews, V.B.; Kiuchi, M.G.; Marisol Lugo Gavidia, L.; Lambert, G.W.; Lambert, E.A.; Herat, L.Y.; Schlaich, M.P. Metabolic dysfunction-associated fatty liver disease (mafld)-a condition associated with heightened sympathetic activation. Int. J. Mol. Sci. 2021, 22, 4241. [Google Scholar] [CrossRef] [PubMed]
- Torre, P.; Motta, B.M.; Sciorio, R.; Masarone, M.; Persico, M. Inflammation and fibrogenesis in mafld: Role of the hepatic immune system. Front. Med. 2021, 8, 781567. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, W.H.; Wu, J.B.; Xiao, W.H. Β-hydroxybutyrate: A crucial therapeutic target for diverse liver diseases. Biomed. Pharmacother. = Biomed. Pharmacother. 2023, 165, 115191. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Reference | Subjects | Independent Variable | Dependent Variable | Main Finding |
|---|---|---|---|---|
| Inokuchi et al. [50] | 20 NGT patients with obesity | NAFLD by computed tomography | Fasting plasma total ketone, βHB | Low fasting total ketone, βHB in NAFLD |
| Croci et al. [51] | 15 lean healthy, 20 NAFLDsubjects with overweight or obesity | NAFLD by liver biopsy | Fasting plasma βHB | Low fasting plasma βHB in NAFLD |
| Männistö et al. [64] | 76 patients with obesity | Steatosis by 1H-MRS, NASH by liver biopsy | Fasting plasma βHB, acetoacetate | Increased ketone bodies in simple steatosis but decreased in NASH |
| Fletcher et al. [53] | 40 non-diabetic patients | NAFLD by 1H-MRS | Fasting plasma ketone, ketogenic pathway measured by 5 stable isotope tracers | Low fasting ketone and ketogenesis in NAFLD |
| Mey et al. [52] | 22 patients with obesity | NAFLD by 1H-MRS | Fasting plasma βHB | Low fasting plasma βHB in NAFLD |
| Kim et al. [62] | 153,076 nondiabetic subjects | Fasting ketonuria | Steatosis by ultrasound Fibrosis by NFS and FIB-4 | Low risk of NAFLD in subjects with ketonuria |
| Lim et al. [63] | 6022 nondiabetic NAFLD patients | Fasting ketonuria | Fibrosis by NFS and FIB-4 | Low risk of advanced fibrosis in subjects with ketonuria |
| Lee et al. [8] | 435 type 2 diabetic patients | Fasting plasma βHB | Non-invasive NAFLD indices | Low risk of NAFLD indices in subjects with intact ketogenesis |
| Kotronen et al. [60] | 58 nondiabetic patients | NAFLD by 1H-MRS | Fasting plasma βHB | Comparable between NAFLD and control |
| Sanyal et al. [57] | 6 NAFLD, 6 NASH, 6 lean healthy subjects | NAFLD by liver biopsy | Fasting plasma βHB | High fasting βHB in NAFLD and NASH |
| Chalasani et al. [59] | 37 nondiabetic patients | NASH by liver biopsy | Fasting plasma βHB | High fasting βHB in NASH |
| Bugianesi et al. [58] | 18 non-obese, non-diabetic patients | NAFLD by liver biopsy | Fasting plasma βHB | High fasting βHB in NAFLD |
| Post et al. [61] | 6297 general population | NAFLD by FLI score | Fasting ketone bodies, including total, βHB, acetoacetate, acetone | High fasting ketone bodies in NAFLD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, J.; Lee, B.-W. Association between Impaired Ketogenesis and Metabolic-Associated Fatty Liver Disease. Biomolecules 2023, 13, 1506. https://doi.org/10.3390/biom13101506
Bae J, Lee B-W. Association between Impaired Ketogenesis and Metabolic-Associated Fatty Liver Disease. Biomolecules. 2023; 13(10):1506. https://doi.org/10.3390/biom13101506
Chicago/Turabian StyleBae, Jaehyun, and Byung-Wan Lee. 2023. "Association between Impaired Ketogenesis and Metabolic-Associated Fatty Liver Disease" Biomolecules 13, no. 10: 1506. https://doi.org/10.3390/biom13101506